

Abstract
F1-20/AP-3 is a synapse-specific phosphoprotein. In this study we characterize the ability of bacterially expressed F1-20/AP-3 to bind and assemble clathrin cages. We find that both of two bacterially expressed alternatively spliced isoforms of F1-20/AP-3 can bind and assemble clathrin as efficiently as preparations of F1-20/AP-3 from bovine brain. This establishes that the clathrin assembly activity found in F1-20/AP-3 preparations from brain extracts is indeed encoded by the cloned gene for F1-20/AP-3. It also demonstrates that post-translation modification is not required for activation of the clathrin binding or assembly function of F1-20/AP-3. Ultrastructural analyses of the clathrin cages assembled by bacterially expressed F1-20/AP-3 reveals a strikingly narrow size distribution. This may be important for the regulation of quanta1 size during neurotransmission. We also express the 33 kD NH2-terminus of F1-20/AP-3 in E. coli, and measure its ability to bind to clathrin triskelia, to bind to clathrin cages, and to assemble clathrin triskelia into clathrin cages. It has been suggested that the 33 kD NH2-terminus of F1-20/AP-3 constitutes a clathrin binding domain. We find that the bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 binds to clathrin triskelia, fails to bind to preassembled clathrin cages, and is not sufficient for clathrin assembly. The finding that the 33 kD NH2-terminus of F1-20/AP-3 binds to clathrin triskelia but fails to assemble clathrin triskelia into clathrin cages is consistent with the published proteolysis studies. The finding that the 33 kD NH2-terminus of F1-20/ AP-3 fails to bind to clathrin cages is novel and potentially important. It is clear from these experiments that the 33 kD NH2-terminus of F1-20/AP-3 is sufficient to carry out some aspects of clathrin binding; however it appears that defining the regions of the protein involved in clathrin binding and assembly may be more complex than originally anticipated.
Keywords: clathrin, endocytosis, synaptic vesicle biogenesis, synaptic vesicle exocytosis, synaptic vesicle recycling
INTRODUCTION
Synaptic transmission is the basic process which underlies cell-cell communication in the nervous system (Jessell and Kandel, 1993). Consequently, there has been considerable interest in understanding how synaptic transmission is effected and regulated at the molecular level. An approach taken by a number of investigators has been to carry out a molecular dissection of the synapse. To date, a large number of synaptic molecules have been successfully cloned and sequenced. These include components of the synaptic cytoskeleton, synaptic vesicle membrane, presynaptic plasma membrane, synaptic basal lamina, and postsynaptic membrane (Sudhof and Jahn, 1991; Hall and Sanes, 1993). Exocytosis and endocytosis, while major steps of synaptic transmission, are also general processes used by essentially all eukaryotic cells. Recently broad based approaches to studying membrane trafficking have converged with the neurobiological approaches (Bennett and Scheller, 1993; Sollner et al., 1993; Whiteheart et al., 1993; Bennett and Scheller, 1994). As part of the effort to molecularly characterize the synapse, we reported the characterization and cloning of the synapse-specific phosphoprotein F1–20 (Sousa et al., 1990, 1992; Zhou et al., 1992). We overexpressed F1–20 and reported the finding that F1–20 is identical to the clathrin assembly protein AP-3 (Zhou et al., 1993). Clathrin-coated vesicles are involved in a large number of membrane trafficking events in cells including receptor-mediated endocytosis (Trowbridge, 1991), traffic between the trans-golgi network and other organelles, and the biogenesis (Regnier-Vigouroux et al., 1991) and recycling of synaptic vesicles (Heuser and Reese, 1973; Maycox et al., 1992; Herskovits et al., 1993; van der Bliek et al., 1993).
A major approach towards elucidating the mechanism of clathrin mediated endocytosis has involved the biochemical characterization of the cytoplasmic coat of clathrin coated vesicles (reviewed in Kirchhausen, 1993). The coats of clathrin coated vesicles were found to consist of clathrin (Pearse, 1975) and a collection of assembly proteins which could mediate the assembly of clathrin into cages (Keen et al., 1979). The assembly proteins AP-1 and AP-2 promote clathrin assembly and are heterotetramers (Robinson, 1992). Two other proteins which promote the assembly of clathrin and are components of clathrin coated vesicles are auxilin (Ahle and Ungewickell, 1990) and AP-3. AP-3 has now been independently described by four different groups as pp155 (Keen and Black, 1986), AP180 (Ahle and Ungewickell, 1986), NP185 (Kohtz and Puszkin, 1988), and F1–20 (Sousa et al., 1990, 1992). PP155, AP180, and NP185 were shown to be the same protein and renamed AP-3 (Murphy et al., 1991). F1–20 and AP-3 were then shown to be identical (Morris et al., 1993; Zhou et al., 1993). The characterization of F1–20/AP-3 and auxilin has lagged behind that of the AP-1 and AP-2 assembly proteins. The “assembly protein fraction” extracted from clathrin coated vesicles (Keen et al., 1979) had actually contained a mixture of AP-1, AP-2, F1–20/ AP-3, and auxilin. However, since F1–20/AP-3 co-migrates with clathrin on SDS-PAGE, it had been assumed to be contaminating clathrin until 1986 (Ahle and Ungewickell, 1986; Keen and Black, 1986). Auxilin comigrates with the 100 kD subunits of the AP-1 and AP-2 complexes and was not recognized as a distinct protein until 1990 (Ahle and Ungewickell, 1990). The one thing all of these proteins have in common is the ability to bind and assemble clathrin. When the clathrin assembly activities of AP-1, AP-2, F1–20/AP-3, and auxilin were compared utilizing a quantitative assembly assay (Lindner and Ungewickell, 1992), it was found that F1–20/ AP-3 is ∼4 times more effective at promoting clathrin assembly than the other assembly proteins. AP-1 and AP-2 are both tetramers which are expressed in many tissues. F1–20/AP-3 and auxilin are both monomers. Western blot analysis revealed that auxilin is found in brain, but not in liver or adrenal gland (Ahle and Ungewickell, 1990). Examination of an even wider range of tissues and cell lines revealed that F1–20/AP-3 is neuronal specific (Kohtz and Puszkin, 1988; Sousa et al., 1992; Morris et al., 1993). F1–20/AP-3 is the only assembly protein to have been shown to be synapse specific (Perry et al., 1991; Sousa et al., 1992). For this reason, we suggested that F1–20/AP-3 is involved in synaptic vesicle biogenesis and recycling (Zhou et al., 1993).
The characterization of the biochemical properties of F1–20/AP-3 have revealed that F1–20/AP-3 functions as a monomer which interacts with clathrin triskelia to assemble them into clathrin cages (Ahle and Ungewickell, 1986; Prasad and Lippoldt, 1988). F1–20/AP-3 binds to clathrin heavy chains independent of light chains (Lindner and Ungewickell, 1991; Murphy and Keen, 1992). Removal of the terminal globular domain of the clathrin heavy chain from clathrin cages by proteolysis abolished the binding of F1-20/AP-3 to the cages; however no binding could be observed to either isolated hubs or isolated terminal domains (Murphy and Keen, 1992). F1–20/AP-3 has also been demonstrated to promote the binding of clathrin to de-coated vesicles (Prasad and Lippoldt, 1988). F1–20/AP-3 is an unusually acidic (Ahle and Ungewickell, 1986; Keen and Black, 1986; Zhou et al., 1992) phosphoprotein (Keen and Black, 1986; Morris et al., 1990; Zhou et al., 1992), which migrates anomalously on SDS-PAGE (Ahle and Ungewickell, 1986; Prasad and Lippoldt, 1988; Murphy et al., 1991; Zhou et al., 1992). A number of different alternatively spliced isoforms of F1–20/AP-3 have been described (Zhou et al., 1992; Morris et al., 1993; Zhou et al., 1993).
The ability of purified bovine brain F1–20/AP-3 to bind and assemble clathrin has been well documented in the literature (Ahle and Ungewickell, 1986; Keen and Black, 1986; Prasad and Lippoldt, 1988; Lindner and Ungewickell, 1992). However, a concern of any biochemical study which utilizes materials prepared from tissue extracts is whether the activity one is measuring is solely attributable to the major protein product of the preparation, or whether other protein components are required which are present in small amounts, and thereby undetected by the method used for the assessment of the purification. For example, AP-3 was itself undetected for some time because it co-migrates with clathrin heavy chain (Ahle and Ungewickell, 1986). Consequently in this study we examine whether bacterially expressed F1–20/AP-3 is capable of binding and assembling clathrin. We find that both of two bacterially expressed alternatively spliced isoforms of F1–20/AP-3 can bind and assemble clathrin as efficiently as bovine brain F1–20/ AP-3. This establishes that the clathrin assembly activity found in F1–20/AP-3 preparations from brain extracts is indeed encoded by the cloned gene for F1–20/AP-3. It also demonstrates that post-trans lational modification is not required for activation of the clathrin binding or assembly function of F1–20/AP-3. Ultrastructural analyses of the clathrin cages assembled by bacterially expressed F1–20/AP-3 reveals a strikingly narrow size distribution. This may be important for the regulation of quantal size during neurotransmission.
The ease with which large amounts of functional F1–20/AP-3 can be prepared from this bacterial expression system should facilitate structure-function studies of this protein. As an example of this we address the question of where the clathrin binding activity of F1–20/AP-3 may reside. Other investigators have assigned the 33 kD NH2-terminus of F1–20/AP-3 as a clathrin binding domain (Murphy et al., 1991; Morris et al., 1993). We find that the bacterially expressed 33 kD NH2-terminus of F1–20/AP-3 binds to clathrin triskelia, fails to bind to preassembled clathrin cages and is not sufficient for clathrin assembly. The finding that the 33 kD NH2-ter-minus of F1–20/AP-3 binds to clathrin triskelia but fails to assemble clathrin triskelia into clathrin cages is consistent with the published proteolysis studies (Murphy et al., 1991). The finding that the 33 kD NH2-terminus of F1–20/AP-3 fails to bind to clathrin cages is novel and potentially important.
MATERIALS AND METHODS
Materials
Bovine brains were obtained from a local slaughterhouse. All chemicals were from USB or Sigma. Protease inhibitors were from Boehringer Mannheim. Cen-tricon-30 and centricon-10 were from Amicon. Quick concentrator-10 and concentrator-30 were from Milli-pore. ECL reagents were from NEN/DuPont. All chromatography resins were from Pharmacia. Bio Image system was from Millipore. All other materials were obtained as described previously (Zhou et al., 1993).
Methods
Overexpression of GST-F1–20/AP-3 (AS15−), GST-F1–20/AP-3 (AS15+), 33 kDa NH2-terminus of F1–20/AP-3, and GST
GST-F1–20/AP-3 (AS15−) was expressed from plasmid pGEX3X–F1–20(AS15−) [originally called 5pGEX3X (Zhou et al., 1993)], as described previously (Zhou et al., 1993). During the course of cloning the entire open reading frame into pBluescript II SK, 20 independent clones were selected. One of them [pBSIISK-F1–20(AS15+)] was detected by restriction mapping as containing the 15 nucleotide (nt) mini-exon, which was confirmed by sequencing. The EcoRV-StuI fragment which contains the 15 nt mini-exon was cut out from this clone and gel purified. The EcoRV-STuI fragment in plasmid pGEX3X–F1–20(AS15−) was replaced by the fragment containing the 15 nt mini exon. This new clone pGEX3X–F1–20(AS15+) was confirmed by restriction mapping. A construct expressing the NH2-ter-minal 33 kDa domain was constructed by PCR using two primers (33k5′ 5′-AGCGAGCTCCCCGGGATGTCG-GGCCAAACGCTCACG-3′ and 33k3′ 5′-GCTCTCG-AGCCCGGGTCACTTACTTAGTGGAGAGGGAGC-3′) flanking the 5′-912 nt of the open reading frame and plasmid pGEX3X–F1–20(AS15−) as the template. Notice that an Xmal site flanking the coding region was introduced into both primers. A stop codon TGA was also introduced into the 3′ primer in front of the Xmal site. The PCR product was gel purified, digested overnight with Xmal and then gel purified again. The digested PCR fragment was subcloned into pGEX3X at the Xmal site. Clones with the correct direction were selected by restriction mapping. Three independent PCR reactions were carried out and their products were each subcloned. All three independent 33 kDa subclones were used in the following experiments.
Three different recombinant plasmids [pGEX3X–F1–20(AS15−); pGEX3X–F1–20(AS15+); pGEX3X–F1–20N33kDa] and parental plasmid pGEX3X without any insert (which will produce E. coli GST protein) were each transformed into BL21 cells. Suitably transformed BL21 cells were grown and induced as described (Zhou et al., 1993) except that BL21 harboring pGEX3X–F1–20(AS15+) was collected 2 hr post IPTG treatment instead of at 6 hr. Unless otherwise specified, the following steps were all carried out at 4°C. Cells were collected by centrifugation at 5,000g for 15 min, resuspended in 0.02 culture volume of E. coli resuspension buffer (130 mM NaC1, 10 mM sodium phosphate, pH 7.4, 0.1 mM PMSF, 100 mM EDTA, 0.1% 2-Mercaptoethanol, 5% Glycerol), and sonicated for 15 sec at 30% power. Triton X-100 was added to a final concentration of 1%; cell debris was pelleted by centrifugation for 5 min at 13,600g. Supernatants were mixed with an equal volume of Glutathione Sepharose 4B (Pharmacia, Piscataway, NJ) pretreated with equilibration buffer (10 mM sodium phosphate, 130 mM NaCl, 0.1 M EDTA, 0.1% 2-Mercaptoethanol, 5% Glycerol, 0.1 mM PMSF, pH 7.4) for 1 hr, followed by extensive washing with equilibration buffer. GST-F1–20/AP-3 (AS15−) and GST-F1–20/AP-3 (AS15+) were eluted by washing with 10 volumes of elution buffer (0.5 M Tris, 2 mM EDTA, 0.1% 2-Mercaptoethanol, 5% Glycerol, 0.1 mM PMSF, 15 mM reduced glutathione, pH 8.0) and concentrated by centrifugation in a centricon-30 Amicon, Beverly, MA) to 4–6 mg/ml. GST was eluted similarly, and concentrated by centrifugation in a centricon-10. The 33 kD NH2-terminus was eluted by incubating the resin in an equal volume of equilibration buffer supplemented with 1 mM CaCl2 and 0.01 mg/ml endoprotease Factor Xa (Boeh-ringer, Mannheim, Germany) at 25°C for 6 hr. The reaction was terminated by the addition of EGTA to 20 mM. This resulted in the removal of GST from the 33 kD NH2-terminus. Free 33 kD NH2-terminus was recovered from the resin by washing with 2 volumes of ice cold equilibration buffer, followed by concentration to 4–6 mg/ml in a centricon-30. All protein solutions were cen-trifuged for 1 hr at 100,000g to remove preformed protein aggregates before use. All expressed proteins were monitored on SDS-PAGE by silver staining. GST-F1–20/AP-3 (AS 15−) and GST-F1–20/AP-3 (AS15+) were also monitored by western blot analysis with the F1–20 Mab as described previously (Zhou et al., 1993). Concentrations of bovine brain F1–20/AP-3 were determined spectrophotometrically using the extinction coefficient 1 A280 = 2 mg/ml (Lindner and Ungewickell, 1991). To correct for proteolysis of the the full-length bacterially expressed proteins, serial dilutions of bovine brain F1–20/AP-3, GST-F1–20/AP-3 (AS15−), and GST-F1–20/ AP-3 (AS15+) were run on 10–15% SDS polyacramide gel followed by silver staining and quantitation by Millipore Bio Image system. Bovine brain F1–20/AP-3 was then used as a standard to determine the concentration of GST-F1–20/AP-3 (AS15−) and GST-F1–20/AP-3 (AS15+). Concentrations of the 33 kD NH2-terminus of F1–20/AP-3 and GST were determined using the BioRad protein assay system.
Preparation of bovine brain clathrin and F1–20/ AP-3
Bovine brain coated vesicle proteins were rapidly prepared as described previously (Zhou et al., 1993). Clathrin and F1–20/AP-3 were further purified from the extract as described (Ahle and Ungewickell, 1986), with the addition of 0.1 mM PMSF to all buffers. SDS-PAGE analysis of the purified clathrin revealed three silver stained bands, corresponding in apparent molecular weight to clathrin heavy chain, and to the two clathrin light chains. Clathrin was determined to be free of contaminating F1–20/AP-3 by western blot analysis with the F1–20 Mab utilizing the ECL detection system as described previously (Zhou et al., 1993). One cycle of assembly-disassembly was carried out as follows. Clathrin triskelia at 4 mg/ml were assembled as described (Morris et al., 1993). Cages were pelleted by centrifugation at 100,000g at 4°C for 1 hr. Cages were then disassembled by incubating in column buffer (0.5 M Tris, 2 mM EDTA, 1 mM DTT, 0.02% NaN3, 5% glycerol, and 0.1 mM PMSF, pH 7.0) at 37°C for 15 min. Protein concentrations were determined spectrophotometrically using extinction coefficients of 1 A280 = 1 mg/ml for clathrin and 1 A280 = 2 mg/ml for F1–20/AP-3 (Lindner and Ungewickell, 1991).
Clathrin cage binding assays
The clathrin cage binding assay was performed as described (Lindner and Ungewickell, 1991; Morris et al., 1993). Clathrin cages were assembled in the presence of calcium containing buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgCl2, and 3 mM CaCl2, 0.1 mM PMSF, pH 6.5) as described (Morris et al., 1993). All proteins used in the following experiment were dialyzed into tartrate buffer (100 mM Na-tartrate, pH 7.0, 10 mM HEPES, 1 mM EGTA, 0.5 mM MgCl2, 0.02% NaN3, 0.1 mM PMSF) or isolation buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgCl2, 0.1 mM PMSF, pH 6.7) before use. Binding of the two full length fusion proteins were carried out by incubating 0.4 mg/ml clathrin cages with the fusion protein at 0.08 mg/ ml, 0.16 mg/ml, or 0.24 mg/ml in 50 |xl tartrate buffer on ice for 45 min. Binding of the 33 kD NH2-terminus of F1–20/AP-3 was carried out by incubating 0.8 mg/ml clathrin cages with 0.24 mg/ml 33 kD NH2-terminus in 50 µl tartrate buffer and isolation buffer respectively on ice for 45 min. All samples were centrifuged at 13,600g for 3 min to remove protein aggregates. Clathrin cages were then collected by ultracentrifugation for 20 min in a Beck-man TLA-100 rotor at 100,000g, followed by analysis of the pellet and supernatant fractions by 10–15% gradient SDS-PAGE. The two full length fusion proteins were detected by western blotting using the F1–20 Mab as described previously (Zhou et al., 1993). The 33 kD NH2-terminus of F1–20/AP-3 and clathrin were detected by Coomassie brilliant blue staining.
Clathrin triskelia binding assay
Clathrin-Sepharose was prepared according to Keen (1987), the final product gave 1.4 mg clathrin/1 ml Sepharose-4B. Binding of the 33 kD NH2-terminus of F1–20/AP-3 to clathrin-Sepharose was performed as described (Murphy et al., 1991), with the following modifications: 0.5 ml clathrin-Sepharose were mixed with 15 µg 33 kD NH2-terminus of F1–20/AP-3 in 500 µl isolation buffer. The mixture was rocked at 4°C for 2 hr. The Sepharose was then allowed to settle and the supernatant was removed (this is called the flow-through); 500 µ1 isolation buffer were used to wash the Sepharose for 15 min at room temperature. The washing was repeated three times. The Sepharose was then eluted three times with 0.5 ml 0.5 M Tris, 0.1 mM PMSF (pH 7.0) at 37°C for 15 min. Flow-through, washes, and eluates were all concentrated using Millipore quick concentrator-10s, and each sample was brought to 20 µl in IX SDS sample buffer. All samples were analyzed by SDS-PAGE, followed by silver staining. As a negative control, underivatized Sepharose-4B was used in the same binding assay with the 33 kD NH2-terminus of F1–20/AP-3. As another negative control, GST protein was used in the same binding assay with clathrin-Sepharose.
Quantitative factor-dependent clathrin assembly assay
The quantitative clathrin assembly assay was carried out as described (Lindner and Ungewickell, 1992; Morris et al., 1993); 2 mg/ml purified bovine brain clathrin triskelia were dialyzed overnight at 4°C against isolation buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgCl2,0.1 mM PMSF, pH 6.7) alone, or in the presence of 2 mg/ml GST as negative controls; 2 mg/ml clathrin triskelia were dialyzed with 2 mg/ml purified bovine brain F1–20/AP-3 as a positive control; 2 mg/ml clathrin triskelia were titrated with the following proteins, respectively, over a concentration range of 0–4 mg/ml: GST-F1–20/ AP-3 (AS15−), GST-F1–20/AP-3 (AS15+), the 33 kD NH2-terminus of F1–20/AP-3, and E. coli GST protein. Samples in triplicate were dialyzed against isolation buffer at 4°C overnight. All samples were centrifuged at 13,600g for 3 min to remove protein aggregates. Clathrin cages were then collected by ultracentrifugation for 20 min in a Beckman TLA-100 rotor at 100,000g, followed by analysis of the pellet and supernatant fractions by 10–15% gradient SDS-PAGE. Proteins were visualized by silver staining. The distribution of clathrin light chains between the pellet and supernatant fractions was used as the criteria of assembly since clathrin heavy chain comigrates with the two full length fusion proteins. Both clathrin light chains were quantitated using the Millipore Bio Image system with 3cx scanner (Millipore, Bedford, MA). Multiple scans at varied loadings were performed to insure linearity; % Assembly equals [pellet/ (pellet + supernatant)] × 100.
Ultrastructural analysis of clathrin cage assembly
Negative staining electron microscopy was performed as described (Crowther and Pearse, 1981). Clathrin cages assembled as described above were diluted with isolation buffer to a final clathrin concentration of 0.4 mg/ml. Samples were immediately deposited on a thin carbon film for 2 min. The film was washed for 20 sec in ddH2O, followed by incubation in 1% aqueous uranyl acetate for 3 min. Specimens were examined in a Zeiss TEM902 electron microscope using an accelerating voltage of 80 kV. Images were taken at a nominal magnification of × 54,504 and × 21,816. Cages diameters were measured on the electron micrographs at both magnifications; 393 cages reassembled in the presence of 2 mM free calcium and 306 cages reassembled in the presence of 2 mg/ml GST-F1–20/AP-3 (AS15−) were measured.
RESULTS
Clathrin Cage Binding Assays
We expressed two alternatively spliced isoforms of F1–20/AP-3 (AS15+ and AS15−), as well as the 33 kD NH2-terminus of F1–20/AP-3 in E. coli. The quality of the full length GST fusion protein and bovine brain AP-3 has been documented (Zhou et al., 1993). All three proteins were expressed at high levels from the expression vector pGEX3X as fusions with the 26 kDa E. coli protein glutathione-S-transferase (GST). When necessary, the GST portion was removed utilizing the protease factor Xa. The ability of these three proteins to bind to pre-assembled clathrin cages was assayed utilizing the cage binding assay as described by Lindner and Un-gewickell (1991). Clathrin cages pellet at 100,000g. F1–20/AP-3, as long as it is soluble and free of aggregates, does not pellet at 100,000g. When F1–20/AP-3 is mixed with the pre-assembled cages, bound F1–20/AP-3 cosediments with the cages at 100,000g, while free F1–20/ AP-3 remains in the supernatant. After running the reactions, the samples are centrifuged briefly at 13,600g to remove any non-specific aggregates that formed during the course of the reaction. Then the reactions are centrifuged at 100,000g to separate bound from free F1–20/ AP-3. In this type of assay, an important control is the demonstration that F1–20/AP-3 is free of 100,000g sedimentable aggregates by taking it through the assay in the absence of clathrin. In the case of the full-length F1–20/ AP-3 protein, we monitored F1–20/AP-3 with an anti-F1–20 Mab, since F1–20/AP-3 co-migrates with clathrin heavy chain on SDS-PAGE and is therefore indistinguishable by Coomassie blue staining (Fig. 1). In the case of the 33 kD NH2-terminus, we monitored it by Coomassie blue staining (Fig. 2). The 33 kDa NH2-terminus was not monitored by immunoblot analysis since it does not contain the determinant recognized by the F1–20 Mab.
Fig. 1.

Bacterially expressed GST-F1-20/AP-3 (AS15−) and GST-F1-20/AP-3 (AS15+) binds to clathrin cages. Either 80 µg/ml (samples 1 and 2), 160 µg/ml (samples 3 and 4), or 240 µg/ml (samples 5 and 6) of either GST-F1-20/AP-3 (AS15−) (A) or F1-20/AP-3 (AS15 +) (B) were incubated in the absence (−) or presence (+) of 400 µg/ml clathrin cages for 45 min at 4°C in tartrate buffer. Following a low-speed spin to remove non-specific aggregates, the clathrin cages were pelleted by ultracentrifugation at 100,000g. The pellet (P) and supernatant (S) fractions were analyzed by SDS-PAGE, followed by western blot analysis with the F1-20 Mab. The uppermost bands are the intact fusion proteins. The lower bands are proteolytic fragments. [Note, we found that F1-20/AP-3 (AS15 +) is more susceptible to aggregation than F1-20/AP-3 (AS15−); hence even though equal amounts of protein were used in the experiments shown in A and B, more aggregates were removed in the low-speed spin in the experiment which utilized F1-20/AP-3 (AS15+) (panel B) than in the experiment which utilized F1-20/AP-3 (AS15−) (A).]
Fig. 2.
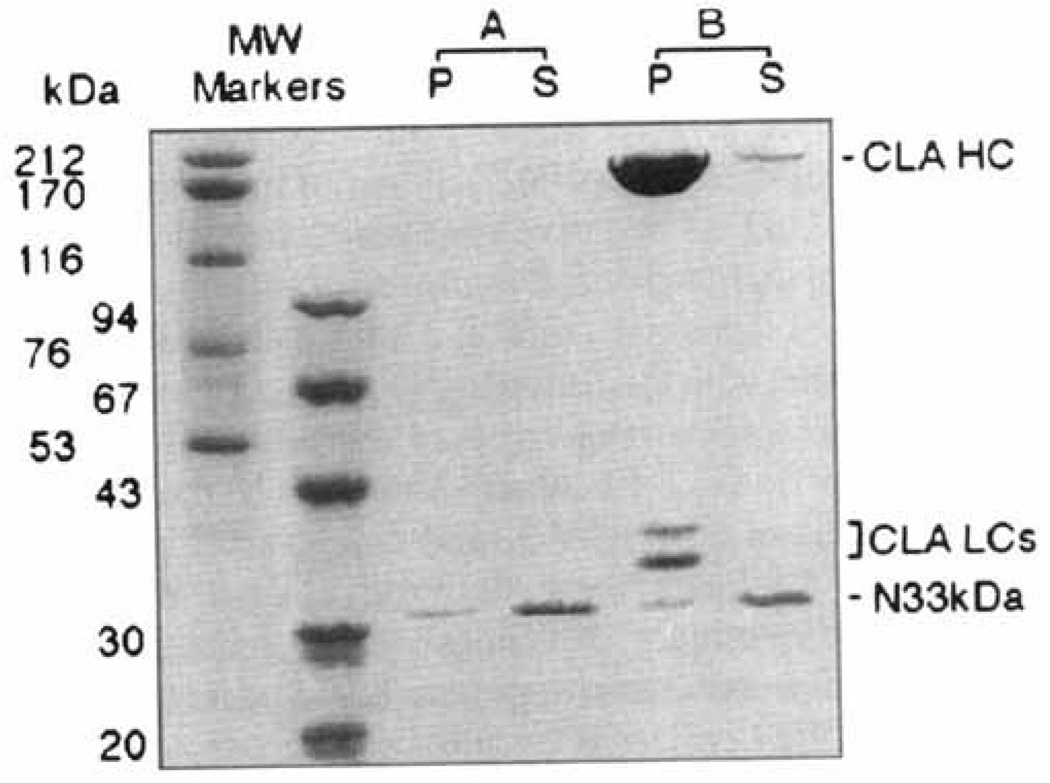
The bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 does not bind to clathrin cages; 240 µg/ml of the bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 was incubated in the absence (A) or presence (B) of 800 µg/ml clathrin cages for 45 min at 4°C in isolation buffer. Following a low-speed spin to remove non-specific aggregates, the clathrin cages were pelleted by ultracentrifugation at 100,000g. The pellet (P) and supernatant (S) fractions were analyzed by SDS-PAGE, followed by Coomassie Blue staining. The positions of clathrin heavy chain (CLA HC), clathrin light chains (CLA LCs), and the 33 kD NH2-terminus (N33kDa) are indicated. Pellets were loaded at triple concentrations. Sizes of the molecular weight markers (Pharmacia) are indicated. When this experiment was repeated with an additional two independent clones expressing the 33 kD NH2-terminus, the results were identical (data not shown).
Figure 1A shows the results obtained with F1–20/ AP-3 (AS15−), the most naturally abundant isoform of F1-20/AP-3. Essentially all of the F1-20/AP-3 remained in the supernatant in reactions that contained 80 µg/ml of F1–20/AP-3 alone (Fig. 1A, sample 1). When 400 µg/ml of clathrin cages were included, essentially all of the F1–20/AP-3 shifted to the pellet (Fig. 1A, sample 2), indicating full binding to the cages. These conditions had been found to be optimal for the binding of bovine F1-20/AP-3 to clathrin cages (Lindner and Ungewickell, 1991). However, as a positive control for the experiments described below with the 33 kD NH2-terminus, we also examined higher concentrations of F1-20/AP-3. At higher protein concentrations some aggregation was unavoidable, but nearly all of the added F1–20/AP-3 remained in the supernatant in reactions lacking clathrin cages (Fig. 1A, reactions 3 and 5) and shifted dramatically to the pellet (Fig. 1A, reactions 4 and 6) in the reactions containing clathrin cages. Note that in the 240 µg/ml reactions some F1-20/AP-3 remained in the supernatant, as the binding sites on the clathrin were saturated. Because F1–20/AP-3 is extremely protease sensitive (Ahle and Ungewickell, 1986; Prasad and Lip-poldt, 1988), multiple immunoreactive bands are always observed. This experiment shows that bacterially expressed F1-20/AP-3 binds clathrin as well as F1-20/AP-3 purified from bovine brain.
We examined whether the presence of the 15 nucleotide mini-exon, which introduces five amino acids into the protein between amino acids 714 and 715, has any effect on clathrin binding. A similar set of cage binding assays was carried out with F1-20/AP-3 (AS15+) (Fig. IB) as had been carried out with F1-20/AP-3 (AS15−) (Fig. 1A). The results were very similar. In reactions lacking clathrin, essentially all of the F1-20/AP-3 remained in the supernatant (Fig. IB, reactions 1,3, and 5). In reactions containing clathrin, F1-20/AP-3 shifted from the supernatant to the pellet (Fig. IB, reactions 2,4, and 6). This indicated that both the AS15− and AS15+ isoforms of F1-20/AP-3 bind clathrin.
We then set out to determine whether the 33 kD NH2-terminus of F1-20/AP-3 was sufficient for clathrin cage binding (Fig. 2). We found no measurable binding of the GST-33 kD-NH2-terminus fusion protein to clathrin cages, even at the highest possible protein concentrations in which the assay was executable. To rule out a potential inhibitory effect by the GST, the experiment shown in Figure 2 was done with protein from which the GST had been removed by factor Xa cleavage. Whether the 33 kDa NH2-terminal of F1-20/AP-3 was incubated in the absence (Fig. 2A), or presence (Fig. 2B) of pre-assembled clathrin cages, essentially all of the protein remained in the supernatant. This indicated that the 33 kDa NH2-terminal of F1-20/AP-3 is not sufficient for clathrin cage binding. This experiment was carried out both in isolation buffer (Fig. 2) and tartrate buffer. Ten independent reactions were carried out, using three independent clones expressing the 33 kDa NH2-terminus of F1–20/AP-3, and the results were always the same (data not shown).
Clathrin Triskelia Binding Assay
We then assayed the binding of the 33 kD NH2-terminus of F1–20/AP-3 to clathrin triskelia as described by Murphy and colleagues (Murphy et al., 1991). We found that the bacterially expressed 33 kD NH2-terminus of F1–20/AP-3 is capable of binding clathrin triskelia. As is shown in Figure 3A, the majority of the 33 kD NH2-terminus is present in the 0.5 M Tris eluates from the clathrin-Sepharose. The possibility that this binding is due to non-specific interactions between the clathrin-Sepharose and the 33 kD NH2-terminus of F1–20/AP-3 was ruled out by two experiments. When an unrelated protein, E.coli GST, was used in the same binding assay, there was no detectable E.coli GST present in the 0.5 M Tris eluate (Fig. 3C). Furthermore, when underivatized Sepharose-4B was used instead of clathrin-Sepharose in the same binding assay, it failed to retain any 33 kD NH2-terminus (Fig. 3B). Three independent reactions were carried out using three clones expressing the 33 kD NH2-terminus of F1–20/AP-3, and the results were essentially the same (data not shown). From these studies, we conclude that the 33 kD NH2-terminus of F1–20/AP-3 binds specifically to clathrin triskelia, consistent with the published proteolysis studies which utilized the bovine protein (Murphy et al., 1991). This also indicated that the bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 is functionally active.
Fig. 3.

The bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 binds specifically to clathrin triskelia; 15 µg of the bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 was incubated with 0.5 ml clathrin-Sepharose in 0.5 ml isolation buffer at 4°C for 2 hr (A), and binding was monitored by batch analysis, as described in Methods. Fraction 1 is the flow-through; fractions 2,3,4 are washes with isolation buffer; and fractions 5,6,7 are eluates with 0.5 M Tris (pH 7.0). All samples were analyzed by SDS-PAGE, followed by silver staining. Negative controls were carried out by incubating 15 µg bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 with 0.5 ml underivatized Sepharose (B), and by incubating 15 µg E. coli GST protein with 0.5 ml clathrin-Sepharose (C).
Clathrin Assembly Assays
Factor-dependent clathrin assembly assays were performed as described (Lindner and Ungewickell, 1992). Clathrin triskelia are dialyzed together with a putative assembly protein in a buffer in which clathrin alone will not assemble into cages. Following the dialysis, samples are first briefly centrifuged at 13,600g to remove nonspecific aggregates and then assembled cages are separated from free triskelia by centrifugation at 100,000g. Supernatant and pellet fractions are analyzed by SDS-PAGE followed by silver staining. While assembly can be monitored by examining the distribution of clathrin heavy and/or light chains, because clathrin heavy chain and F1-20/AP-3 co-migrate, we followed assembly by monitoring the doublet of light chains. Quantitative analysis of all data shown in Figures 4 and 5 was performed using a Millipore BioImage System with 3cx scanner.
Fig. 4.

Bacterially expressed GST-F1-20/AP-3 (AS15−) and GST-F1-20/AP-3 (AS15+) assemble clathrin triskelia into cages, while the bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 is not sufficient for clathrin assembly; 2 mg/ml clathrin triskelia were dialyzed overnight at 4°C against Isolation buffer alone (A), or with the addition of either 2 mg/ml GST (B), 2 mg/ml purified bovine brain F1-20/AP-3 (C), 2 mg/ml GST-F1-20/AP-3 (AS15−) (D), 2 mg/ml GST-F1-20/AP-3 (AS15+) (E), or 2 mg/ml 33 kD NH2-terminus of F1-20/AP-3 (F). The three test proteins at 2 mg/ml were also dialyzed overnight at 4°C against isolation buffer without clathrin triskelia: GST-F1-20/AP-3 (AS 15−) (G), GST-F1-20/AP-3 (AS15+) (H). Following a low-speed spin to remove non-specific aggregates, newly assembled clathrin cages were pelleted by ultracentrifugation at 100,000g. The pellet and supernatant fractions were analyzed by SDS-PAGE, followed by silver staining. The distribution of the clathrin light chains between the pellet and supernatant fractions were used to evaluate assembly, since the clathrin heavy chain co-migrates with F1-20/AP-3.
Fig. 5.

Quantitative factor-dependent assembly of clathrin triskelia; 2 mg/ml clathrin triskelia were dialyzed overnight at 4°C against isolation buffer alone, or with the addition of the indicated concentrations of either GST-F1-20/AP-3 (AS15−) (open triangle), GST-F1-20/AP-3 (AS15+) (solid circle), the 33 kD NH2-terminus of F1-20/AP-3 (solid triangle), or the E. coli GST protein (open circle). Following a low-speed spin to remove non-specific aggregates, newly assembled clathrin cages were pelleted by ultracentrifugation at 100,000g. The pellet and supernatant fractions were analyzed by SDS-PAGE, followed by silver staining. The distribution of the clathrin light chains between the pellet and supernatant fractions were quantitated using a Millipore BioImage System with 3cx scanner. Multiple scans at varied loadings were performed to ensure linearity. Each data point on the plot is the average assembly determined from three independent assembly assays. The error bars indicate one standard deviation from this average.
When clathrin triskelia at 2 mg/ml were sedimented following dialysis in the absence of assembly proteins, 89% of the clathrin was found in the supernatant (Fig. 4A). Likewise, when clathrin triskelia were sedimented following dialysis in the presence of 2 mg/ml GST, 91% of the clathrin was found in the supernatant (Fig. 4B). When clathrin triskelia were sedimented following dialysis in the presence of 2 mg/ml purified bovine F1-20/ AP-3, 47% of the clathrin was found in the supernatant and 53% of the clathrin was found in the pellet, indicative of cage formation (i.e., clathrin assembly) (Fig. 4C). When clathrin triskelia were sedimented following dialysis in the presence of 2 mg/ml bacterially expressed GST-F1–20/AP-3 (AS15−) (Fig. 4D), or 2 mg/ml bacterially expressed GST-F1–20/AP-3 (AS15+) (Fig. 4E), 56% and 60% of the clathrin was found in the respective pellets, indicative of clathrin assembly comparable to that of F1-20/AP-3 purified from bovine brain (Fig. 4C). When clathrin triskelia were sedimented following dialysis in the presence of 2 mg/ml bacterially expressed 33 kD NH2-terminus of F1–20/AP-3 (Fig. 4F), 91% of the clathrin remained in the supernatant, as in the negative controls (Fig. 4A,B).
Experiments similar to those shown in Figure 4 were carried out over a wider range of protein concentrations in triplicate and plotted in Figure 5 in order to provide a more quantitative assessment of the ability of the three bacterially expressed proteins to assemble clathrin. Both GST-F1–20/AP-3 (AS15−) and GST-F1–20/AP-3 (AS15+) reach peak assembly activity between 1 and 2 mg/ml. The 33 kD NH2-terminus of F1–20/AP-3 does not show statistically significant assembly activity above the levels seen by clathrin alone or clathrin mixed with E. coli GST protein, even at 4 mg/ml. We conclude that bacterially expressed F1–20/AP-3 is able to assemble clathrin, while the 33 kD NH2-terminus is not sufficient for clathrin assembly.
Ultrastructural Analysis of Assembled Clathrin Cages
We analyzed the morphology of the cages assembled by the bacterially expressed proteins F1-20/AP-3 (AS15−) and F1–20/AP-3 (AS15+) (Fig. 6) by negative staining electron microscopy. When clathrin triskelia were dialyzed in the standard buffer used for measuring factor-dependent assembly, no distinct clathrin cages were observed in the electron micrographs (Fig. 6A). When clathrin triskelia were dialyzed in a calcium containing buffer that promotes factor-independent assembly, clathrin cages were observed, although their size is highly variable (Fig. 6B). When clathrin triskelia were dialyzed in factor-dependent assembly reactions containing either F1–20/AP-3 (AS 15−) (Fig. 6C) or F1–20/AP-3 (AS15+) (Fig. 6D), clathrin cages of an extremely narrow size distribution were observed. When clathrin triskelia were dialyzed in factor-dependent assembly reactions containing either GST (Fig. 6E) or the NH2-terminal 33 kDa domain of F1–20/AP-3 (Fig. 6F), no cages were observed. In order to provide a quantitative assessment of the morphology we measured and plotted the distribution of cage diameters from a large number of fields (Fig. 7). The diameters of the clathrin cages that were assembled in the factor-independent assembly reaction range from 60–120 nm (Fig. 7, upper panel). Cages assembled by bacterially expressed GST-F1–20/ AP-3 (AS15−) (Fig. 7, lower panel) are predominantly 70–90 nm in diameter.
Fig. 6.

Ultrastructural analysis of assembled clathrin cages. The morphology of the products of the assembly assays depicted in Figure 3 were evaluated by negative staining electron microscopy. They were compared with the products of a factor-independent assembly assay in which 2 mM free Ca++ was added to initiate clathrin assembly (B). Products are shown from the factor-dependent assembly assays containing either no added protein (A), GST-F1-20/AP-3 (AS15−) (C), GST-F1-20/AP-3 (AS15+) (D), GST (E), or the 33 kD NH2-terminus of F1-20/AP-3 (F). Scale bar, 130 nm.
Fig. 7.

Size distribution of the assembled clathrin cages. Diameters of 393 cages assembled in the factor-independent clathrin assembly assay in which 2 mM free Ca++ was added to initiate clathrin assembly were measured (top). Diameters of 306 cages assembled in the factor-dependent clathrin assembly assay with GST-F1-20/AP-3 (AS15−) were measured (bottom).
DISCUSSION
It was clear from the previously reported studies (Morris et al., 1993; Zhou et al., 1993) that purified preparations of AP-3 contained a protein that was identical to F1-20. However, it had not yet been demonstrated that the biological activities of clathrin binding and assembly ascribed to that protein preparation were directly attributable to the gene product of the F1-20 cDNA. Our finding that bacterially expressed F1-20 binds and assembles clathrin (Figs. 1, 4–6) constitutes the first demonstration that the gene product of the cDNA we originally described as F1-20 (Zhou et al., 1992) has the same biological activity as the protein described as AP-3 (Murphy et al., 1991). In addition, this finding also suggests that post-translational modification of F1-20/AP-3 is not important for clathrin binding and assembly, since the bacterially expressed protein is unlikely to be correctly post-translationally modified. It is known that F1–20/AP-3 is phosphorylated in vivo (Keen and Black, 1986; Zhou et al., 1992), predominantly on serine residues. F1–20/AP-3 has also been shown to be phosphorylated in vitro by an endogenous coated vesicle associated protein kinase activated by polylysine (Morris et al., 1990).
We had previously described two distinct isoforms of F1-20 cDNA which differed by the presence of 15 nucleotides (Zhou et al., 1992). RNAase protection analysis and RT-PCR amplification of mouse brain RNA revealed that both isoforms are present in cellular RNA (Zhou et al., 1992). The predominant cellular form of the RNA lacks the 15 nucleotide mini-exon (Zhou et al., 1992). We found that F1–20/AP-3 expressed from recombinant plasmids, either containing [pGEX3X–F1-20(AS15+)] or lacking [(pGEX3X–F1-20(AS15−)], this mini-exon, have similar clathrin binding and assembly properties (Figs. 1, 4–6). This suggests that alternative RNA splicing of the 15 nucleotide mini-exon does not directly modulate the clathrin binding or assembly activities of F1–20/AP-3.
Other investigators have assigned the 33 kD NH2-terminus of F1–20/AP-3 as a clathrin binding domain (Murphy et al., 1991; Morris et al., 1993). This was first based on the finding that the purified 33 kDa trypsin fragment bound to clathrin triskelia (Murphy et al., 1991), and the subsequent finding that this fragment corresponded to the NH2-terminus (Morris et al., 1993), although it was noted that this fragment failed to assemble clathrin triskelia into clathrin cages (Murphy et al., 1991). It was also found that in clathrin cage binding assays utilizing an unfractionated trypsin digest that 70— 80% of the 33 kD fragments, and ∼50% of the 50 kD (apparent MW= 107 kD) central fragments were bound to preassembled clathrin cages (Morris et al., 1993). The 50 kD and 33 kD fragments had been observed to associate non-covalently (Murphy et al., 1991). Therefore, we chose a system in which we could evaluate the binding and assembly activities of the 33 kD NH2-terminus, free of any other regions of the protein. We found that the bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 binds to clathrin triskelia (Fig. 3A), fails to bind to preassembled clathrin cages (Fig. 2), and is not sufficient for clathrin assembly (Figs. 4–6). The finding that the bacterially expressed 33 kD NH2-terminus of F1-20/AP-3 binds to clathrin triskelia but fails to assemble clathrin triskelia into clathrin cages is consistent with the published proteolysis studies (Murphy et al., 1991). The finding that the bacterially expressed 33 kD NH2-termi-nus of F1-20/AP-3 fails to bind to clathrin cages is novel and potentially important. It is clear from these experiments that the 33 kD NH2-terminus of F1-20/AP-3 is sufficient to carry out some aspects of clathrin binding; however it appears that defining the regions of the protein involved in clathrin binding and assembly may be more complex than originally anticipated. Therefore, we have initiated a scanning mutagenesis study in order to identify all of the residues of the protein which participate in clathrin binding and assembly.
While there are differences between the bovine 33 kD NH2-terminal fragment, and the bacterially expressed 33 kD NH2-terminus, we do not think that these differences effect our conclusions for the following reasons. Initially we made our observations with a fusion between GST and the 33 kD NH2-terminus. It was unlikely that fusion with GST had an inhibitory effect, since the two full-length fusion proteins GST-F1–20/AP-3 (AS15−) and GST-F 1–20/AP-3 (AS15+) both displayed clathrin binding and assembly activities similar to bovine brain F1–20/AP-3 (Figs. 1, 4–6). Nevertheless, we ruled out this possibility by removing the GST by cleavage with Factor Xa. We found that the bacterially expressed 33 kD NH2-terminus of F1–20/AP-3 was not able to bind to preassembled clathrin cages (Fig. 2), or to assemble clathrin triskelia into cages (Figs. 4–6). To rule out that a mutation had been inadvertently introduced in the construction of the recombinant plasmid pGEX3X–F1-20N33kDa, three independent PCR reactions were carried out and their products were each subcloned. All three independent subclones were used in the experiments described (Figs. 2–6), and all three gave identical results. It is also possible that the 33 kD NH2-terminus is capable of binding clathrin cages, but that it must be correctly post-translationally modified. We think this possibility is unlikely, since the two bacterially expressed full-length proteins, which would be expected to lack the same post-translational modifications, are able to bind clathrin cages and assemble clathrin. Another possibility is that the 33 kD NH2-terminus is capable of binding clathrin cages, but that when it is expressed as an isolated domain in bacteria it does not fold correctly. We think this possibility is unlikely, since the 33 kD NH2-terminus is expressed in bacteria in a soluble form at levels comparable to the two bacterially expressed full-length proteins, and it is able to bind clathrin triskelia (Fig. 3). Generally when proteins are not correctly folded, they are more likely to be insoluble, and more susceptible to proteolytic attack. In our previously reported studies (Zhou et al., 1993) we demonstrated that both bacterially expressed F1–20/AP-3 and bovine F1-20/AP-3 yield similar stable fragments following protease digestion, suggesting that their folding is similar. In unpublished studies, we found that the bacterially expressed 33 kD NH2-terminus is resistant to trypsin digestion, as had been reported for the NH2-terminal fragment of bovine F1–20/AP-3 (Murphy et al., 1991). Thus we conclude that the bacterially expressed 33 kD NH2-terminus of F1–20/AP-3 is not sufficient for clathrin cage binding or clathrin assembly.
We found that the sizes of clathrin cages assembled in the absence of F1–20/AP-3 range from 60–120 nm in diameter, while the sizes of clathrin cages assembled in the presence of F1–20/AP-3 range from 70–90 nm in diameter (Figs. 6,7). This is consistent with the findings of other investigators that bovine F1–20/AP-3 interacts with clathrin triskelia to assemble them into a homogeneous population of clathrin cages (Ahle and Ungewickell, 1986; Prasad and Lippoldt, 1988). This strengthens the conclusion that the complete biochemical activity of the protein preparation AP-3 is attributable to the gene product of the F1-20 cDNA. Furthermore, we suggest that the consequences of F1–20/AP-3 restricting the available range of clathrin cage sizes may be important for regulating quantal release during neurotransmission. We hypothesized that one function of the synapse-specific clathrin assembly protein F1-20/AP-3 is to direct clathrin assembly at the pre-synaptic plasma membrane and/or synaptic endosomal compartment during synaptic vesicle biogenesis and recycling (Zhou et al., 1993). Since mature synaptic vesicles are derived from clathrin coated-vesicles (Maycox et al., 1992) we expect that the size of a mature synaptic vesicle would be proportional to the size of its precursor clathrin coated vesicle. It should be appreciated that a two-fold variation in synaptic vesicle diameter leads to an eight-fold difference in synaptic vesicle volume, and (we expect) an eight-fold variation in neurotransmitter content. In this regard, it has long been noted that a striking feature of synaptic vesicles is their extremely narrow size distribution, consistent with the quantal theory of neurotransmitter packaging (Gray and Willis, 1970). Therefore, we hypothesize that an important function of the synapse-specific clathrin assembly protein F1–20/AP-3 is to limit the range of vesicle sizes, and therefore increase the uniformity of quantal release.
ACKNOWLEDGMENTS
This work was supported by grant NS29051 to EML. We gratefully acknowledge Ms. Nancy Hrinya Tannery for providing outstanding technical assistance. We also thank Dr. Bob Duda and Mr. Tom Harper for providing advice and assistance with the electron microscopy.
REFERENCES
- Ahle S, Ungewickell E. Purification and properties of a new clathrin assembly protein. Embo J. 1986;5:3143–3149. doi: 10.1002/j.1460-2075.1986.tb04621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahle S, Ungewickell E. Auxilin, a newly identified clathrin-associated protein in coated vesicles from bovine brain. J Cell Biol. 1990;111:19–29. doi: 10.1083/jcb.111.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Scheller RH. The molecular machinery for secretion is conserved from yeast to neurons. Proc Natl Acad Sci USA. 1993;90:2559–2563. doi: 10.1073/pnas.90.7.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Scheller RH. A molecular description of synaptic vesicle membrane trafficking. Annu Rev Biochem. 1994;63:63–100. doi: 10.1146/annurev.bi.63.070194.000431. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Pearse BM. Assembly and packing of clathrin into coats. J Cell Biol. 1981;91:790–797. doi: 10.1083/jcb.91.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EG, Willis RA. On synaptic vesicles, complex vesicles and dense projections. Brain Res. 1970;24:149–168. doi: 10.1016/0006-8993(70)90097-1. [DOI] [PubMed] [Google Scholar]
- Hall ZW, Sanes JR. Synaptic structure and development: The neuromuscular junction. Cell. 1993;72(suppl):99–121. doi: 10.1016/s0092-8674(05)80031-5. [DOI] [PubMed] [Google Scholar]
- Herskovits JS, Burgess CC, Obar RA, Vallee RB. Effects of mutant rat dynamin on endocytosis. J Cell Biol. 1993;122:565–578. doi: 10.1083/jcb.122.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM, Kandel ER. Synaptic transmission: A bidirectional and self-modifiable form of cell-cell communication. Cell. 1993;72(suppl):1–30. doi: 10.1016/s0092-8674(05)80025-x. [DOI] [PubMed] [Google Scholar]
- Keen JH. Clathrin assembly proteins: Affinity purification and a model for coat assembly. J Cell Biol. 1987;105:1989–1998. doi: 10.1083/jcb.105.5.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen JH, Black MM. The phosphorylation of coated membrane proteins in intact neurons. J Cell Biol. 1986;102:1325–1333. doi: 10.1083/jcb.102.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen JH, Willingham MC, Pastan IH. Clathrin-coated vesicles: Isolation, dissociation and factor-dependent reassociation of clathrin baskets. Cell. 1979;16:303–312. doi: 10.1016/0092-8674(79)90007-2. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T. Coated pits and coated vesicles-sorting it all out. Curr Opinion Structural Biol. 1993;3:182–188. [Google Scholar]
- Kohtz DS, Puszkin S. A neuronal protein (NP185) associated with clathrin-coated vesicles: Characterization of NP185 with monoclonal antibodies. J Biol Chem. 1988;263:7418–7425. [PubMed] [Google Scholar]
- Lindner R, Ungewickell E. Light-chain-independent binding of adaptors, AP180, and auxilin to clathrin. Biochemistry. 1991;30:9097–9101. doi: 10.1021/bi00101a027. [DOI] [PubMed] [Google Scholar]
- Lindner R, Ungewickell E. Clathrin-associated proteins of bovine brain coated vesicles: An analysis of their number and assembly-promoting activity. J Biol Chem. 1992;267:16567–16573. [PubMed] [Google Scholar]
- Maycox PR, Link E, Reetz A, Morris SA, Jahn R. Clathrin- coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. J Cell Biol. 1992;118:1379–1388. doi: 10.1083/jcb.118.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Mann A, Ungewickell E. Analysis of 100–180-kDa phosphoproteins in clathrin-coated vesicles from bovine brain. J Biol Chem. 1990;265:3354–3357. [PubMed] [Google Scholar]
- Morris SA, Schroder S, Plessmann U, Weber K, Ungewickell E. Clathrin assembly protein AP180: Primary structure, domain organization and identification of a clathrin binding site. EMBO J. 1993;12:667–675. doi: 10.1002/j.1460-2075.1993.tb05700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Keen JH. Recognition sites for clathrin-associated proteins AP-2 and AP-3 on clathrin triskelia. J Biol Chem. 1992;267:10850–10855. [PubMed] [Google Scholar]
- Murphy JE, Pleasure IT, Puszkin S, Prasad K, Keen JH. Clathrin assembly protein AP-3. The identity of the 155K protein, AP 180, and NP185 and demonstration of a clathrin binding domain. J Biol Chem. 1991;266:4401–4408. [PubMed] [Google Scholar]
- Pearse BM. Coated vesicles from pig brain: purification and biochemical characterization. J Mol Biol. 1975;97:93–98. doi: 10.1016/s0022-2836(75)80024-6. [DOI] [PubMed] [Google Scholar]
- Perry DG, Hanson V, Benuck ML, Puszkin S. Neuronal protein NP185 in avian and murine cerebellum: Expression during development and evidence for its presence in nerve endings. J Histochem Cytochem. 1991;39:1461–1470. doi: 10.1177/39.11.1918924. [DOI] [PubMed] [Google Scholar]
- Prasad K, Lippoldt RE. Molecular characterization of the AP180 coated vesicle assembly protein. Biochemistry. 1988;27:6098–6104. doi: 10.1021/bi00416a040. [DOI] [PubMed] [Google Scholar]
- Regnier-Vigouroux A, Tooze SA, Huttner WB. Newly synthesized synaptophysin is transported to synaptic-like mi-crovesicles via constitutive secretory vesicles and the plasma membrane. EMBO J. 1991;10:3589–3601. doi: 10.1002/j.1460-2075.1991.tb04925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. Adaptins. Trends Cell Biol. 1992;2:293–297. doi: 10.1016/0962-8924(92)90118-7. [DOI] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Ger-omanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Sousa R, Tannery NH, Lafer EM. Two new monoclonal antibodies provide immunohistochemical evidence for the unique biochemical similarity of the mouse globus pallidus, entope-duncular nucleus and substantia nigra pars reticulata. Neuro-science. 1990;34:403–410. doi: 10.1016/0306-4522(90)90149-x. [DOI] [PubMed] [Google Scholar]
- Sousa R, Tannery NH, Zhou S, Lafer EM. Characterization of a novel synapse-specific protein. I. Development expression and cellular localization of the F1-20 protein and mRNA. J Neurosci. 1992;12:2130–2143. doi: 10.1523/JNEUROSCI.12-06-02130.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Jahn R. Proteins of synaptic vesicles involved in exocytosis and membrane recycling. Neuron. 1991;6:665–677. doi: 10.1016/0896-6273(91)90165-v. [DOI] [PubMed] [Google Scholar]
- Trowbridge IS. Endocytosis and signals for internalization [published erratum appears in Curr Opin Cell Biol 3:1062] Curr Opin Cell Biol. 1991;3:634–641. doi: 10.1016/0955-0674(91)90034-v. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrow SA, Rothman JE. SNAP family of NSF attachment proteins includes a brain-specific isoform. Nature. 1993;362:353–355. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
- Zhou S, Sousa R, Tannery NH, Lafer EM. Characterization of a novel synapse-specific protein. II. cDNA cloning and sequence analysis of the F1-20 protein. J Neurosci. 1992;12:2144–55. doi: 10.1523/JNEUROSCI.12-06-02144.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Tannery NH, Yang J, Puszkin S, Lafer EM. The synapse-specific phosphoprotein F1-20 is identical to the clathrin assembly protein AP-3. J Biol Chem. 1993;268:12655–12662. [PubMed] [Google Scholar]