

ABSTRACT
Type IV pili (T4P) are dynamic protein filaments that mediate bacterial adhesion, biofilm formation, and twitching motility. The highly conserved PilMNOP proteins form an inner membrane alignment subcomplex required for function of the T4P system, though their exact roles are unclear. Three potential interaction interfaces for PilNO were identified: core-core, coiled coils (CC), and the transmembrane segments (TMSs). A high-confidence PilNO heterodimer model was used to select key residues for mutation, and the resulting effects on protein-protein interactions were examined both in a bacterial two-hybrid (BTH) system and in their native Pseudomonas aeruginosa context. Mutations in the oppositely charged CC regions or the TMS disrupted PilNO heterodimer formation in the BTH assay, while up to six combined mutations in the core failed to disrupt the interaction. When the mutations were introduced into the P. aeruginosa chromosome at the pilN or pilO locus, specific changes at each of the three interfaces—including core mutations that failed to disrupt interactions in the BTH system—abrogated surface piliation and/or impaired twitching motility. Unexpectedly, specific CC mutants were hyperpiliated but nonmotile, a hallmark of pilus retraction defects. These data suggest that PilNO participate in both the extension and retraction of T4P. Our findings support a model of multiple, precise interaction interfaces between PilNO; emphasize the importance of studying protein function in a minimally perturbed context and stoichiometry; and highlight potential target sites for development of small-molecule inhibitors of the T4P system.
IMPORTANCE Pseudomonas aeruginosa is an opportunistic pathogen that uses type IV pili (T4P) for host attachment. The T4P machinery is composed of four cell envelope-spanning subcomplexes. PilN and PilO heterodimers are part of the alignment subcomplex and essential for T4P function. Three potential PilNO interaction interfaces (the core-core, coiled-coil, and transmembrane segment interfaces) were probed using site-directed mutagenesis followed by functional assays in an Escherichia coli two-hybrid system and in P. aeruginosa. Several mutations blocked T4P assembly and/or motility, including two that revealed a novel role for PilNO in pilus retraction, while other mutations affected extension dynamics. These critical PilNO interaction interfaces represent novel targets for small-molecule inhibitors with the potential to disrupt T4P function.
INTRODUCTION
Alarming increases in antibiotic resistance have prompted the search for alternative strategies to combat bacterial infection (1). Genomic strategies designed to identify essential bacterial targets have been informative but less successful for drug development than originally predicted due to high mutation rates and resistance (2). Alternative strategies—such as phage therapy and the use of “antivirulence” drugs that target bacterial factors involved in pathogenesis—show promise since they can effectively disarm bacteria, which are then more easily cleared by the host immune system. The targeting of nonvital bacterial processes decreases selection pressure, making mutation-based resistance to these therapies less likely (3). Investigations of the type III secretion system (T3SS), where promising molecules—some of which showed broad-spectrum activity for various bacterial pathogens—were identified through whole-cell-based high-throughput screens, have had success with this strategy (4–6).
Type IV pili (T4P) are common bacterial virulence factors (7–9) and an attractive target for therapeutics. They are long, thin (5- to 8-nm-diameter), hair-like appendages which extend from the bacterial surface and promote attachment, cell-cell aggregation, biofilm formation, and twitching motility (10–15). T4P are produced by a wide variety of bacteria and archaea, including the opportunistic pathogen Pseudomonas aeruginosa, which infects individuals with severe burns, cystic fibrosis, or immunodeficiencies (16). Mutants lacking T4P are impaired in host colonization and infectivity (7). T4P-mediated twitching motility is a form of flagellum-independent surface translocation that results from cycles of pilus extension, adhesion, and retraction (10, 17). Upon pilus retraction, the cell body is winched toward the point of attachment (10, 17, 18).
The T4P system in P. aeruginosa is comprised of four subcomplexes that together span the entire Gram-negative cell envelope (Fig. 1A) (11). The outer membrane (OM) subcomplex is composed of an oligomer of 12 to 14 secretin subunits, PilQ (19), bound to a pilotin protein, PilF, forming a pore through which the pilus exits the cell (20, 21). There are two inner membrane (IM) subcomplexes—the motor and alignment subcomplexes—that together make up the IM platform. The motor subcomplex is composed of a platform protein, PilC, and associated cytoplasmic ATPases (PilBTU in P. aeruginosa) thought to provide the mechanical forces that lead to pilus extension/retraction (22, 23). The secretin and motor subcomplexes are linked by the alignment subcomplex (PilMNOP), which may also have a role in secretin gating (24, 25). The final subcomplex is the pilus, a helical filament of major and minor pilin subunits plus the PilY1 adhesin (13, 18, 26–28). Together with a number of regulatory proteins whose functions are incompletely understood (29–32), these subcomplexes form a fully functional T4P system.
FIG 1.

The type IV pilus system in P. aeruginosa. (A) Components of the type IV pilus proteins are labeled according to P. aeruginosa nomenclature. Components include the outer membrane (OM) secretin subcomplex (red), the inner membrane (IM) motor subcomplex (green), the IM alignment subcomplex (dark blue), and the pilus fiber (purple). (Adapted from reference 11.) PG, peptidoglycan. (B) Cartoon representation of two alignment subcomplex components, PilN (blue) and PilO (gray), indicating the approximate locations of residue substitutions (black). The three regions examined in this study, i.e., the core region, the coiled-coil region, and the transmembrane segment (TMS), are indicated on right. ch, chimera.
The alignment subcomplex is composed of four proteins. PilM is a cytoplasmic protein with an actin-like fold, similar to bacterial cytoskeleton proteins FtsA and MreB (25, 33). A narrow crevice on PilM binds tightly to a highly conserved motif in the cytoplasmic N terminus of PilN in P. aeruginosa (34) and Thermus thermophilus (33). PilN and PilO are bitopic IM proteins with similar predicted topologies (34). Both have short cytoplasmic N termini, a single transmembrane segment (TMS), two extended α-helices connected by a short linker forming a coiled coil (CC), and a C-terminal core domain composed of dual ferredoxin-like αββ motifs (see Fig. S1 in the supplemental material) (34). In the crystal structure of a PilO homodimer lacking 68 residues at its N terminus, the truncated coiled-coil segment is folded up against the core domain (34). A recent analysis of the crystal structure of a C-terminal fragment (residues 94 to 207) of T. thermophilus PilN revealed a dual αββ motif with an extra α-helix insertion that is predicted to be missing in some orthologs—including P. aeruginosa PilN—resulting in a secondary structure order of αβαβ-αββ (35). PilN and PilO form stable heterodimers and are dependent upon one another for stability in vivo (34). The fourth component of the alignment subcomplex is the inner membrane lipoprotein PilP, which binds the PilNO heterodimer through its extended N-terminal segment (24). Previous studies reported an interaction between PilP and PilQ in N. meningitidis (36), and, more recently, interaction of the C-terminal β-domain of P. aeruginosa PilP with the N0 domain of PilQ was demonstrated by copurification experiments (24). Together, the PilMNOP proteins connect the inner and outer membrane components of the T4P system and are critical for its function.
Disruption of a key protein homodimer by small molecules was shown to block formation of the type IV secretion (T4S) system and to diminish virulence of Brucella spp. (37, 38). Toward our goal of designing inhibitors of T4P function in P. aeruginosa, here we examined the contributions of the core, coiled-coil, and transmembrane segments of the PilNO heterodimer to their interaction and function. Point mutations in the coiled-coil region disrupted PilNO heterodimer formation in a bacterial two-hybrid (BTH) assay, as did swapping of their transmembrane segments. In contrast, up to six combined point mutations in the core region had no effect. When the same mutations were introduced at their native loci on the P. aeruginosa chromosome, many—including those that failed to disrupt PilNO interactions in the BTH assay— altered twitching motility and surface piliation. Chromosomal mutations thus provided key information about T4P system dynamics that could not be predicted from the results of the pairwise BTH assay. Our findings support a model in which multiple, precise PilNO interaction interfaces are critical for subcomplex dynamics and highlight potential target sites for inhibitor design.
MATERIALS AND METHODS
Strains, media, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material, and primers used in this study are listed in Table S2. Escherichia coli and P. aeruginosa were grown at 37°C in LB Lennox broth (LB; Bioshop) supplemented when necessary with antibiotics at the following final concentrations (μg · ml−1): ampicillin (Ap), 100; carbenicillin (Cb), 200; kanamycin (Kn), 50; gentamicin (Gm), 15 for Escherichia coli and 30 for P. aeruginosa, unless otherwise specified. Plasmids were transformed by heat shock into chemically competent E. coli cells. All constructs were verified by DNA sequencing (MOBIX—McMaster University).
BTH β-galactosidase activity assay.
Chemically competent E. coli strain BTH101 was cotransformed with various combinations of pUT18C and pKT25 protein fusions and tested for activity using a 96-well plate-based assay as previously described (39) with modifications. Briefly, BTH101 cells cotransformed with PilN and PilO fusion constructs were grown at 30°C in LB supplemented with Ap and Kn with shaking at 200 rpm overnight. LB containing antibiotics and 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG; Sigma-Aldrich) was inoculated with a 1:5 dilution of an overnight culture and grown at 30°C, with shaking at 260 rpm, to an optical density at 600 nm (OD600) of ∼0.6 and standardized. Cells were harvested by centrifugation for 3 min at 2,292 × g in a microcentrifuge, washed with 500 μl 1× phosphate-buffered saline (PBS; 140 mM NaCl, 2.68 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4), and resuspended in 400 μl of the same solution. The cells were lysed using MP Bio Lysing Matrix silica beads and a FastPrep-24 instrument (MP Biomedicals) for 20 s following the manufacturer's instructions. Tubes were centrifuged to remove the matrix and cell debris, and the supernatant was collected. A 50-μl volume of the supernatant was added to a 96-well microplate containing 50 μl of 2× assay buffer (200 mM sodium phosphate buffer [pH 7.3], 2 mM MgCl2, 100 mM β-mercaptoethanol, 1.33 mg/ml ONPG [o-nitrophenyl-β-d-galactopyranoside; Sigma-Aldrich]) per well. The plate was incubated for 30 min at 37°C, on a rocking platform. The reaction was stopped with the addition of 150 μl of 1 M sodium carbonate, and absorbance at 420 nm and 550 nm was measured using a Multiscan GO microplate reader (Thermo Scientific). The specific β-galactosidase activity was converted to Miller units using the following equation (where A is absorbance and T is time in minutes):
All assays were performed in triplicate, and at least three independent experiments were performed for each transformant. A one-way analysis of variance (ANOVA) test was performed followed by a Dunnett posttest to compare each interaction pair to the positive control.
Generation of pilN and pilO mutations on the P. aeruginosa chromosome.
pilN and pilO mutants were constructed using a Flp-FRT (FLP recombination target) system as previously described (40). Briefly, suicide vectors which contained the target gene, pilN or pilO, containing the desired mutations plus the flanking genes (pEX18Gm::pilMNO or pEX18Gm::pilNOP, respectively), were introduced into E. coli SM10 cells. The constructs were transferred by conjugation at a 1:9 ratio of P. aeruginosa to E. coli. The mixed culture was pelleted for 3 min at 2,292 × g in a microcentrifuge, and the pellet was resuspended in 50 μl of LB, spot-plated on LB agar, and incubated overnight at 37°C. P. aeruginosa PAK strains which contained either pilN::FRT or pilO::FRT mutations were used as recipients for pEX18Gm::pilMNO or pEX18Gm::pilNOP constructs, respectively, in mating experiments (41). After mating experiments were performed, cells were scraped from the LB agar plate and resuspended in 1 ml of LB and the E. coli SM10 donor was counterselected by plating on Pseudomonas isolation agar (PIA; Difco) containing Gm (100 μg · ml−1). Gm-resistant P. aeruginosa isolates were streaked on LB no-salt plates with sucrose (1% [wt/vol] Bacto tryptone, 0.5% [wt/vol] Bacto yeast extract, 5% [wt/vol] sucrose, 1.5% agar) and then incubated for 16 h at 30°C. Selected colonies were cultured in parallel on LB agar with and without Gm. Gm-sensitive colonies were screened by PCR using pilN or pilO primer pairs to confirm replacement of the FRT-disrupted gene, and PCR products of the expected size were sequenced to confirm incorporation of the desired mutations.
Generation of pilT mutants.
pilT mutants were constructed using the Flp-FRT recombination system as previously described (40). Briefly, a suicide vector containing the target gene (pilT) disrupted by a gentamicin resistance cassette flanked by FRT sites (pEX18AP::pilT::GmFRT) (42) was introduced into chemically competent E. coli SM10 cells. The constructs were transferred to P. aeruginosa PAK strains containing pilN mutations (ES132-133VA, MR141-142KL, or EV157-158AD) by biparental mating as described above. E. coli SM10 was counterselected by plating on PIA containing Gm (100 μg · ml−1). Gm-resistant isolates were selected and streaked on LB no-salt plates with sucrose and then incubated for 16 h at 30°C. Selected colonies were plated in parallel on LB plates supplemented with either Cb or Gm, and Gm-resistant, Cb-sensitive colonies were selected. Removal of the integrated Gm cassette by Flp recombinase-catalyzed excision was achieved by conjugally transferring the Flp-expressing pFLP2 from E. coli SM10 into P. aeruginosa as previously described (40). E. coli SM10 was counterselected by plating on PIA containing Cb. pFLP2 was cured by streaking Cb-resistant isolates on LB no-salt plates containing 5% (wt/vol) sucrose and incubating for 16 h at 30°C. Select colonies were streaked in parallel on LB, LB plus Cb, and LB plus Gm plates. Cb- and Gm-sensitive colonies were selected, and the pilT::FRT mutation was confirmed by PCR and DNA sequencing.
Twitching motility assays.
Twitching assays were performed as previously described (43). Briefly, single colonies were stab inoculated to the bottom of a 1% LB agar plate. The plates were incubated for 36 h at 37°C. After incubation, the agar was carefully removed and the adherent bacteria were stained with 1% (wt/vol) crystal violet dye, followed by washing with water to remove unbound dye. Twitching zone areas were measured using ImageJ software (NIH) (44). All experiments were performed in triplicate with at least three independent replicates.
Sheared surface protein preparation.
Surface pili and flagella were analyzed as described previously (43). Briefly, strains of interest were streaked in a grid-like pattern on LB agar plates and incubated at 37°C for 16 h. By the use of a glass coverslip, the bacteria were gently scraped from the surface of the agar and resuspended in 4.5 ml of PBS. Surface appendages were sheared by vortex mixing for 30 s. Suspensions were transferred to 1.5 ml Eppendorf tubes and pelleted by centrifugation for 5 min at 11,688 × g. The supernatant was transferred to fresh tubes and centrifuged for 20 min to remove the remaining cellular debris. The supernatant was transferred to new tubes, and a 1/10 volume of 5 M NaCl and 30% (wt/vol) polyethylene glycol 8000 (PEG 8000; Sigma-Aldrich) were added to precipitate soluble proteins. Tubes were incubated on ice for 90 min. Proteins were collected by centrifugation for 30 min at 11,688 × g, and the supernatant was discarded. The pellet was resuspended in 150 μl of SDS sample buffer (125 mM Tris [pH 6.8], 2% β-mercaptoethanol, 20% glycerol, 4% SDS, 0.001% bromophenol blue). Samples were boiled for 10 min and separated using 15% SDS-PAGE. Proteins were visualized by staining with Coomassie brilliant blue (0.1% Coomassie brilliant blue R-250, 50% methanol, 10% glacial acetic acid).
Preparation of whole-cell lysates.
Cultures were grown overnight at 37°C in LB supplemented with appropriate antibiotics to an OD600 of 0.6. A 1-ml aliquot of cells was collected by centrifugation at 2,292 × g for 3 min in a microcentrifuge. The cell pellet was resuspended in 100 μl of SDS sample buffer and boiled for 10 min. Whole-cell lysate samples were separated on 15% SDS-PAGE and subjected to Western blot analysis.
Western blot analysis.
Whole-cell lysate samples were separated on 15% SDS-PAGE and transferred to nitrocellulose membranes for 1 h at 225 mA. Membranes were blocked using 5% (wt/vol) low-fat skim milk powder–PBS for 1 h at room temperature on a shaking platform, followed by incubation with the appropriate antisera for 2 h at room temperature at the following dilutions: PilM, 1/2,500; PilNOP, 1/1,000 (each); PilA, 1/5,000. The membranes were washed twice in PBS for 5 min and then incubated in goat anti-rabbit IgG alkaline phosphatase-conjugated secondary antibody (Bio-Rad) at a dilution of 1/3,000 for 1 h at room temperature. The membranes were washed twice in PBS for 5 min and visualized with alkaline phosphatase developing reagent (Bio-Rad) following the manufacturer's protocol.
RESULTS
PilN and PilO dimer formation in the BTH assay.
Using a bacterial adenylate cyclase two-hybrid (BTH) system amenable to examining potential interactions of membrane-bound proteins (39, 45), we tested interactions between both truncated and full-length versions of PilN and PilO (Fig. 2A). Truncated constructs encompassed the core and CC domains but lacked the TMS and cytoplasmic N termini (Fig. 2B; see also Fig. S1 in the supplemental material). Stable expression of all fusions was confirmed via Western blotting with specific antisera to PilN or PilO (see Fig. S2). Interactions between the periplasmic regions of PilN and PilO (PilNΔ44 and PilOΔ51, respectively)—previously established by copurification and gel filtration (34, 46)—were confirmed, as were interactions between the full-length proteins (Fig. 2A). In addition to forming heterodimers with PilNΔ44, the PilOΔ51 fragment formed homodimers. Unexpectedly, full-length PilO did not homodimerize in the BTH assay, with β-galactosidase activities similar to those seen with the negative control (Fig. 2A). Neither truncated nor full-length PilN formed homodimers in this assay (Fig. 2A).
FIG 2.
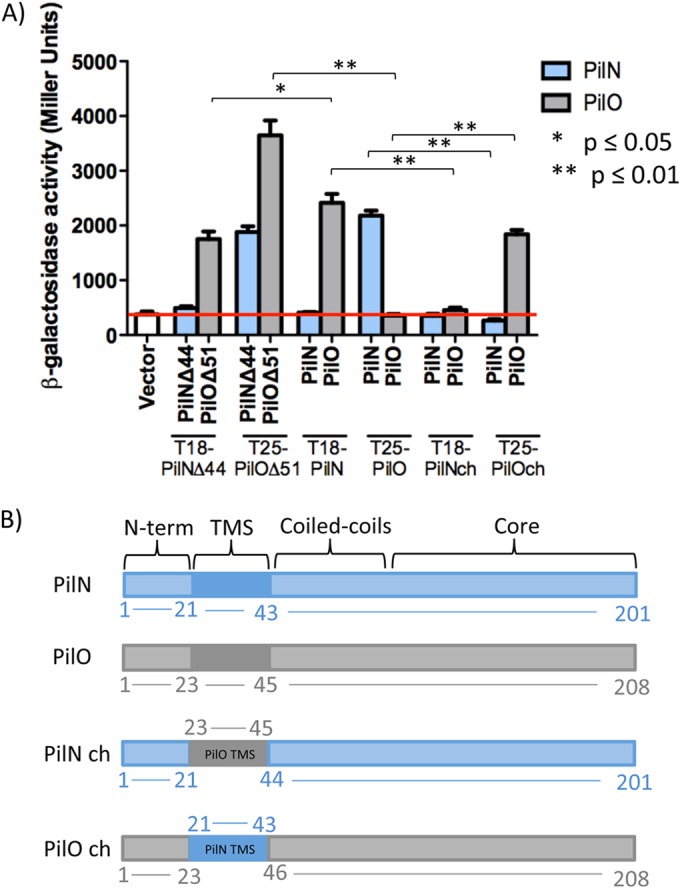
Chimeric proteins disrupt PilNO heterodimer formation in the BTH system. (A) N-terminal truncations of PilN and PilO (PilNΔ44 and PilOΔ51) and full-length versions of PilN and PilO and the PilN chimera (PilN ch) and the PilO chimera (PilO ch) were fused at their N termini to the T18 or T25 fragment of adenylate cyclase from Bordetella pertussis. PilN and PilO interactions were detected using both truncated and full-length versions, whereas only the truncated version of PilO formed homodimers. Swapping the TMS from PilO for that of PilN (PilO chimera) resulted in a loss of interaction with PilN but restored the interaction with PilO, whereas the PilN chimera (PilN with the TMS of PilO) was unable to interact with either full-length PilN or PilO. Experiments were performed in triplicate (n = 3). Bars represent the means ± standard errors. The red line indicates the β-galactosidase activity of the vector-only negative control (white bar). *, P ≤ 0.05; **, P ≤ 0.01. (B) Cartoon schematic of the PilN (blue), PilO (gray), and PilN chimera and PilO chimera constructs used in this study. Boundaries of the transmembrane segment (TMS), coiled coils, and core are indicated. N-term, N terminus.
The transmembrane segments of PilNO modulate their interaction in the BTH assay.
Although we showed previously (46) that the periplasmic regions of PilN and PilO are sufficient for their interaction, evidence from the Neisseria T4P system and the related type II secretion (T2S) system of Dickeya dadantii (for GspLM, the functional equivalents of PilMNO) indicated that TMSs likely participate in PilNO interactions (47, 48). Contributions of the P. aeruginosa PilN and PilO TMS to their interactions were examined using chimeras. The PilN chimera construct consisted of full-length PilN in which the TMS (residues 21 to 43) was replaced with that of PilO (residues 23 to 45). In the PilO chimera, the TMS (residues 23 to 45) was swapped for that of PilN (residues 21 to 43) (Fig. 2B). Although homodimerization of full-length PilO was negative in the BTH assay, the PilO chimera could interact with wild-type PilO but lost the ability to interact with PilN (Fig. 2A). Full-length PilN did not homodimerize, and the PilN chimera failed to interact with either PilN or PilO in this assay (Fig. 2A).
Point mutations in the coiled coils of PilN or PilO disrupt their interaction in the BTH assay.
Attempts to purify soluble P. aeruginosa PilN for structural studies, or to obtain an X-crystal structure of the PilNO heterodimer, have so far been unsuccessful. While a structure of a T. thermophilus PilN periplasmic fragment is available (35), its primary sequence identity with P. aeruginosa PilN is very low, and the arrangements of their secondary structure elements differ, precluding use of T. thermophilus PilN as a template to produce a high-confidence structural model. Instead, the truncated P. aeruginosa PilO homodimer structure (34) was used to generate a high-confidence Phyre2 (49) model of a PilNO heterodimer (Fig. 3A). The model was used to design a series of single or double point substitutions meant to disrupt hydrophobic or charged interactions at the core-core or coiled-coil interfaces (Fig. 1B and Tables 1 and 2). Because the PilNO heterodimer, like the PilO homodimer, is predicted to have a large buried surface area—2,082 Å2 for the PilOΔ68 homodimer (34)—some mutations were generated in pairs to rapidly exclude those with no effect. Residues at the predicted core-core interface between PilN α4 and PilO β4 or between PilN β3 and PilO α4, with estimated distances of less than 12 Å between Cα atoms, were selected for mutagenesis (Fig. 3B and Tables 1 and 2). Coiled-coil residues were selected based on their conservation in orthologs from other T4P-expressing bacteria (Fig. 3C and Tables 1 and 2). The mutations were introduced into the full-length T18-PilN and T25-PilO constructs, and stable expression of mutant fusions was verified by Western blotting (see Fig. S2 in the supplemental material). In the BTH assay, none of the core mutations (PilN ES132-133VA, MR141-142KL, and EV156-157AD) nor a combination of all these mutations (PilN Triple) disrupted the PilNO interaction (Fig. 4, gray bars). In the coiled-coil region, PilN L81K and LD64-65KK and PilO M92K disrupted the PilNO interaction (Fig. 4, black bars). These data suggested that, in addition to the TMS, the coiled-coil region was involved in PilNO interactions.
FIG 3.

Model of a PilNO heterodimer. (A) A ribbon diagram representation mapping the N and C termini, secondary structure elements, and structural domains of PilNΔ57 (blue) and PilOΔ68 (gray) using a Phyre2 (49) model of PilN generated using the PilOΔ68 homodimer (2RJZ [34]) as a template. (B) Closeup of the core-core domain, which consists of PilN α-helices 3 and 4 and β-strands 1 to 4 and PilO α-helices 3 and 4 and β-strands 1 to 5. The residues on PilN chosen for substitution are shown as follows: for the α4 helix, E132, S133, M141, and R142; for the β3 strand, E157 and V158. (C) Closeup of the PilNO coiled coils, which consist of α-helices 1 and 2. The residues chosen for substitution are shown as follows: for the PilN α1 helix, L64 and D65; for the α2 helix, L81 and L82; for the PilO α2 helix, M92.
TABLE 1.
Summary of PilN mutants and their phenotypes
| PilN mutation | Location | BTH PilO interaction | Twitching motility | Surface pili |
|---|---|---|---|---|
| ES132-133VA | Core | Yes | Yes | Yes |
| MR141-142KL | Core | Yes | No | No |
| EV157-158AD | Core | Yes | No | No |
| Triple mutation (ESMREV132-133–141-142–157-158VAKLAD) | Core | Yes | No | No |
| L81K | Coiled coils | No | Yes—reduced | Yes—hyperpiliated |
| L81A | Coiled coils | Yes | Yes | Yes |
| L82K | Coiled coils | Yes | Yes | Yes |
| LD64-65KK | Coiled coils | No | No | Yes—reduced |
| PilN chimera | TMS | No | No | Yes—reduced |
TABLE 2.
Summary of PilO mutants and their phenotypes
| PilO mutation | Location | BTH PilN interaction | Twitching motility | Surface pili |
|---|---|---|---|---|
| M92A | Coiled coils | Yes | Yes | Yes |
| M92K | Coiled coils | No | Yes—reduced | Yes—hyperpiliated |
| PilO chimera | TMS | No | Yes—reduced | Yes |
FIG 4.

Specific mutations in the coiled-coil regions disrupt PilNO interaction. Core mutations corresponding to PilN ES132-133VA, MR141-142KL, and EV157-158AD, individually and in combination (Triple), failed to disrupt interactions between full-length PilN and PilO in the BTH assay. Mutations in the coiled coils PilN L81K and LD64-65KK and PilO M92K disrupted the interaction, whereas PilN L81A and L82K and PilO M92A had no effect. Experiments were performed in triplicate (n = 3). Bars represent the means ± standard errors. The red line indicates β-galactosidase activity of the vector-only negative control (Vector). The positive control was the full-length T18-PilN and T25-PilO interaction (white bar labeled “PilN”).
Substitutions in PilN and PilO disrupt the dynamics of the T4P system.
PilMNOP are encoded in a single operon, and their stability is sensitive to perturbations in stoichiometry (41). To retain their native context, all point substitutions and the chimeric mutations swapping the TMSs of PilN and PilO were integrated into the P. aeruginosa chromosome at their original loci. After the stable expression of all alignment subcomplex proteins and the major pilin PilA was verified using specific antisera (see Fig. S3A in the supplemental material), the mutants were assessed for surface pilus expression and twitching motility.
We predicted that mutations failing to disrupt heterodimerization in the BTH assay (PilN ES132-133VA, MR141-142KL, EV157-158AD, Triple, L81A, and L82K and PilO M92A) would confer a wild-type phenotype, while those that prevented interactions (PilN L81K and LD64-65KK and the PilN chimera and PilO M92K and the PilO chimera) would prevent pilus assembly and, thus, motility. Mutants PilN ES132-133VA, L81A, and L82K and PilO M92A had phenotypes similar to the wild-type phenotype, but, surprisingly, core mutants PilN MR141-142KL and EV157-158AD—and consequently, the Triple mutant that contains those mutations—were unable to twitch due to a lack of pili (Fig. 5A). Coiled-coil mutants PilN L81K and PilO M92K that failed to interact in the BTH assay unexpectedly had wild-type or greater levels of pili but only ∼60% of the wild-type level of twitching motility, a statistically significant decrease (P ≤ 0.01) (Fig. 5A; see also Fig. S4 in the supplemental material). These phenotypes were consistent with defects in pilus retraction. The PilN LD64-65KK coiled-coil mutant and the PilN chimera had only a small amount of pili and no twitching motility, while the PilO chimera had levels of pili similar to the wild-type level but was significantly impaired in its ability to twitch, at ∼50% of the level seen with the wild-type control (P ≤ 0.01) (Fig. 5A; see also Fig. S4). These data indicate that specific residues at the core-core and coiled-coil interfaces, and in the TMS, play diverse roles in function of the PilN and PilO proteins and that interaction (or lack thereof) in the BTH assay does not necessarily correlate with dysfunction in the native context.
FIG 5.

Specific pilN and pilO point mutations on the P. aeruginosa PAK chromosome affect piliation and twitching motility. (A) PilN and PilO mutants plus wild-type (wt), nonpiliated (pilA), and hyperpiliated (pilT) strains and negative controls (pilN and pilO, respectively) were tested for twitching motility (top) and sheared surface pili (bottom). Columns, twitching motility zones from each mutant stained with 1% (wt/vol) crystal violet; rows, sheared surface proteins separated on a 15% SDS-PAGE gel stained with Coomassie brilliant blue to visualize pilins (top) and flagellins (loading control) (bottom) for each PilN and PilO mutant. The interaction (+) or lack of interaction (−) for each mutant with its wt partner in the BTH assay is summarized below the gel images. (B) Single point substitutions were created for mutants MR141-142KL, EV157-158AD, and LD64-65KK (M141K and R142L, E157A and V158D, and L64K and D65K, respectively) and the mutants subjected to twitching motility assays (top) and sheared surface pilin assays (bottom) as described above.
The core domain of PilN is more sensitive to perturbation than the coiled-coil region.
To further dissect the residues in the PilN MR141-142KL, EV157-158AD, and LD64-65KK mutants that were responsible for aberrant function, single mutations from each pair were introduced into the P. aeruginosa chromosome at the pilN locus. Single substitutions had no effect on the stable expression of alignment subcomplex components or intracellular levels of PilA (see Fig. S3B in the supplemental material). Single mutants M141K, R142L, E157A, and V158D recapitulated the double-mutant phenotype, with no pili and motility, while single mutants L64K and D65K had wild-type levels of pili and twitching motility (Fig. 5B; see also Fig. S4) and regained the ability to interact with PilO in the BTH assay (see Fig. S5). Therefore, single substitutions in the core domain of PilN independently disrupted T4P function, while the combination of coiled-coil mutations was necessary to produce the observed functional defects.
Nonpiliated mutants assemble pili in a retraction-deficient background.
In a pilT mutant lacking the retraction ATPase, any pili that are assembled become trapped on the surface of the bacteria (50). In a retraction-deficient background, mutants lacking individual components of the alignment subcomplex assemble fewer pili than a pilT control (23). We hypothesized that PilN MR141-142KL and EV157-158AD mutants may assemble pili in a retraction-deficient background, similarly to a pilN deletion mutant. The PilT retraction ATPase was inactivated in PilN ES132-133VA, MR141-142KL, and EV157-158AD mutants. As predicted, PilN MR141-142KL and EV157-158AD assembled a small amount of pili when retraction was disabled (Fig. 6), a lesser amount than the PilN ES132-133VA control (which has pili; Fig. 5A) or the wild type. Interestingly, the PilN point mutants assembled fewer pili than a mutant in which pilN was completely deleted (Fig. 6). These data suggest that small changes in the PilN MR141–142KL and EV157-158AD mutant interface with PilO severely restrict pilus assembly and that the resulting imbalance between extension and retraction leads to bald cells.
FIG 6.
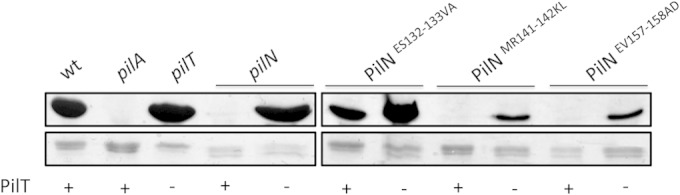
PilN ES132-133VA and MR141-142KL assemble pili in retraction-deficient backgrounds. PilN mutants ES132-133VA, MR141-142KL, and EV157-158AD were tested in a retraction-deficient (pilT) background for their ability to assemble pili. Mutants were compared to wild-type (wt), nonpiliated (pilA), and hyperpiliated (pilT) strains and to a PilN mutant (pilN) and a PilN/PilT double mutant (pilN/pilT). Sheared surface proteins were separated on a 15% SDS-PAGE gel stained with Coomassie brilliant blue to visualize pilins (top) and flagellins (loading control) (bottom) for each PilN mutant.
DISCUSSION
Various functions for the PilMNOP T4P alignment subcomplex have been proposed, including a possible role in relaying signals from the cytoplasmic motor components to the secretin complex in the OM (24, 25). Here we provide evidence suggesting that the highly conserved components PilN and PilO contribute to both extension and retraction of the pilus fiber. PilNO interactions are precise—because they can be altered by mutation of single residues, resulting in loss of function—and specific—since extension or retraction can be independently perturbed, depending on the location and nature of the mutation.
Structural and functional similarities between the T2S and T4P systems have long been recognized (51). Despite minimal sequence identity between orthologous components of the alignment subcomplexes in the two systems, atomic resolution structures revealed a high degree of structural similarity, suggesting conservation of function (33, 34, 46, 52–54). Previous studies suggested that multiple segments of GspL and GspM, the T2S equivalents of PilMN and PilO, respectively, participate in their interaction (47, 48). Our BTH assay results confirmed the presence of heterodimeric interactions between the periplasmic domains of PilN and PilO (core plus coiled coil) (see Fig. S1 in the supplemental material), as well as the full-length, membrane-bound forms (41, 46). The increased β-galactosidase activity observed for full-length versus truncated PilNO suggested that the TMSs contribute to the interaction. Although full-length PilO did not homodimerize in the BTH assay, a PilO chimera with the PilN TMS gained the ability to interact with full-length PilO, while losing the ability to interact with PilN (Fig. 2A), suggesting that the TMS modulates the interaction. Failure of PilO to homodimerize in the BTH assay may relate to the low isoelectric point (pI 4.2) of its coiled-coil region (34), which could cause charge repulsion, increasing the distance between the T18 and T25 fusion tags and preventing enzyme reconstitution. A similar phenomenon could explain the lack of PilN homodimerization in the BTH assay due the high (10.2) pI in this region of the protein (34), although it seems less likely given that even the truncated form of PilN failed to interact with itself (Fig. 2A). The PilN chimera (which has the TMS of PilO) failed to interact with full-length PilO, or with full-length PilN (Fig. 2A). Given that their TMSs should be compatible, failure of the PilN chimera to interact with PilN (Fig. 2A) did not match our prediction but suggests that additional contacts in the coiled-coil and core regions are required to stabilize TMS interactions. These findings are consistent with BTH data for N. meningitidis T4P components, in that the PilN ortholog interacts with PilO but not with itself, and with previous genetic and biochemical data showing that PilN is misfolded and unstable in the absence of PilO (34, 41, 46, 48). In contrast, PilN from the T. thermophilus T4P system (35) and GspL in the T2S system (54–56) form homodimers. In the T. thermophilus model, homodimers of PilMN were hypothesized to form the initial transmembrane platform for assembly, followed by docking of PilO onto PilN to form PilNO heterodimers (35). Partner switching between the homo- and heterodimer states of GspL and GspM (PilMN and PilO, respectively) in the T2S system has been reported and has functional implications (47, 56). Whether the interactions among components of the T4P alignment subcomplex are dynamic is under investigation.
Defects in pilus retraction caused by inactivation of the retraction ATPase, PilT, result in a hyperpiliated phenotype (50). Loss of the PilT paralog PilU (57), or specific point mutations in the antiretraction factor PilY1 (58) or the platform protein PilC (23), can also result in piliated cells with reduced motility. We identified new mutations in PilNO (PilN L81K and PilO M92K) that result in the distinctive combination of reduced twitching motility and increased piliation, suggesting that they affect pilus retraction. These apposing residues are located in the α2-helices of the coiled-coil 2 (CC-2) region (Fig. 3C), which were previously hypothesized to be key interaction interfaces due to their large charge differential (34). The residues of interest likely form a key hydrophobic contact, since PilN L81A—but not L81K—retains wild-type function; similarly, PilO M92A has normal function, while M92K disrupts twitching (Fig. 5A).
Substituting the TMS of PilN for that of PilO in P. aeruginosa caused a marked reduction in motility (see Fig. S4 in the supplemental material) without decreasing piliation (Fig. 5A), also characteristic of a retraction defect. Although T4P dynamics were clearly impaired, the PilO chimera retained the ability to heterodimerize with PilN in P. aeruginosa, as evidenced by the stability of the proteins (see Fig. S3A) and their ability to assemble pili (Fig. 5A). When the TMS of PilN was replaced with that of PilO, the PilN chimera was unable to homo- or heterodimerize in the BTH assay and in P. aeruginosa led to trace amounts of surface pili and no motility. The PilN chimera was detrimental to T4P assembly, potentially due to decreased interaction with its cytoplasmic partner, PilM (see Fig. S6).
The contributions of the TMS of inner membrane components to their interactions have been largely overlooked in the T4P and T2S systems due to past focus on biochemical and structural analysis of soluble cytoplasmic or periplasmic fragments. In the T2S system of D. dadantii, multiple interactions between the TMSs of GspL and GspM, plus a third integral IM protein, GspC (the equivalent of PilP), were demonstrated using pulldown assays, BTH assays, and Cys cross-linking (47). The mechanism of T2S pseudopilus disassembly is unknown, but in the case of T4P, the phenotypes of the coiled-coil and TMS mutants support the conclusion that precise PilNO interactions contribute to pilus disassembly, as well as assembly.
Multiple mutations at the PilNO core-core interface had no effect on interaction in the pairwise BTH assay, so we were surprised when four of six mutations at this surface severely reduced pilus assembly when introduced into P. aeruginosa (Fig. 4 and 5A). These results validate the utility of the PilNO Phyre2 model in selection of functionally significant residues and stress the importance of testing the effects of mutations in a native, multiprotein complex context. PilN mutations MR141-142KL and EV157-158AD are located on α4 and β3, respectively, while the PilN ES132-133VA pair, which had no effect on function, is located in an unstructured region at the top of α4 (Fig. 3B). Loss of function might relate to subtle shifts in the core-core interface, such that it is no longer correctly oriented for optimal pilus assembly. Alternatively, these mutations could affect interactions of the PilNO heterodimer with the lipoprotein, PilP, which has a long unstructured N-terminal region that is unstable in the absence of the PilNO heterodimer (46). PilP was stable in all mutants examined (see Fig. S3 in the supplemental material), but the MR141-142KL and EV157-158AD substitutions may affect its interactions with the PilNO heterodimer, reducing pilus assembly. PilN MR141-142KL and EV157-158AD double mutants and their single-mutant derivatives assemble pili on the surface only when retraction is blocked (Fig. 6), implying inefficient pilus extension, or a reduced ability to counteract retraction (23, 59). Correct orientation of PilNOP interfaces could be important for optimal PilP and PilQ interactions and control of secretin gating (36, 46, 60).
The PilN LD64-65KK mutation located in the first α-helix (CC-1 region) (Fig. 3C) disrupted PilNO interactions in the BTH assay and, in P. aeruginosa, decreased the amount of surface piliation and twitching motility (Fig. 4 and 5A), without affecting the stability of the alignment subcomplex proteins (see Fig. S3A in the supplemental material). This defect was less pronounced than those caused by the core mutations. Mutation of both residues in PilN LD64-65KK was necessary to produce the observed phenotype (Fig. 5B), suggesting that this part of the coiled-coil region is more permissive for substitution; of note, its contribution to function would have been missed had only single-residue substitutions been tested.
From these data, we propose a model in which the TMSs of PilN and PilO initiate their heterodimerization. PilN and PilO contacts are subsequently stabilized through interactions of their oppositely charged coiled-coil regions, as predicted for EpsLM in the Vibrio T2S system (34). The PilNO heterodimer then interacts with PilP (46), creating, with PilM bound to PilN′s N terminus (33, 34), a transenvelope complex upon PilP's interaction with PilQ (24). Without the correct contacts in these regions, cytoplasmic components such as PilM may be unable to accurately relay conformational changes arising from the activity of IM platform components, including the PilB and PilT ATPases. This model could explain why the PilN chimera was more detrimental to the function of the system than the PilO chimera, as the TMS swap in PilN might alter relay of conformational changes from PilM. In the T2S and the type IVb pilus (T4bP) systems, PilB-like ATPases have been cocrystallized with their PilM homologs, confirming interactions between these components (61–63). Alternatively, communication between the PilMNOP subcomplex and PilQ could be altered such that the secretin is not appropriately gated to allow for normal pilus assembly/disassembly dynamics (37, 38). Finally, since interaction between components of the alignment subcomplex and pilin subunits has been confirmed in both T2S and T4P systems (24, 35, 48, 64), changes in those interactions may also contribute to loss of function.
Together, our findings suggest that precise PilNO interactions play a critical role in T4P dynamics, supporting the idea that the alignment subcomplex is not simply a static connector of inner and outer membrane components. Depending on their location and nature, small changes can cause a range of effects from subtle (less piliation, reduced motility) to drastic (no piliation or hyperpiliation, loss of motility). The data emphasize the importance of studying protein-protein interactions in their native context and stoichiometry, since chromosomal mutations provided key information on dynamics that could not have been predicted from BTH assay results. Our discovery that mutating even a single residue at a critical contact point can render the T4P system dysfunctional is encouraging from the perspective of drug development. Small-molecule disruption of protein-protein interactions has been successful in the T3S and T4S systems (4–6, 65, 66) but can be challenging due to the potentially large interaction interfaces involved. This work shows that, despite their large predicted buried surface areas, there are critical interfaces of PilNO that can be targeted to block piliation and/or motility without completely disrupting their interactions, suggesting that inhibitors could have a high likelihood of success.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an operating grant (MOP-93585) from the Canadian Institutes of Health Research to L.L.B. and P.L.H. P.L.H. is the recipient of a Tier I Canada Research Chair in Structural Biology.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00220-15.
REFERENCES
- 1.Levy SB, Marshall B. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med 10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 2.Coates AR, Hu Y. 2007. Novel approaches to developing new antibiotics for bacterial infections. Br J Pharmacol 152:1147–1154. doi: 10.1038/sj.bjp.0707432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clatworthy AE, Pierson E, Hung DT. 2007. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol 3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 4.Keyser P, Elofsson M, Rosell S, Wolf-Watz H. 2008. Virulence blockers as alternatives to antibiotics: type III secretion inhibitors against Gram-negative bacteria. J Intern Med 264:17–29. doi: 10.1111/j.1365-2796.2008.01941.x. [DOI] [PubMed] [Google Scholar]
- 5.Kauppi AM, Nordfelth R, Uvell H, Wolf-Watz H, Elofsson M. 2003. Targeting bacterial virulence: inhibitors of type III secretion in Yersinia. Chem Biol 10:241–249. doi: 10.1016/S1074-5521(03)00046-2. [DOI] [PubMed] [Google Scholar]
- 6.Bailey L, Gylfe A, Sundin C, Muschiol S, Elofsson M, Nordstrom P, Henriques-Normark B, Lugert R, Waldenstrom A, Wolf-Watz H, Bergstrom S. 2007. Small molecule inhibitors of type III secretion in Yersinia block the Chlamydia pneumoniae infection cycle. FEBS Lett 581:587–595. doi: 10.1016/j.febslet.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Hahn HP. 1997. The type-4 pilus is the major virulence-associated adhesin of Pseudomonas aeruginosa–a review. Gene 192:99–108. doi: 10.1016/S0378-1119(97)00116-9. [DOI] [PubMed] [Google Scholar]
- 8.Bieber D, Ramer SW, Wu CY, Murray WJ, Tobe T, Fernandez R, Schoolnik GK. 1998. Type IV pili, transient bacterial aggregates, and virulence of enteropathogenic Escherichia coli. Science 280:2114–2118. doi: 10.1126/science.280.5372.2114. [DOI] [PubMed] [Google Scholar]
- 9.Farinha MA, Conway BD, Glasier LM, Ellert NW, Irvin RT, Sherburne R, Paranchych W. 1994. Alteration of the pilin adhesin of Pseudomonas aeruginosa PAO results in normal pilus biogenesis but a loss of adherence to human pneumocyte cells and decreased virulence in mice. Infect Immun 62:4118–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bradley DE. 1973. Basic characterization of a Pseudomonas aeruginosa pilus-dependent bacteriophage with a long noncontractile tail. J Virol 12:1139–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burrows LL. 2012. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu Rev Microbiol 66:493–520. doi: 10.1146/annurev-micro-092611-150055. [DOI] [PubMed] [Google Scholar]
- 12.Burrows LL. 2005. Weapons of mass retraction. Mol Microbiol 57:878–888. doi: 10.1111/j.1365-2958.2005.04703.x. [DOI] [PubMed] [Google Scholar]
- 13.Mattick JS. 2002. Type IV pili and twitching motility. Annu Rev Microbiol 56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- 14.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 15.Pelicic V. 2008. Type IV pili: e pluribus unum? Mol Microbiol 68:827–837. doi: 10.1111/j.1365-2958.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- 16.Govan JR, Deretic V. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60:539–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skerker JM, Berg HC. 2001. Direct observation of extension and retraction of type IV pili. Proc Natl Acad Sci U S A 98:6901–6904. doi: 10.1073/pnas.121171698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craig L, Pique ME, Tainer JA. 2004. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- 19.Burkhardt J, Vonck J, Averhoff B. 2011. Structure and function of PilQ, a secretin of the DNA transporter from the thermophilic bacterium Thermus thermophilus HB27. J Biol Chem 286:9977–9984. doi: 10.1074/jbc.M110.212688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koo J, Tammam S, Ku SY, Sampaleanu LM, Burrows LL, Howell PL. 2008. PilF is an outer membrane lipoprotein required for multimerization and localization of the Pseudomonas aeruginosa Type IV pilus secretin. J Bacteriol 190:6961–6969. doi: 10.1128/JB.00996-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koo J, Tang T, Harvey H, Tammam S, Sampaleanu L, Burrows LL, Howell PL. 2013. Functional mapping of PilF and PilQ in the Pseudomonas aeruginosa type IV pilus system. Biochemistry 52:2914–2923. doi: 10.1021/bi3015345. [DOI] [PubMed] [Google Scholar]
- 22.Chiang P, Habash M, Burrows LL. 2005. Disparate subcellular localization patterns of Pseudomonas aeruginosa type IV pilus ATPases involved in twitching motility. J Bacteriol 187:829–839. doi: 10.1128/JB.187.3.829-839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takhar HK, Kemp K, Kim M, Howell PL, Burrows LL. 2013. The platform protein is essential for type IV pilus biogenesis. J Biol Chem 288:9721–9728. doi: 10.1074/jbc.M113.453506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tammam S, Sampaleanu LM, Koo J, Manoharan K, Daubaras M, Burrows LL, Howell PL. 2013. PilMNOPQ from the Pseudomonas aeruginosa type IV pilus system form a transenvelope protein interaction network that interacts with PilA. J Bacteriol 195:2126–2135. doi: 10.1128/JB.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayers M, Howell PL, Burrows LL. 2010. Architecture of the type II secretion and type IV pilus machineries. Future Microbiol 5:1203–1218. doi: 10.2217/fmb.10.76. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen Y, Jackson SG, Aidoo F, Junop M, Burrows LL. 2010. Structural characterization of novel Pseudomonas aeruginosa type IV pilins. J Mol Biol 395:491–503. doi: 10.1016/j.jmb.2009.10.070. [DOI] [PubMed] [Google Scholar]
- 27.Rudel T, Scheurerpflug I, Meyer TF. 1995. Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature 373:357–359. doi: 10.1038/373357a0. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen Y, Sugiman-Marangos S, Harvey H, Bell SD, Charlton CL, Junop MS, Burrows LL. 2014. Pseudomonas aeruginosa minor pilins prime type IVa pilus assembly and promote surface display of the PilY1 adhesin. J Biol Chem doi: 10.1074/jbc.M114.616904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strom MS, Lory S. 1993. Structure-function and biogenesis of the type IV pili. Annu Rev Microbiol 47:565–596. doi: 10.1146/annurev.mi.47.100193.003025. [DOI] [PubMed] [Google Scholar]
- 30.Beatson SA, Whitchurch CB, Sargent JL, Levesque RC, Mattick JS. 2002. Differential regulation of twitching motility and elastase production by Vfr in Pseudomonas aeruginosa. J Bacteriol 184:3605–3613. doi: 10.1128/JB.184.13.3605-3613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darzins A. 1994. Characterization of a Pseudomonas aeruginosa gene cluster involved in pilus biosynthesis and twitching motility: sequence similarity to the chemotaxis proteins of enterics and the gliding bacterium Myxococcus xanthus. Mol Microbiol 11:137–153. doi: 10.1111/j.1365-2958.1994.tb00296.x. [DOI] [PubMed] [Google Scholar]
- 32.Leighton TL, Buensuceso R, Howell PL, Burrows LL. 2015. Biogenesis of Pseudomonas aeruginosa type IV pili and regulation of their function. Environ Microbiol doi: 10.1111/1462-2920.12849. [DOI] [PubMed] [Google Scholar]
- 33.Karuppiah V, Derrick JP. 2011. Structure of the PilM-PilN inner membrane type IV pilus biogenesis complex from Thermus thermophilus. J Biol Chem 286:24434–24442. doi: 10.1074/jbc.M111.243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sampaleanu LM, Bonanno JB, Ayers M, Koo J, Tammam S, Burley SK, Almo SC, Burrows LL, Howell PL. 2009. Periplasmic domains of Pseudomonas aeruginosa PilN and PilO form a stable heterodimeric complex. J Mol Biol 394:143–159. doi: 10.1016/j.jmb.2009.09.037. [DOI] [PubMed] [Google Scholar]
- 35.Karuppiah V, Collins RF, Thistlethwaite A, Gao Y, Derrick JP. 2013. Structure and assembly of an inner membrane platform for initiation of type IV pilus biogenesis. Proc Natl Acad Sci U S A 110:E4638–E4647. doi: 10.1073/pnas.1312313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balasingham SV, Collins RF, Assalkhou R, Homberset H, Frye SA, Derrick JP, Tonjum T. 2007. Interactions between the lipoprotein PilP and the secretin PilQ in Neisseria meningitidis. J Bacteriol 189:5716–5727. doi: 10.1128/JB.00060-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichow SL, Korotkov KV, Hol WG, Gonen T. 2010. Structure of the cholera toxin secretion channel in its closed state. Nat Struct Mol Biol 17:1226–1232. doi: 10.1038/nsmb.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korotkov KV, Gonen T, Hol WG. 2011. Secretins: dynamic channels for protein transport across membranes. Trends Biochem Sci 36:433–443. doi: 10.1016/j.tibs.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Battesti A, Bouveret E. 2012. The bacterial two-hybrid system based on adenylate cyclase reconstitution in Escherichia coli. Methods 58:325–334. doi: 10.1016/j.ymeth.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 40.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 41.Ayers M, Sampaleanu LM, Tammam S, Koo J, Harvey H, Howell PL, Burrows LL. 2009. PilM/N/O/P proteins form an inner membrane complex that affects the stability of the Pseudomonas aeruginosa type IV pilus secretin. J Mol Biol 394:128–142. doi: 10.1016/j.jmb.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 42.Asikyan ML, Kus JV, Burrows LL. 2008. Novel proteins that modulate type IV pilus retraction dynamics in Pseudomonas aeruginosa. J Bacteriol 190:7022–7034. doi: 10.1128/JB.00938-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kus JV, Tullis E, Cvitkovitch DG, Burrows LL. 2004. Significant differences in type IV pilin allele distribution among Pseudomonas aeruginosa isolates from cystic fibrosis (CF) versus non-CF patients. Microbiology 150:1315–1326. doi: 10.1099/mic.0.26822-0. [DOI] [PubMed] [Google Scholar]
- 44.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol 187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tammam S, Sampaleanu LM, Koo J, Sundaram P, Ayers M, Chong PA, Forman-Kay JD, Burrows LL, Howell PL. 2011. Characterization of the PilN, PilO and PilP type IVa pilus subcomplex. Mol Microbiol 82:1496–1514. doi: 10.1111/j.1365-2958.2011.07903.x. [DOI] [PubMed] [Google Scholar]
- 47.Lallemand M, Login FH, Guschinskaya N, Pineau C, Effantin G, Robert X, Shevchik VE. 2013. Dynamic interplay between the periplasmic and transmembrane domains of GspL and GspM in the type II secretion system. PLoS One 8:e79562. doi: 10.1371/journal.pone.0079562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Georgiadou M, Castagnini M, Karimova G, Ladant D, Pelicic V. 2012. Large-scale study of the interactions between proteins involved in type IV pilus biology in Neisseria meningitidis: characterization of a subcomplex involved in pilus assembly. Mol Microbiol 84:857–873. doi: 10.1111/j.1365-2958.2012.08062.x. [DOI] [PubMed] [Google Scholar]
- 49.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 50.Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, Koomey M. 1998. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Microbiol 29:321–330. doi: 10.1046/j.1365-2958.1998.00935.x. [DOI] [PubMed] [Google Scholar]
- 51.Bose N, Payne SM, Taylor RK. 2002. Type 4 pilus biogenesis and type II-mediated protein secretion by Vibrio cholerae occur independently of the TonB-facilitated proton motive force. J Bacteriol 184:2305–2309. doi: 10.1128/JB.184.8.2305-2309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abendroth J, Bagdasarian M, Sandkvist M, Hol WG. 2004. The structure of the cytoplasmic domain of EpsL, an inner membrane component of the type II secretion system of Vibrio cholerae: an unusual member of the actin-like ATPase superfamily. J Mol Biol 344:619–633. doi: 10.1016/j.jmb.2004.09.062. [DOI] [PubMed] [Google Scholar]
- 53.Abendroth J, Rice AE, McLuskey K, Bagdasarian M, Hol WG. 2004. The crystal structure of the periplasmic domain of the type II secretion system protein EpsM from Vibrio cholerae: the simplest version of the ferredoxin fold. J Mol Biol 338:585–596. doi: 10.1016/j.jmb.2004.01.064. [DOI] [PubMed] [Google Scholar]
- 54.Abendroth J, Mitchell DD, Korotkov KV, Johnson TL, Kreger A, Sandkvist M, Hol WG. 2009. The three-dimensional structure of the cytoplasmic domains of EpsF from the type 2 secretion system of Vibrio cholerae. J Struct Biol 166:303–315. doi: 10.1016/j.jsb.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLaughlin LS, Haft RJ, Forest KT. 2012. Structural insights into the type II secretion nanomachine. Curr Opin Struct Biol 22:208–216. doi: 10.1016/j.sbi.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Py B, Loiseau L, Barras F. 2001. An inner membrane platform in the type II secretion machinery of Gram-negative bacteria. EMBO Rep 2:244–248. doi: 10.1093/embo-reports/kve042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whitchurch CB, Mattick JS. 1994. Characterization of a gene, pilU, required for twitching motility but not phage sensitivity in Pseudomonas aeruginosa. Mol Microbiol 13:1079–1091. doi: 10.1111/j.1365-2958.1994.tb00499.x. [DOI] [PubMed] [Google Scholar]
- 58.Orans J, Johnson MD, Coggan KA, Sperlazza JR, Heiniger RW, Wolfgang MC, Redinbo MR. 2010. Crystal structure analysis reveals Pseudomonas PilY1 as an essential calcium-dependent regulator of bacterial surface motility. Proc Natl Acad Sci U S A 107:1065–1070. doi: 10.1073/pnas.0911616107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carbonnelle E, Helaine S, Nassif X, Pelicic V. 2006. A systematic genetic analysis in Neisseria meningitidis defines the Pil proteins required for assembly, functionality, stabilization and export of type IV pili. Mol Microbiol 61:1510–1522. doi: 10.1111/j.1365-2958.2006.05341.x. [DOI] [PubMed] [Google Scholar]
- 60.Lee HM, Wang KC, Liu YL, Yew HY, Chen LY, Leu WM, Chen DC, Hu NT. 2000. Association of the cytoplasmic membrane protein XpsN with the outer membrane protein XpsD in the type II protein secretion apparatus of Xanthomonas campestris pv. campestris. J Bacteriol 182:1549–1557. doi: 10.1128/JB.182.6.1549-1557.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abendroth J, Murphy P, Sandkvist M, Bagdasarian M, Hol WG. 2005. The X-ray structure of the type II secretion system complex formed by the N-terminal domain of EpsE and the cytoplasmic domain of EpsL of Vibrio cholerae. J Mol Biol 348:845–855. doi: 10.1016/j.jmb.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 62.Yamagata A, Milgotina E, Scanlon K, Craig L, Tainer JA, Donnenberg MS. 2012. Structure of an essential type IV pilus biogenesis protein provides insights into pilus and type II secretion systems. J Mol Biol 419:110–124. doi: 10.1016/j.jmb.2012.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu C, Korotkov KV, Hol WG. 2014. Crystal structure of the full-length ATPase GspE from the Vibrio vulnificus type II secretion system in complex with the cytoplasmic domain of GspL. J Struct Biol 187:223–235. doi: 10.1016/j.jsb.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gray MD, Bagdasarian M, Hol WG, Sandkvist M. 2011. In vivo cross-linking of EpsG to EpsL suggests a role for EpsL as an ATPase-pseudopilin coupling protein in the type II secretion system of Vibrio cholerae. Mol Microbiol 79:786–798. doi: 10.1111/j.1365-2958.2010.07487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith MA, Coincon M, Paschos A, Jolicoeur B, Lavallee P, Sygusch J, Baron C. 2012. Identification of the binding site of Brucella VirB8 interaction inhibitors. Chem Biol 19:1041–1048. doi: 10.1016/j.chembiol.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 66.Paschos A, den Hartigh A, Smith MA, Atluri VL, Sivanesan D, Tsolis RM, Baron C. 2011. An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infect Immun 79:1033–1043. doi: 10.1128/IAI.00993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.