

Abstract
Potassium is the most abundant cation in the intracellular fluid, and maintaining the proper distribution of potassium across the cell membrane is critical for normal cell function. Long-term maintenance of potassium homeostasis is achieved by alterations in renal excretion of potassium in response to variations in intake. Understanding the mechanism and regulatory influences governing the internal distribution and renal clearance of potassium under normal circumstances can provide a framework for approaching disorders of potassium commonly encountered in clinical practice. This paper reviews key aspects of the normal regulation of potassium metabolism and is designed to serve as a readily accessible review for the well informed clinician as well as a resource for teaching trainees and medical students.
Keywords: renal physiology, potassium, tubular transport, collecting duct, aldosterone
Introduction
Potassium plays a key role in maintaining cell function. Almost all cells possess an Na+-K+-ATPase, which pumps Na+ out of the cell and K+ into the cell and leads to a K+ gradient across the cell membrane (K+in>K+out) that is partially responsible for maintaining the potential difference across the membrane. This potential difference is critical to the function of cells, particularly in excitable tissues, such as nerve and muscle. The body has developed numerous mechanisms for defense of serum K+. These mechanisms serve to maintain a proper distribution of K+ within the body as well as regulate the total body K+ content.
Internal Balance of K+
The kidney is primarily responsible for maintaining total body K+ content by matching K+ intake with K+ excretion. Adjustments in renal K+ excretion occur over several hours; therefore, changes in extracellular K+ concentration are initially buffered by movement of K+ into or out of skeletal muscle. The regulation of K+ distribution between the intracellular and extracellular space is referred to as internal K+ balance. The most important factors regulating this movement under normal conditions are insulin and catecholamines (1).
After a meal, the postprandial release of insulin functions to not only regulate the serum glucose concentration but also shift dietary K+ into cells until the kidney excretes the K+ load re-establishing K+ homeostasis. These effects are mediated through insulin binding to cell surface receptors, which stimulates glucose uptake in insulin-responsive tissues through the insertion of the glucose transporter protein GLUT4 (2,3). An increase in the activity of the Na+-K+-ATPase mediates K+ uptake (Figure 1). In patients with the metabolic syndrome or CKD, insulin-mediated glucose uptake is impaired, but cellular K+ uptake remains normal (4,5), demonstrating differential regulation of insulin-mediated glucose and K+ uptake.
Figure 1.

The cell model illustrates β2-adrenergic and insulin-mediated regulatory pathways for K+ uptake. β2-Adrenergic and insulin both lead to K+ uptake by stimulating the activity of the Na+-K+-ATPase pump primarily in skeletal muscle, but they do so through different signaling pathways. β2-Adrenergic stimulation leads to increased pump activity through a cAMP- and protein kinase A (PKA)–dependent pathway. Insulin binding to its receptor leads to phosphorylation of the insulin receptor substrate protein (IRS-1), which, in turn, binds to phosphatidylinositide 3-kinase (PI3-K). The IRS-1–PI3-K interaction leads to activation of 3-phosphoinositide–dependent protein kinase-1 (PDK1). The stimulatory effect of insulin on glucose uptake and K+ uptake diverge at this point. An Akt-dependent pathway is responsible for membrane insertion of the glucose transporter GLUT4, whereas activation of atypical protein kinase C (aPKC) leads to membrane insertion of the Na+-K+-ATPase pump (reviewed in ref. 3).
Catecholamines regulate internal K+ distribution, with α-adrenergic receptors impairing and β-adrenergic receptors promoting cellular entry of K+. β2-Receptor–induced stimulation of K+ uptake is mediated by activation of the Na+-K+-ATPase pump. These effects play a role in regulating the cellular release of K+ during exercise (6).
Under normal circumstances, exercise is associated with movement of intracellular K+ into the interstitial space in skeletal muscle. Increases in interstitial K+ can be as high as 10–12 mM with severe exercise. Accumulation of K+ is a factor limiting the excitability and contractile force of muscle accounting for the development of fatigue (7,8). Additionally, increases in interstitial K+ play a role in eliciting rapid vasodilation, allowing for blood flow to increase in exercising muscle (9). During exercise, release of catecholamines through β2 stimulation limits the rise in extracellular K+ concentration that otherwise occurs as a result of normal K+ release by contracting muscle. Although the mechanism is likely to be multifactorial, total body K+ depletion may blunt the accumulation of K+ into the interstitial space, limiting blood flow to skeletal muscle and accounting for the association of hypokalemia with rhabdomyolysis.
Changes in plasma tonicity and acid–base disorders also influence internal K+ balance. Hyperglycemia leads to water movement from the intracellular to extracellular compartment. This water movement favors K+ efflux from the cell through the process of solvent drag. In addition, cell shrinkage causes intracellular K+ concentration to increase, creating a more favorable concentration gradient for K+ efflux. Mineral acidosis, but not organic acidosis, can be a cause of cell shift in K+. As recently reviewed, the general effect of acidemia to cause K+ loss from cells is not because of a direct K+-H+ exchange, but, rather, is because of an apparent coupling resulting from effects of acidosis on transporters that normally regulate cell pH in skeletal muscle (10) (Figure 2).
Figure 2.

The effect of metabolic acidosis on internal K+ balance in skeletal muscle. (A) In metabolic acidosis caused by inorganic anions (mineral acidosis), the decrease in extracellular pH will decrease the rate of Na+-H+ exchange (NHE1) and inhibit the inward rate of Na+-3HCO3 cotransport (NBCe1 and NBCe2). The resultant fall in intracellular Na+ will reduce Na+-K+-ATPase activity, causing a net loss of cellular K+. In addition, the fall in extracellular HCO3 concentration will increase inward movement of Cl− by Cl-HCO− exchange, further enhancing K+ efflux by K+-Cl− cotransport. (B) Loss of K+ from the cell is much smaller in magnitude in metabolic acidosis caused by an organic acidosis. In this setting, there is a strong inward flux of the organic anion and H+ through the monocarboxylate transporter (MCT; MCT1 and MCT4). Accumulation of the acid results in a larger fall in intracellular pH, thereby stimulating inward Na+ movement by way of Na+-H+ exchange and Na+-3HCO3 cotransport. Accumulation of intracellular Na+ maintains Na+-K+-ATPase activity, thereby minimizing any change in extracellular K+ concentration.
Intracellular K+ serves as a reservoir to limit the fall in extracellular K+ concentrations occurring under pathologic conditions where there is loss of K+ from the body. The efficiency of this effect was shown by military recruits undergoing training in the summer (11). These subjects were able to maintain a near-normal serum K+ concentration despite daily sweat K+ loses of >40 mmol and an 11-day cumulative total body K+ deficit of approximately 400 mmol.
Studies in rats using a K+ clamp technique afforded insight into the role of skeletal muscle in regulating extracellular K+ concentration (12). With this technique, insulin is administered at a constant rate, and K+ is simultaneously infused at a rate designed to prevent any drop in plasma K+ concentration. The amount of K+ administered is presumed to be equal to the amount of K+ entering the intracellular space of skeletal muscle.
In rats deprived of K+ for 10 days, the plasma K+ concentration decreased from 4.2 to 2.9 mmol/L. Insulin-mediated K+ disappearance declined by more than 90% compared with control values. This decrease in K+ uptake was accompanied by a >50% reduction in both the activity and expression of muscle Na+-K+-ATPase, suggesting that decreased pump activity might account for the decrease in insulin effect. This decrease in muscle K+ uptake, under conditions of K+ depletion, may limit excessive falls in extracellular K+ concentration that occur under conditions of insulin stimulation. Concurrently, reductions in pump expression and activity facilitate the ability of skeletal muscle to buffer declines in extracellular K+ concentrations by donating some component of its intracellular stores.
There are differences between skeletal and cardiac muscle in the response to chronic K+ depletion. Although skeletal muscle readily relinquishes K+ to minimize the drop in plasma K+ concentration, cardiac tissue K+ content remains relatively well preserved. In contrast to the decline in activity and expression of skeletal muscle Na+-K+-ATPase, cardiac Na+-K+-ATPase pool size increases in K+-deficient animals. This difference explains the greater total K+ clearance capacity after the acute administration of intravenous KCl to rats fed a K+-free diet for 2 weeks compared with K+-replete controls (13,14). Cardiac muscle accumulates a considerable amount of K+ in the setting of an acute load. When expressed on a weight basis, the cardiac capacity for K+ uptake is comparable with that of skeletal muscle under conditions of K+ depletion and may actually exceed skeletal muscle under control conditions.
Renal Potassium Handling
Potassium is freely filtered by the glomerulus. The bulk of filtered K+ is reabsorbed in the proximal tubule and loop of Henle, such that less than 10% of the filtered load reaches the distal nephron. In the proximal tubule, K+ absorption is primarily passive and proportional to Na+ and water (Figure 3). K+ reabsorption in the thick ascending limb of Henle occurs through both transcellular and paracellular pathways. The transcellular component is mediated by K+ transport on the apical membrane Na+-K+-2Cl− cotransporter (Figure 4). K+ secretion begins in the early distal convoluted tubule and progressively increases along the distal nephron into the cortical collecting duct (Figure 5). Most urinary K+ can be accounted for by electrogenic K+ secretion mediated by principal cells in the initial collecting duct and the cortical collecting duct (Figure 6). An electroneutral K+ and Cl− cotransport mechanism is also present on the apical surface of the distal nephron (15). Under conditions of K+ depletion, reabsorption of K+ occurs in the collecting duct. This process is mediated by upregulation in the apically located H+-K+-ATPase on α-intercalated cells (16) (Figure 7).
Figure 3.

A cell model for K+ transport in the proximal tubule. K+ reabsorption in the proximal tubule primarily occurs through the paracellular pathway. Active Na+ reabsorption drives net fluid reabsorption across the proximal tubule, which in turn, drives K+ reabsorption through a solvent drag mechanism. As fluid flows down the proximal tubule, the luminal voltage shifts from slightly negative to slightly positive. The shift in transepithelial voltage provides an additional driving force favoring K+ diffusion through the low-resistance paracellular pathway. Experimental studies suggest that there may be a small component of transcellular K+ transport; however, the significance of this pathway is not known. K+ uptake through the Na+-K+-ATPase pump can exit the basolateral membrane through a conductive pathway or coupled to Cl−. An apically located K+ channel functions to stabilize the cell negative potential, particularly in the setting of Na+-coupled cotransport of glucose and amino acids, which has a depolarizing effect on cell voltage.
Figure 4.

A cell model for K+ transport in the thick ascending limb of Henle. K+ reabsorption occurs by both paracellular and transcellular mechanisms. The basolateral Na+-K+-ATPase pump maintains intracellular Na+ low, thus providing a favorable gradient to drive the apically located Na+-K+-2Cl− cotransporter (an example of secondary active transport). The apically located renal outer medullary K+ (ROMK) channel provides a pathway for K+ to recycle from cell to lumen, and ensures an adequate supply of K+ to sustain Na+-K+-2Cl− cotransport. This movement through ROMK creates a lumen-positive voltage, providing a driving force for passive K+ reabsorption through the paracellular pathway. Some of the K+ entering the cell through the cotransporter exits the cell across the basolateral membrane, accounting for transcellular K+ reabsorption. K+ can exit the cell through a conductive pathway or in cotransport with Cl−. ClC-Kb is the primary pathway for Cl− efflux across the basolateral membrane.
Figure 5.

A cell model for K+ transport in the distal convoluted tubule (DCT). In the early DCT, luminal Na+ uptake is mediated by the apically located thiazide-sensitive Na+-Cl− cotransporter. The transporter is energized by the basolateral Na+-K+-ATPase, which maintains intracellular Na+ concentration low, thus providing a favorable gradient for Na+ entry into the cell through secondary active transport. The cotransporter is abundantly expressed in the DCT1 but progressively declines along the DCT2. ROMK is expressed throughout the DCT and into the cortical collecting duct. Expression of the epithelial Na+ channel (ENaC), which mediates amiloride-sensitive Na+ absorption, begins in the DCT2 and is robustly expressed throughout the downstream connecting tubule and cortical collecting duct. The DCT2 is the beginning of the aldosterone-sensitive distal nephron (ASDN) as identified by the presence of both the mineralocorticoid receptor and the enzyme 11β-hydroxysteroid dehydrogenase II. This enzyme maintains the mineralocorticoid receptor free to only bind aldosterone by metabolizing cortisol to cortisone, the latter of which has no affinity for the receptor. Electrogenic-mediated K+ transport begins in the DCT2 with the combined presence of ROMK, ENaC, and aldosterone sensitivity. Electroneutral K+-Cl− cotransport is present in the DCT and collecting duct. Conditions that cause a low luminal Cl− concentration increase K+ secretion through this mechanism, which occurs with delivery of poorly reabsorbable anions, such as sulfate, phosphate, or bicarbonate.
Figure 6.

The cell that is responsible for K+ secretion in the initial collecting duct and the cortical collecting duct is the principal cell. This cell possesses a basolateral Na+-K+-ATPase that is responsible for the active transport of K+ from the blood into the cell. The resultant high cell K+ concentration provides a favorable diffusion gradient for movement of K+ from the cell into the lumen. In addition to establishing a high intracellular K+ concentration, activity of this pump lowers intracellular Na+ concentration, thus maintaining a favorable diffusion gradient for movement of Na+ from the lumen into the cell. Both the movements of Na+ and K+ across the apical membrane occur through well defined Na+ and K+ channels.
Figure 7.

Reabsorption of HCO3 in the distal nephron is mediated by apical H+ secretion by the α-intercalated cell. Two transporters secrete H+, a vacuolar H+-ATPase and an H+-K+-ATPase. The H+-K+-ATPase uses the energy derived from ATP hydrolysis to secrete H+ into the lumen and reabsorb K+ in an electroneutral fashion. The activity of the H+-K+-ATPase increases in K+ depletion and, thus, provides a mechanism by which K+ depletion enhances both collecting duct H+ secretion and K+ absorption.
Under most homeostatic conditions, K+ delivery to the distal nephron remains small and is fairly constant. By contrast, the rate of K+ secretion by the distal nephron varies and is regulated according to physiologic needs. The cellular determinants of K+ secretion in the principal cell include the intracellular K+ concentration, the luminal K+ concentration, the potential (voltage) difference across the luminal membrane, and the permeability of the luminal membrane for K+. Conditions that increase cellular K+ concentration, decrease luminal K+ concentration, or render the lumen more electronegative will increase the rate of K+ secretion. Conditions that increase the permeability of the luminal membrane for K+ will increase the rate of K+ secretion. Two principal determinants of K+ secretion are mineralocorticoid activity and distal delivery of Na+ and water.
Aldosterone is the major mineralocorticoid in humans and affects several of the cellular determinants discussed above, leading to stimulation of K+ secretion. First, aldosterone increases intracellular K+ concentration by stimulating the activity of the Na+-K+-ATPase in the basolateral membrane. Second, aldosterone stimulates Na+ reabsorption across the luminal membrane, which increases the electronegativity of the lumen, thereby increasing the electrical gradient favoring K+ secretion. Lastly, aldosterone has a direct effect on the luminal membrane to increase K+ permeability (17).
A second principal determinant affecting K+ secretion is the rate of distal delivery of Na+ and water. Increased distal delivery of Na+ stimulates distal Na+ absorption, which will make the luminal potential more negative and, thus, increase K+ secretion. Increased flow rates also increase K+ secretion. When K+ is secreted in the collecting duct, the luminal K+ concentration rises, which decreases the diffusion gradient and slows additional K+ secretion. At higher luminal flow rates, the same amount of K+ secretion will be diluted by the larger volume such that the rise in luminal K+ concentration will be less. Thus, increases in the distal delivery of Na+ and water stimulate K+ secretion by lowering luminal K+ concentration and making the luminal potential more negative.
Two populations of K+ channels have been identified in the cells of the cortical collecting duct. The renal outer medullary K+ (ROMK) channel is considered to be the major K+-secretory pathway. This channel is characterized by having low conductance and a high probability of being open under physiologic conditions. The maxi-K+ channel (also known as the large-conductance K+ [BK] channel) is characterized by a large single channel conductance and quiescence in the basal state and activation under conditions of increased flow (18). In addition to increased delivery of Na+ and dilution of luminal K+ concentration, recruitment of maxi-K+ channels contributes to flow-dependent increased K+ secretion. Renal K+ channels are subjects of extensive reviews (19–21).
The effect of increased tubular flow to activate maxi-K+ channels may be mediated by changes in intracellular Ca2+ concentration (22). The channel is Ca2+-activated, and an acute increase in flow increases intracellular Ca2+ concentrations in the principal cell. It has been suggested that the central cilium (a structure present in principal cells) may facilitate transduction of signals of increased flow to increased intracellular Ca2+ concentration. In cultured cells, bending of primary cilia results in a transient increase in intracellular Ca2+, an effect blocked by antibodies to polycystin 2 (23). Although present in nearly all segments of the nephron, the maxi-K channel has been identified as the mediator of flow-induced K+ secretion in the distal nephron and cortical collecting duct (24).
Development of hypokalemia in type II Bartter syndrome illustrates the importance of maxi-K+ channels in renal K+ excretion (25). Patients with type II Bartter syndrome have a loss-of-function mutation in ROMK manifesting with clinical features of the disease in the perinatal period. ROMK provides the pathway for recycling of K+ across the apical membrane in the thick ascending limb of Henle. This recycling generates a lumen-positive potential that drives the paracellular reabsorption of Ca2+ and Mg2+ and provides luminal K+ to the Na+-K+-2Cl− cotransporter (Figure 4).
Mutations in ROMK decrease NaCl and fluid reabsorption in the thick limb, mimicking a loop diuretic effect, which causes volume depletion. Despite the increase in distal Na+ delivery, K+ wasting is not consistently observed, because ROMK is also the major K+-secretory pathway for regulated K+ excretion in the collecting duct. In fact, in the perinatal period, infants with this form of Bartter syndrome often exhibit a transient hyperkalemia consistent with loss of function of ROMK in the collecting duct. However, over time, these patients develop hypokalemia as a result of increased flow-mediated K+ secretion through maxi-K+ channels. Studies in an ROMK-deficient mouse model of type II Bartter syndrome are consistent with this mechanism (26). The transient hyperkalemia observed in the perinatal period is likely related to the fact that ROMK channels are functionally expressed earlier than maxi-K+ channels during the course of development.
In this regard, growing infants and children are in a state of positive K+ balance, which correlates with growth and increasing cell number. Early in development, there is a limited capacity of the distal nephron to secrete K+ because of a paucity of both apically located ROMK and maxi-K+ channels. The increase in K+-secretory capacity with maturation is initially a result of increased expression of ROMK. Several weeks later, maxi-K+ channel expression develops, allowing for flow-mediated K+ secretion to occur (reviewed in ref. 27). The limitation in distal K+ secretion is channel-specific, because the electrochemical gradient favoring K+ secretion, as determined by activity of the Na+-K+-ATPase and Na+ reabsorption, is not limiting. Additionally, increased flow rates are accompanied by appropriate increases in Na+ reabsorption and intracellular Ca2+ concentrations in the distal nephron, despite the absence of stimulatory effect on K+ secretion (28). Activity of the H+- K+-ATPase, which couples K+ reabsorption to H+ secretion in intercalated cells, is similar in newborns and adults. K+ reabsorption through this pump, combined with decreased expression of K+-secretory channels, helps maintain a state of positive K+ balance during somatic growth after birth. These features of distal K+ handling by the developing kidney are a likely explanation for the high incidence of nonoliguric hyperkalemia in preterm infants (29).
Another physiologic state characterized by a period of positive K+ balance is pregnancy, where approximately 300 mEq K+ is retained (30). High circulating levels of progesterone may play a role in this adaptation through stimulatory effects on K+ and H+ transport by the H+-K+ α2-ATPase isoform in the distal nephron (31).
In addition to stimulating maxi-K+ channels, increased tubular flow has been shown to stimulate Na+ absorption through the epithelial Na+ channel (ENaC) in the collecting duct. This increase in absorption not only is because of increased delivery of Na+, but also seems to be the result of mechanosensitive properties intrinsic to the channel. Increased flow creates a shear stress that activates ENaCs by increasing channel open probability (32,33).
It has been hypothesized that biomechanical regulation of renal tubular Na+ and K+ transport in the distal nephron may have evolved as a response to defend against sudden increases in extracellular K+ concentration that occur in response to ingestion of K+-rich diets typical of early vertebrates (22). According to this hypothesis, an increase in GFR after a protein-rich meal would lead to an increase in distal flow activating the ENaC, increasing intracellular Ca2+ concentration, and activating maxi-K+ channels. These events would enhance K+ secretion, thus providing a buffer to guard against development of hyperkalemia.
In patients with CKD, loss of nephron mass is counterbalanced by an adaptive increase in the secretory rate of K+ in remaining nephrons such that K+ homeostasis is generally well maintained until the GFR falls below 15–20 ml/min (34). The nature of the adaptive process is thought to be similar to the adaptive process that occurs in response to high dietary K+ intake in normal subjects (35). Chronic K+ loading in animals augments the secretory capacity of the distal nephron, and, therefore, renal K+ excretion is significantly increased for any given plasma K+ level. Increased K+ secretion under these conditions occurs in association with structural changes characterized by cellular hypertrophy, increased mitochondrial density, and proliferation of the basolateral membrane in cells in the distal nephron and principal cells of the collecting duct. Increased serum K+ and mineralocorticoids independently initiate the amplification process, which is accompanied by an increase in Na+-K+-ATPase activity.
Aldosterone Paradox
Under conditions of volume depletion, activation of the renin-angiotensin system leads to increased aldosterone release. The increase in circulating aldosterone stimulates renal Na+ retention, contributing to the restoration of extracellular fluid volume, but occurs without a demonstrable effect on renal K+ secretion. Under condition of hyperkalemia, aldosterone release is mediated by a direct effect of K+ on cells in the zona glomerulosa. The subsequent increase in circulating aldosterone stimulates renal K+ secretion, restoring the serum K+ concentration to normal, but does so without concomitant renal Na+ retention.
The ability of aldosterone to signal the kidney to stimulate salt retention without K+ secretion in volume depletion and stimulate K+ secretion without salt retention in hyperkalemia has been referred to as the aldosterone paradox (36). In part, this ability can be explained by the reciprocal relationship between urinary flow rates and distal Na+ delivery with circulating aldosterone levels. Under conditions of volume depletion, proximal salt and water absorption increase, resulting in decreased distal delivery of Na+ and water. Although aldosterone levels are increased, renal K+ excretion remains fairly constant, because the stimulatory effect of increased aldosterone is counterbalanced by the decreased delivery of filtrate to the distal nephron. Under condition of an expanded extracellular fluid volume, distal delivery of filtrate is increased as a result of decreased proximal tubular fluid reabsorption. Once again, renal K+ excretion remains relatively constant in this setting, because circulating aldosterone levels are suppressed. It is only under pathophysiologic conditions that increased distal Na+ and water delivery are coupled to increased aldosterone levels. Renal K+ wasting will occur in this setting (37) (Figure 8).
Figure 8.

Under normal circumstances, delivery of Na+ to the distal nephron is inversely associated with serum aldosterone levels. For this reason, renal K+ excretion is kept independent of changes in extracellular fluid volume. Hypokalemia caused by renal K+ wasting can be explained by pathophysiologic changes that lead to coupling of increased distal Na+ delivery and aldosterone or aldosterone-like effects. When approaching the hypokalemia caused by renal K+ wasting, one must determine whether the primary disorder is an increase in mineralocorticoid activity or an increase in distal Na+ delivery. EABV, effective arterial blood volume.
Renal K+ secretion also remains stable during changes in flow rate resulting from variations in circulating vasopressin. In this regard, vasopressin has a stimulatory effect on renal K+ secretion (38,39). This kaliuretic property may serve to oppose a tendency to K+ retention under conditions of antidiuresis when a low-flow rate-dependent fall in distal tubular K+ secretion might otherwise occur. In contrast, suppressed endogenous vasopressin leads to decreased activity of the distal K+-secretory mechanism, thus limiting excessive K losses under conditions of full hydration and water diuresis.
Although the inverse relationship between aldosterone levels and distal delivery of salt and water serves to keep renal K+ excretion independent of volume status, recent reviews have suggested a more complex mechanism centered on the with no lysine [K] 4 (WNK4) protein kinase in the distal nephron (40,41). WNK4 is one of four members of a family of serine-threonine kinases each encoded by a different gene and characterized by the atypical placement of the catalytic lysine residue that is present in most other protein kinases. Inactivating mutations in WNK4 lead to development of pseudohypoaldosteronism type II (PHAII; Gordon syndrome). This disorder is inherited in an autosomal dominant fashion and is characterized by hypertension and hyperkalemia (42). Circulating aldosterone levels are low, despite the presence of hyperkalemia. Thiazide diuretics are particularly effective in treating both the hypertension and hyperkalemia (43).
Wild-type WNK4 acts to reduce surface expression of the thiazide-sensitive Na+-Cl− cotransporter and also stimulates clathrin-dependent endocytosis of ROMK in the collecting duct (44,45). The inactivating mutation of WNK4 responsible for PHAII leads to increased cotransporter activity and further stimulates endocytosis of ROMK. The net effect is increased NaCl reabsorption combined with decreased K+ secretion. Mutated WNK4 also enhances paracellular Cl− permeability caused by increased phosphorylation of claudins, which are tight junction proteins involved in regulating paracellular ion transport (46). In addition to increasing Na+ retention, this change in permeability further impairs K+ secretion, because the lumen-negative voltage, which normally serves as a driving force for K+ secretion, is dissipated.
Because development of hypertension and hyperkalemia resulting from the PHAII-mutated WNK4 protein can be viewed as an exaggerated response to a reduction in extracellular fluid volume (salt retention without increased K+ secretion), it has been proposed that wild-type WNK4 may act as a molecular switch determining balance between renal NaCl reabsorption and K+ secretion (45,47). Under conditions of volume depletion, the switch would be altered in a manner reminiscent of the PHAII mutant such that NaCl reabsorption is increased, but K+ secretion is further inhibited. However, when increased serum K+ concentration occurs in the absence of volume depletion, WNK4 alterations result in maximal renal K+ secretion without Na+ retention.
Angiotensin II (AII) has emerged as an important modulator of this switch. Under conditions of volume depletion, AII and aldosterone levels are increased (Figure 9). In addition to effects leading to enhanced NaCl reabsorption in the proximal tubule, AII activates the Na+-Cl− cotransporter in a WNK4-dependent manner, and it is primarily located in the initial part of the distal convoluted tubule (DCT; DCT1) (48,49). AII also activates ENaC, which is found in the aldosterone-sensitive distal nephron (ASDN) comprised of the second segment of the DCT (DCT2), the connecting tubule, and the collecting duct (50). The activation of ENaC by AII is additive to that of aldosterone (51). In this manner, AII and aldosterone act in concert to stimulate Na+ retention. At the same time, AII inhibits ROMK by both WNK4-dependent and -independent mechanisms (52,53). This inhibitory effect on ROMK along with decreased Na+ delivery to the collecting duct brought about by AII stimulation of Na+ reabsorption in the proximal nephron, and DCT1 allows for simultaneous Na+ conservation without K+ wasting.
Figure 9.
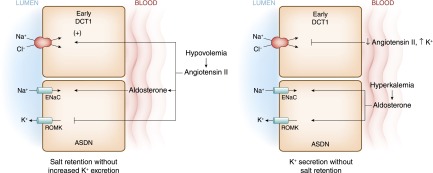
The aldosterone paradox refers to the ability of the kidney to stimulate NaCl retention with minimal K+ secretion under conditions of volume depletion and maximize K+ secretion without Na+ retention in hyperkalemia. With volume depletion (left panel), increased circulating angiotensin II (AII) levels stimulate the Na+-Cl− cotransporter in the early DCT. In the ASDN, AII along with aldosterone stimulate the ENaC. In this latter segment, AII exerts an inhibitory effect on ROMK, thereby providing a mechanism to maximally conserve salt and minimize renal K+ secretion. When hyperkalemia or increased dietary K+ intake occurs with normovolemia (right panel), low circulating levels of AII or direct effects of K+ lead to inhibition of Na+-Cl− cotransport activity along with increased activity of ROMK. As a result, Na+ delivery to the ENaC is optimized for the coupled electrogenic secretion of K+ through ROMK. As discussed in the text, with no lysine [K] 4 (WNK4) proteins are integrally involved in the signals by which the paradox is brought about. It should be emphasized the WNK proteins are part of a complex signaling network still being fully elucidated. The interested reader is referred to several recent reviews and advancements on this subject (48,51,91–93).
Hyperkalemia, or an increase in dietary K+ intake, can increase renal K+ secretion independent of change in mineralocorticoid activity and without causing volume retention. This effect was shown in Wistar rats fed a diet very low in NaCl and K+ for several days and given a pharmacologic dose of deoxycorticosterone to ensure a constant and nonvariable effect of mineralocorticoids (54,55). After a KCl load administered into the peritoneal cavity, two distinct phases were noted. In the first 2 hours, there was a large increase in the rate of renal K+ excretion that was largely caused by an increase in the K+ concentration in the cortical collecting duct. During this early phase, flow through the collecting duct increased only slightly, suggesting that changes in K+ concentration were largely caused by an increase in K+-secretory capacity of the collecting duct. This effect would be consistent with known effects of dietary supplementation of K+ to increase channel density of both ROMK and maxi-K+ channels (56).
In the subsequent 4 hours, renal K+ excretion continued to be high, but during this second phase, the kaliuresis was mostly accounted for by increased flow through the collecting duct. The increased flow was attributed to an inhibitory effect of increased interstitial K+ concentration on reabsorption of NaCl in the upstream ascending limb of Henle, an effect supported by microperfusion studies in the past (57,58). The timing of the two phases is presumably important, because higher flows would be most effective in promoting kaliuresis only after establishment of increased channel density. Although older studies are consistent with decreased Na+ absorption in the thick limb and proximal nephron after increased K+ intake, inhibitory effects in these high-capacity segments lack the precision and timing necessary to ensure that downstream delivery of Na+ is appropriate to maximally stimulate K+ secretion and at the same time, not be excessive, predisposing to volume depletion, particularly in the setting of a low Na+ diet (57–59).
The low-capacity nature of the DCT and its location immediately upstream from the ASDN make this segment a more likely site for changes in dietary K+ intake to modulate Na+ transport and ensure that downstream delivery of Na+ is precisely the amount needed to ensure maintenance of K+ homeostasis without causing unwanted effects on volume. In this regard, increased dietary K+ intake leads to an inhibitory effect on Na+ transport in this segment and does so through effects on WNK1, another member of the WNK family of kinases (60,61). WNK1 is ubiquitously expressed throughout the body in multiple spliced forms. By contrast, a shorter WNK1 transcript lacking the amino terminal 1–437 amino acids of the long transcript is highly expressed in the kidney but not other tissues, and it is referred to as kidney-specific WNK1 (KS-WNK1). KS-WNK1 is restricted to the DCT and part of the connecting duct and functions as a physiologic antagonist to the actions of long WNK1. Changes in the ratio of KS-WNK1 and long WNK1 in response to dietary K+ contribute to the physiologic regulation of renal K+ excretion (62–65).
Under normal circumstances, long WNK1 prevents the ability of WNK4 to inhibit activity of the Na+-Cl− cotransporter in the DCT. Thus, increased activity of long WNK1 leads to a net increase in NaCl reabsorption. Dietary K+ loading increases the abundance of KS-WNK1. Increased KS-WNK1 antagonizes the inhibitory effect of long WNK1 on WNK4. The net effect is inhibition of Na+-Cl− cotransport in the DCT and increased Na+ delivery to more distal parts of the tubule. In addition, increased KS-WNK1 antagonizes the effect of long WNK1 to stimulate endocytosis of ROMK. Furthermore, KS-WNK1 exerts a stimulatory effect on the ENaC. Thus, increases in KS-WNK1 in response to dietary K+ loading facilitate K+ secretion through the combined effects of increased Na+ delivery through downregulation of Na+-Cl− cotransport in the DCT, increased electrogenic Na+ reabsorption through the ENaC, and greater abundance of ROMK.
Increased aldosterone levels in response to a high K+ diet lead to effects that complement the effects of KS-WNK1 (66,67). The serum- and glucocorticoid-dependent protein kinase (SGK1) is an immediate transcriptional target of aldosterone binding to the mineralocorticoid receptor. Activation of SGK1 leads to phosphorylation of WNK4, resulting in a loss of the ability of WNK4 to inhibit ROMK and the ENaC (66,68). Aldosterone-induced activation of SGK1 also leads to increased ENaC expression and activity by causing the phosphorylation of ubiquitin protein ligase Nedd4–2. Phosphorylated Nedd4–2 results in less retrieval of ENaC from the apical membrane (69). It should be emphasized that the absence of AII is a critical factor in the ability of high K+ intake to bring about the changes necessary to facilitate K+ secretion without excessive Na+ reabsorption.
Role in Hypertension
Changes in KS-WNK1 and long WNK1 that occur in response to dietary K+ intake affect renal Na+ handling in a way that may be of importance in the observed relationship between dietary K+ intake and hypertension. Epidemiologic studies established that K+ intake is inversely related to the prevalence of hypertension (70). In addition, K+ supplements and avoidance of hypokalemia lowers BP in hypertensive subjects. By contrast, BP increases in hypertensive subjects placed on a low K+ diet. This increase in BP is associated with increased renal Na+ reabsorption (71).
K+ deficiency increases the ratio of long WNK1 to KS-WNK1. Long WNK1 is associated with increased retrieval of ROMK, thus providing an appropriate response to limit K+ secretion. However, long WNK1 also leads to a stimulatory effect on ENaC activity as well as releasing the inhibitory effect of WNK4 on Na+ reabsorption mediated by the NaCl cotransporter in the DCT (72,73). These effects suggest that reductions in K+ secretion under conditions of K+ deficiency will occur at the expense of increased Na+ retention.
Renal conservation of K+ and Na+ under conditions of K+ deficiency may be considered an evolutionary adaptation, because dietary K+ and Na+ deficiency likely occurred together for early humans (74). However, such an effect is potentially deleterious in our present setting, because evolution has seen a large increase in the ratio of dietary intake of Na+ versus K+. The effects of an increased ratio of WNK1 to KS-WNK1 in the kidney under conditions of modern day high Na+/low K+ diet could be central to the pathogenesis of salt-sensitive hypertension (75).
Enteric Sensor of K+
There is evidence to support the existence of enteric solute sensors capable of responding to dietary Na+, K+, and phosphate that signal the kidney to rapidly alter ion excretion or reabsorption (76–78). In experimental animals, and using protocols to maintain identical plasma K+ concentration, the kaliuretic response to a K+ load is greater when given as a meal compared with an intravenous infusion (79). These studies suggest that dietary K+ intake through a splanchnic sensing mechanism can signal increases in renal K+ excretion independent of changes in plasma K+ concentration or aldosterone (reviewed in ref. 80).
Although the precise signaling mechanism is not known, recent studies suggest that the renal response may be because of rapid and nearly complete dephosphorylation of the Na+-Cl− cotransporter in the DCT, causing decreased activity of the transporter and, thus, enhancing delivery of Na+ to the ASDN (81,82). In these studies, gastric delivery of K+ led to dephosphorylation of the cotransporter within minutes independent of aldosterone and based on in vitro studies, independent of changes in extracellular K+ concentration. The temporally associated increase in renal K+ excretion results from a more favorable electrochemical driving force caused by the downstream shift in Na+ reabsorption from the DCT to the ENaC in the ASDN as well as increased maxi-K+ channel K+ secretion brought on by increased flow. This rapid natriuretic response to increases in dietary K+ intake is consistent with the BP-lowering effect of K+-rich diets discussed earlier.
Circadian Rhythm of K+ Secretion
During a 24-hour period, urinary K+ excretion varies in response to changes in activity and fluctuations in K+ intake caused by the spacing of meals. However, even when K+ intake and activity are evenly spread over a 24-hour period, there remains a circadian rhythm whereby K+ excretion is lower at night and in the early morning hours and then increases in the afternoon (83–86). This circadian pattern results from changes in intratubular K+ concentration in the collecting duct as opposed to variations in urine flow rate (87).
In the mouse distal nephron, a circadian rhythm exists for gene transcripts that encode proteins involving K+ secretion (88). Gene expression of ROMK is greater during periods of activity, whereas expression of the H+-K+-ATPase is higher during rest, which correspond to periods when renal K+ excretion is greater and less, respectively (89). Changes in plasma aldosterone levels may play a contributory role, because circadian rhythm of glucocorticoid synthesis and secretion has been described in the adrenal gland. In addition, expression of clock genes within cells of the distal nephron suggests that a pacemaker function regulating K+ transport may be an intrinsic component of the kidney that is capable of operating independent of outside influence.
The clinical significance of this rhythmicity in K+ and other electrolyte secretions is not known. Evidence suggests that dysregulation of circadian rhythms may contribute to a lack of nocturnal decline in BP, with eventual development of sustained hypertension as well as accelerated CKD and cardiovascular disease (85,90).
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Palmer BF: A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 56: 387–393, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Foley K, Boguslavsky S, Klip A: Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 50: 3048–3061, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Ho K: A critically swift response: Insulin-stimulated potassium and glucose transport in skeletal muscle. Clin J Am Soc Nephrol 6: 1513–1516, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Nguyen TQ, Maalouf NM, Sakhaee K, Moe OW: Comparison of insulin action on glucose versus potassium uptake in humans. Clin J Am Soc Nephrol 6: 1533–1539, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvestrand A, Wahren J, Smith D, DeFronzo RA: Insulin-mediated potassium uptake is normal in uremic and healthy subjects. Am J Physiol 246: E174–E180, 1984 [DOI] [PubMed] [Google Scholar]
- 6.Williams ME, Gervino EV, Rosa RM, Landsberg L, Young JB, Silva P, Epstein FH: Catecholamine modulation of rapid potassium shifts during exercise. N Engl J Med 312: 823–827, 1985 [DOI] [PubMed] [Google Scholar]
- 7.Clausen T, Nielsen OB: Potassium, Na+,K+-pumps and fatigue in rat muscle. J Physiol 584: 295–304, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenna MJ, Bangsbo J, Renaud JM: Muscle K+, Na+, and Cl disturbances and Na+-K+ pump inactivation: Implications for fatigue. J Appl Physiol (1985) 104: 288–295, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Clifford PS: Skeletal muscle vasodilatation at the onset of exercise. J Physiol 583: 825–833, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aronson PS, Giebisch G: Effects of pH on potassium: New explanations for old observations. J Am Soc Nephrol 22: 1981–1989, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knochel JP, Dotin LN, Hamburger RJ: Pathophysiology of intense physical conditioning in a hot climate. I. Mechanisms of potassium depletion. J Clin Invest 51: 242–255, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonough AA, Youn JH: Role of muscle in regulating extracellular [K+]. Semin Nephrol 25: 335–342, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Bundgaard H, Kjeldsen K: Potassium depletion increases potassium clearance capacity in skeletal muscles in vivo during acute repletion. Am J Physiol Cell Physiol 283: C1163–C1170, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Bundgaard H: Potassium depletion improves myocardial potassium uptake in vivo. Am J Physiol Cell Physiol 287: C135–C141, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Velázquez H, Ellison DH, Wright FS: Chloride-dependent potassium secretion in early and late renal distal tubules. Am J Physiol 253: F555–F562, 1987 [DOI] [PubMed] [Google Scholar]
- 16.DuBose TD, Jr., Codina J, Burges A, Pressley TA: Regulation of H(+)-K(+)-ATPase expression in kidney. Am J Physiol 269: F500–F507, 1995 [DOI] [PubMed] [Google Scholar]
- 17.Stokes JB: Mineralocorticoid effect on K+ permeability of the rabbit cortical collecting tubule. Kidney Int 28: 640–645, 1985 [DOI] [PubMed] [Google Scholar]
- 18.Palmer LG, Frindt G: High-conductance K channels in intercalated cells of the rat distal nephron. Am J Physiol Renal Physiol 292: F966–F973, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Hebert SC, Desir G, Giebisch G, Wang W: Molecular diversity and regulation of renal potassium channels. Physiol Rev 85: 319–371, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang WH: Regulation of ROMK (Kir1.1) channels: New mechanisms and aspects. Am J Physiol Renal Physiol 290: F14–F19, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grimm PR, Sansom SC: BK channels in the kidney. Curr Opin Nephrol Hypertens 16: 430–436, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Satlin LM, Carattino MD, Liu W, Kleyman TR: Regulation of cation transport in the distal nephron by mechanical forces. Am J Physiol Renal Physiol 291: F923–F931, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J: Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129–137, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Welling PA: Regulation of renal potassium secretion: Molecular mechanisms. Semin Nephrol 33: 215–228, 2013 [DOI] [PubMed] [Google Scholar]
- 25.Pluznick JL, Sansom SC: BK channels in the kidney: Role in K(+) secretion and localization of molecular components. Am J Physiol Renal Physiol 291: F517–F529, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Bailey MA, Cantone A, Yan Q, MacGregor GG, Leng Q, Amorim JB, Wang T, Hebert SC, Giebisch G, Malnic G: Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of Type II Bartter’s syndrome and in adaptation to a high-K diet. Kidney Int 70: 51–59, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Gurkan S, Estilo GK, Wei Y, Satlin LM: Potassium transport in the maturing kidney. Pediatr Nephrol 22: 915–925, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Woda CB, Miyawaki N, Ramalakshmi S, Ramkumar M, Rojas R, Zavilowitz B, Kleyman TR, Satlin LM: Ontogeny of flow-stimulated potassium secretion in rabbit cortical collecting duct: Functional and molecular aspects. Am J Physiol Renal Physiol 285: F629–F639, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Thayyil S, Kempley ST, Sinha A: Can early-onset nonoliguric hyperkalemia be predicted in extremely premature infants? Am J Perinatol 25: 129–133, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Lindheimer MD, Richardson DA, Ehrlich EN, Katz AI: Potassium homeostasis in pregnancy. J Reprod Med 32: 517–522, 1987 [PubMed] [Google Scholar]
- 31.Elabida B, Edwards A, Salhi A, Azroyan A, Fodstad H, Meneton P, Doucet A, Bloch-Faure M, Crambert G: Chronic potassium depletion increases adrenal progesterone production that is necessary for efficient renal retention of potassium. Kidney Int 80: 256–262, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Morimoto T, Liu W, Woda C, Carattino MD, Wei Y, Hughey RP, Apodaca G, Satlin LM, Kleyman TR: Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. Am J Physiol Renal Physiol 291: F663–F669, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Carattino MD, Sheng S, Kleyman TR: Mutations in the pore region modify epithelial sodium channel gating by shear stress. J Biol Chem 280: 4393–4401, 2005 [DOI] [PubMed] [Google Scholar]
- 34.van Ypersele de Strihou C: Potassium homeostasis in renal failure. Kidney Int 11: 491–504, 1977 [DOI] [PubMed] [Google Scholar]
- 35.Stanton BA: Renal potassium transport: Morphological and functional adaptations. Am J Physiol 257: R989–R997, 1989 [DOI] [PubMed] [Google Scholar]
- 36.Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G: Aldosterone paradox: Differential regulation of ion transport in distal nephron. Physiology (Bethesda) 26: 115–123, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Palmer BF: A physiologic-based approach to the evaluation of a patient with hypokalemia. Am J Kidney Dis 56: 1184–1190, 2010 [DOI] [PubMed] [Google Scholar]
- 38.Field MJ, Stanton BA, Giebisch GH: Influence of ADH on renal potassium handling: A micropuncture and microperfusion study. Kidney Int 25: 502–511, 1984 [DOI] [PubMed] [Google Scholar]
- 39.Cassola AC, Giebisch G, Wang W: Vasopressin increases density of apical low-conductance K+ channels in rat CCD. Am J Physiol 264: F502–F509, 1993 [DOI] [PubMed] [Google Scholar]
- 40.Kahle KT, Ring AM, Lifton RP: Molecular physiology of the WNK kinases. Annu Rev Physiol 70: 329–355, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Kahle KT, Rinehart J, Giebisch G, Gamba G, Hebert SC, Lifton RP: A novel protein kinase signaling pathway essential for blood pressure regulation in humans. Trends Endocrinol Metab 19: 91–95, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z: Pseudohypoaldosteronism type II: Marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab 87: 3248–3254, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Gordon RD: The syndrome of hypertension and hyperkalaemia with normal GFR. A unique pathophysiological mechanism for hypertension? Clin Exp Pharmacol Physiol 13: 329–333, 1986 [DOI] [PubMed] [Google Scholar]
- 44.Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S: Molecular pathogenesis of pseudohypoaldosteronism type II: Generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5: 331–344, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP: Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, Suda S, Hayama A, Sasaki S, Uchida S: Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc Natl Acad Sci U S A 101: 4690–4694, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kahle KT, Wilson FH, Lifton RP: Regulation of diverse ion transport pathways by WNK4 kinase: A novel molecular switch. Trends Endocrinol Metab 16: 98–103, 2005 [DOI] [PubMed] [Google Scholar]
- 48.Castañeda-Bueno M, Gamba G: Mechanisms of sodium-chloride cotransporter modulation by angiotensin II. Curr Opin Nephrol Hypertens 21: 516–522, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G: Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci U S A 109: 7929–7934, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun P, Yue P, Wang WH: Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol 302: F679–F687, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Lubbe N, Zietse R, Hoorn EJ: Effects of angiotensin II on kinase-mediated sodium and potassium transport in the distal nephron. Curr Opin Nephrol Hypertens 22: 120–126, 2013 [DOI] [PubMed] [Google Scholar]
- 52.Wei Y, Zavilowitz B, Satlin LM, Wang WH: Angiotensin II inhibits the ROMK-like small conductance K channel in renal cortical collecting duct during dietary potassium restriction. J Biol Chem 282: 6455–6462, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yue P, Sun P, Lin DH, Pan C, Xing W, Wang W: Angiotensin II diminishes the effect of SGK1 on the WNK4-mediated inhibition of ROMK1 channels. Kidney Int 79: 423–431, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halperin ML, Cheema-Dhadli S, Lin SH, Kamel KS: Control of potassium excretion: A Paleolithic perspective. Curr Opin Nephrol Hypertens 15: 430–436, 2006 [DOI] [PubMed] [Google Scholar]
- 55.Cheema-Dhadli S, Lin SH, Keong-Chong C, Kamel KS, Halperin ML: Requirements for a high rate of potassium excretion in rats consuming a low electrolyte diet. J Physiol 572: 493–501, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Najjar F, Zhou H, Morimoto T, Bruns JB, Li HS, Liu W, Kleyman TR, Satlin LM: Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am J Physiol Renal Physiol 289: F922–F932, 2005 [DOI] [PubMed] [Google Scholar]
- 57.Stokes JB: Consequences of potassium recycling in the renal medulla. Effects of ion transport by the medullary thick ascending limb of Henle’s loop. J Clin Invest 70: 219–229, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sufit CR, Jamison RL: Effect of acute potassium load on reabsorption in Henle’s loop in the rat. Am J Physiol 245: F569–F576, 1983 [DOI] [PubMed] [Google Scholar]
- 59.Brandis M, Keyes J, Windhager EE: Potassium-induced inhibition of proximal tubular fluid reabsorption in rats. Am J Physiol 222: 421–427, 1972 [DOI] [PubMed] [Google Scholar]
- 60.Cheng CJ, Truong T, Baum M, Huang CL: Kidney-specific WNK1 inhibits sodium reabsorption in the cortical thick ascending limb. Am J Physiol Renal Physiol 303: F667–F673, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL: Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum Mol Genet 20: 855–866, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Reilly M, Marshall E, Macgillivray T, Mittal M, Xue W, Kenyon CJ, Brown RW: Dietary electrolyte-driven responses in the renal WNK kinase pathway in vivo. J Am Soc Nephrol 17: 2402–2413, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Wade JB, Fang L, Liu J, Li D, Yang CL, Subramanya AR, Maouyo D, Mason A, Ellison DH, Welling PA: WNK1 kinase isoform switch regulates renal potassium excretion. Proc Natl Acad Sci U S A 103: 8558–8563, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lazrak A, Liu Z, Huang CL: Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci U S A 103: 1615–1620, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cope G, Murthy M, Golbang AP, Hamad A, Liu CH, Cuthbert AW, O’Shaughnessy KM: WNK1 affects surface expression of the ROMK potassium channel independent of WNK4. J Am Soc Nephrol 17: 1867–1874, 2006 [DOI] [PubMed] [Google Scholar]
- 66.Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP: An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci U S A 104: 4025–4029, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Náray-Fejes-Tóth A, Snyder PM, Fejes-Tóth G: The kidney-specific WNK1 isoform is induced by aldosterone and stimulates epithelial sodium channel-mediated Na+ transport. Proc Natl Acad Sci U S A 101: 17434–17439, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin DH, Yue P, Rinehart J, Sun P, Wang Z, Lifton R, Wang WH: Protein phosphatase 1 modulates the inhibitory effect of With-no-Lysine kinase 4 on ROMK channels. Am J Physiol Renal Physiol 303: F110–F119, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Palmer BF, Alpern RJ: Liddle’s syndrome. Am J Med 104: 301–309, 1998 [DOI] [PubMed] [Google Scholar]
- 70.Appel LJ, Brands MW, Daniels SR, Karanja N, Elmer PJ, Sacks FM; American Heart Association: Dietary approaches to prevent and treat hypertension: A scientific statement from the American Heart Association. Hypertension 47: 296–308, 2006 [DOI] [PubMed] [Google Scholar]
- 71.Krishna GG, Kapoor SC: Potassium depletion exacerbates essential hypertension. Ann Intern Med 115: 77–83, 1991 [DOI] [PubMed] [Google Scholar]
- 72.Vallon V, Wulff P, Huang DY, Loffing J, Völkl H, Kuhl D, Lang F: Role of Sgk1 in salt and potassium homeostasis. Am J Physiol Regul Integr Comp Physiol 288: R4–R10, 2005 [DOI] [PubMed] [Google Scholar]
- 73.Rozansky DJ: The role of aldosterone in renal sodium transport. Semin Nephrol 26: 173–181, 2006 [DOI] [PubMed] [Google Scholar]
- 74.Eaton SB: The ancestral human diet: What was it and should it be a paradigm for contemporary nutrition? Proc Nutr Soc 65: 1–6, 2006 [DOI] [PubMed] [Google Scholar]
- 75.Huang CL, Kuo E: Mechanisms of disease: WNK-ing at the mechanism of salt-sensitive hypertension. Nat Clin Pract Nephrol 3: 623–630, 2007 [DOI] [PubMed] [Google Scholar]
- 76.Thomas L, Kumar R: Control of renal solute excretion by enteric signals and mediators. J Am Soc Nephrol 19: 207–212, 2008 [DOI] [PubMed] [Google Scholar]
- 77.Michell AR, Debnam ES, Unwin RJ: Regulation of renal function by the gastrointestinal tract: Potential role of gut-derived peptides and hormones. Annu Rev Physiol 70: 379–403, 2008 [DOI] [PubMed] [Google Scholar]
- 78.Lee FN, Oh G, McDonough AA, Youn JH: Evidence for gut factor in K+ homeostasis. Am J Physiol Renal Physiol 293: F541–F547, 2007 [DOI] [PubMed] [Google Scholar]
- 79.Oh KS, Oh YT, Kim SW, Kita T, Kang I, Youn JH: Gut sensing of dietary K⁺ intake increases renal K⁺ excretion. Am J Physiol Regul Integr Comp Physiol 301: R421–R429, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Youn JH: Gut sensing of potassium intake and its role in potassium homeostasis. Semin Nephrol 33: 248–256, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J: Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013 [DOI] [PubMed] [Google Scholar]
- 82.McDonough AA, Youn JH: Need to quickly excrete K(+)? Turn off NCC. Kidney Int 83: 779–782, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Firsov D, Tokonami N, Bonny O: Role of the renal circadian timing system in maintaining water and electrolytes homeostasis. Mol Cell Endocrinol 349: 51–55, 2012 [DOI] [PubMed] [Google Scholar]
- 84.Firsov D, Bonny O: Circadian regulation of renal function. Kidney Int 78: 640–645, 2010 [DOI] [PubMed] [Google Scholar]
- 85.Bonny O, Firsov D: Circadian regulation of renal function and potential role in hypertension. Curr Opin Nephrol Hypertens 22: 439–444, 2013 [DOI] [PubMed] [Google Scholar]
- 86.Gumz ML, Rabinowitz L: Role of circadian rhythms in potassium homeostasis. Semin Nephrol 33: 229–236, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steele A, deVeber H, Quaggin SE, Scheich A, Ethier J, Halperin ML: What is responsible for the diurnal variation in potassium excretion? Am J Physiol 267: R554–R560, 1994 [DOI] [PubMed] [Google Scholar]
- 88.Zuber AM, Centeno G, Pradervand S, Nikolaeva S, Maquelin L, Cardinaux L, Bonny O, Firsov D: Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci U S A 106: 16523–16528, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Salhi A, Centeno G, Firsov D, Crambert G: Circadian expression of H,K-ATPase type 2 contributes to the stability of plasma K⁺ levels. FASEB J 26: 2859–2867, 2012 [DOI] [PubMed] [Google Scholar]
- 90.Gumz ML, Stow LR, Lynch IJ, Greenlee MM, Rudin A, Cain BD, Weaver DR, Wingo CS: The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J Clin Invest 119: 2423–2434, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, Chiga M, Kikuchi E, Nomura N, Mori Y, Matsuo H, Murata T, Nomura S, Asano T, Kawaguchi H, Nonoyama S, Rai T, Sasaki S, Uchida S: Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep 3: 858–868, 2013 [DOI] [PubMed] [Google Scholar]
- 92.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP: Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci U S A 110: 7838–7843, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH: The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol 22: 605–614, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]