

ABSTRACT
Oxidative stresses triggered by reactive oxygen species (ROS) that damage various cellular components are unavoidable for virtually all living organisms. In defense, microorganisms have evolved sophisticated mechanisms to sense, respond to, and battle against ROS. Shewanella oneidensis, an important research model for applied and environmental microbes, employs OxyR to mediate the response to H2O2 by derepressing the production of the major H2O2 scavenger KatB as a major means toward these goals. Surprisingly, despite enhanced H2O2 degradation, the oxyR mutant carries a viability deficiency phenotype (plating defect), which can be suppressed by the addition of exogenous iron species. Experiments showed that the defect was not due to iron starvation. Rather, multiple lines of evidence suggested that H2O2 generated abiotically in lysogeny broth (LB) is responsible for the defect by quickly killing mutant cells. We then showed that the iron species suppressed the plating defect by two distinct mechanisms, either as an H2O2 scavenger without involving living cells or as an environmental cue to stimulate an OxyR-independent response to help cells cope with oxidative stress. Based on the suppression of the plating defect by overproduction of H2O2 scavengers in vivo, we propose that cellular components that are vulnerable to H2O2 and responsible for the defect may reside outside the cytoplasm.
IMPORTANCE In bacteria, OxyR is the major regulator controlling the cellular response to H2O2. The loss of OxyR results in reduced viability in many species, but the underlying mechanism is unknown. We showed in S. oneidensis that this defect was due to H2O2 generated abiotically in LB. We then showed that this defect could be corrected by the addition of Fe2+ or catalase to the LB or increased intracellular production of catalase. Further analyses revealed that Fe2+ was able not only to decompose H2O2 directly but also to stimulate the activity of OxyR-independent H2O2-scavenging enzymes. Our data indicate that iron species play a previously underappreciated role in protecting cells from H2O2 in environments.
INTRODUCTION
Oxidative stress, one of the unavoidable crises for the vast majority of aerobic organisms, is essentially an imbalance between the production of reactive oxygen species (ROS) and the ability of cells to counteract or detoxify their harmful effects (1). ROS include superoxide (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (·OH), which cause damage to biomolecules such as lipids, proteins, and DNA (1). As an uncharged species that penetrates membranes, H2O2 is life threatening by itself and, more importantly, by reacting with Fe2+ to generate the deadly toxic ·OH, a process called the Fenton reaction (2). H2O2 is ubiquitous because of the existence of diverse ways for its generation, both biologically and abiotically (3). H2O2, in concert with O2−, is a metabolic by-product of cellular oxygen respiration, mostly through the accidental auto-oxidation of nonrespiratory flavoproteins and the turnover of committed oxidases (4, 5). Additionally, many organisms synthesize H2O2 and secrete it into their environment to suppress the growth of potentially competing and/or detrimental microbes (3). Moreover, H2O2 is formed by chemical processes without the involvement of living cells during the interaction of reduced metals and sulfur species with oxygen and, also, by photochemical reactions (3, 6). It is estimated that H2O2 is generated through aerobic respiration in Escherichia coli at a rate of 10 to 15 μM/s during growth in air-saturated glucose medium, whereas the amount of H2O2 produced by abiotic processes is as much as 20 μM (3, 7). In defense, organisms have evolved various strategies to cope with H2O2. The most effective and efficient strategy is to express enzymes that can directly decompose the oxidant, including alkylhydroperoxide reductase (Ahp), catalases, and various peroxidases (8). Ahp serves as the major scavenger when intracellular H2O2 levels are low, while catalases dominate when H2O2 levels are high. Although a large number of peroxidases have been identified, our understanding of them is much less complete, presumably because their effects are less apparent (9). Another strategy exploited by cells is to express ROS-resistant isoenzymes or pathways to replace the sensitive ones, such as activation of fumarase C and aconitase A, as well as induction of the alternative SUF Fe-S cluster synthesis system, in addition to the ISC housekeeping system (10, 11). Besides these mechanisms, repair systems like the base excision repair (BER) pathway and SOS are activated to address damaged components after the stress has been removed and cellular metabolism resumed (1).
In E. coli and many other bacteria, OxyR, a LysR family transcriptional regulator, is the predominant regulator mediating the cellular response to H2O2 (1). The activation of OxyR relies on the oxidation of two conserved cysteine residues and the formation of an intramolecular disulfide bond when H2O2 levels are over the limit (12). Upon activation, the regulator turns on dozens of genes, including the H2O2 scavenger genes ahpCF and katG (catalase gene), as well as the iron storage protein gene dps (13, 14). Conceivably, E. coli cells lacking the oxyR gene are hypersensitive to H2O2 (15, 16). While this phenotype is regarded as the hallmark for the oxyR mutants, the impacts of loss of the gene appear to be broad and diverse (17–20). The oxyR mutants are highly susceptible to redox-cycling agents, such as medadione and paraquat, which induce O2− generation, but not to organic peroxides, such as tert butyl hydroperoxide (21). In addition, the viability deficiency phenotype (plating defect) was often observed with the oxyR mutants (17, 21, 22). However, the growth rate of cells in liquid medium is not significantly affected by the oxyR mutations in general (22, 23). An exception to the E. coli OxyR model is the OxyR homolog of Neisseria species, whose loss results in enhanced resistance to H2O2 killing (24). This is because Neisseria OxyR acts as both a repressor of catalase expression and an activator for other members of its regulon (25, 26).
Members of the genus Shewanella, a group of Gram-negative facultative gammaproteobacteria, inhabit redox-stratified environments and are renowned for their versatile respiratory abilities (27). These bacteria are more sensitive to H2O2 than E. coli, in concert with the high susceptibility to UV and ionizing radiation, understandings derived from studies of the model species Shewanella oneidensis (28–30). Not surprisingly, S. oneidensis has an OxyR homolog, which is demonstrated to be the main regulator mediating the cellular response to H2O2 (29). However, several lines of evidence show that this protein has properties significantly different from those of its counterparts studied to date. First, although S. oneidensis OxyR acts as both an activator and a repressor, like the Neisseria OxyR proteins (26), it controls a much larger number of genes. Upon OxyR activation, S. oneidensis mainly relies on derepression of katB (encoding the major catalase) and dps genes to alleviate oxidative damage by degrading H2O2 and by sequestering the intracellular free iron, respectively (29). As a result, an S. oneidensis oxyR mutant degrades H2O2 more rapidly than the wild type. Meanwhile, a number of genes, including ahpCF and those encoding peroxidases, are positively controlled by OxyR, but their contribution to protection against oxidative stress is much less evident. Second, to date, S. oneidensis OxyR has been the only one that could not functionally complement E. coli OxyR, suggesting that this OxyR has evolved some unique structural features. Third, despite its enhanced ability to decompose H2O2, the oxyR mutant bears the plating defect, a phenotype observed in bacteria whose OxyR proteins act as a positive regulator for catalase genes (17, 22, 23). Fourth, the plating defect can be suppressed by supplying rather than limiting exogenous iron. This is in sharp contrast to the established strategy utilized by many other bacteria to combat oxidative stress, where Fe2+ binding and iron storage are induced during H2O2 stress, leading to reduced free iron levels (2).
The goal of this study was to elucidate the mechanism underlying the colony development defect of the S. oneidensis oxyR mutant. We found that this defect was neither critically linked to aerobic respiration, a major process generating H2O2 intracellularly, nor attributable to iron starvation. Rather, cells lacking OxyR became more vulnerable to H2O2 and died quickly on lysogeny broth (LB) plates because of H2O2 generated abiotically. We then developed evidence suggesting that iron functioned both as an H2O2 scavenger without involving living cells and as an environmental cue to stimulate an OxyR-independent response to help cells to combat oxidative stress. Furthermore, suppression of the plating defect by overproduction of H2O2 scavengers in vivo suggested that the components responsible may be localized outside the cytoplasm.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
All bacterial strains and plasmids used in this study can be found in Table 1. Information about all of the primers used in this study is given in Table S1 in the supplemental material. E. coli and S. oneidensis were grown in lysogeny broth (LB; Difco, Detroit, MI) under aerobic conditions at 37 and 30°C for genetic manipulation. For testing of the plating defect, both LB and the defined MS medium [KCl, 1.34 mM; NaH2PO4, 5 mM; Na2SO4, 0.7 mM; NaCl, 52 mM; piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 3 mM; NH4Cl, 28 mM; sodium lactate, 30 mM; MgSO4, 1 mM; CaCl2, 0.27 mM; and FeCl2, 3.6 μM, pH 7.0] were used. When necessary, the following chemicals were added to the growth medium: 2,6-diaminopimelic acid (DAP), 0.3 mM; ampicillin, 50 μg/ml; kanamycin, 50 μg/ml; gentamicin, 15 μg/ml; and streptomycin, 100 μg/ml.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| E. coli strains | ||
| DH5α | Host for cloning | Laboratory stock |
| WM3064 | ΔdapA, donor strain for conjugation | W. Metcalf, UIUC |
| S. oneidensis strains | ||
| MR-1 | Wild type | Laboratory stock |
| HG0956-8 | ΔahpCF, derived from MR-1 | 29 |
| HG1070 | ΔkatB, derived from MR-1 | 29 |
| HG1328 | ΔoxyR, derived from MR-1 | 29 |
| HG3CAT | ΔkatB ΔkatG1 ΔkatG2, derived from MR-1 | 29 |
| HGACAT | Δahp ΔkatG1 ΔkatG2, derived from MR-1 | This study |
| HG0956-8-1070 | ΔkatB ΔahpCF, derived from MR-1 | This study |
| HG0976-1070 | ΔkatB Δohr, derived from MR-1 | This study |
| HG1070-2178 | ΔkatB ΔccpA, derived from MR-1 | This study |
| Plasmids | ||
| pHGM01 | Apr Gmr Cmr, att-based suicide vector | 31 |
| pHG101 | Kmr, promoterless broad-host vector | 32 |
| pHGE-Ptac | Kmr, IPTG-inducible expression vector | 33 |
| pHGEI01 | Kmr, integrative lacZ reporter vector | 34 |
| pBBR-Cre | Spr, helper plasmid for antibiotic cassette removal | 56 |
| pHGE-Ptac-ahpCF | Vector for inducible expression of Ahp | This study |
| pHGE-Ptac-katB | Vector for inducible expression of KatB | This study |
| pHGEI01-ryhB | pHGEI01 containing the ryhB promoter | This study |
Mutagenesis and complementation of mutant strains.
In-frame deletion strains of S. oneidensis were constructed using the att-based fusion PCR method as described previously (31). In brief, two fragments flanking the gene of interest were amplified by PCR with primers containing attB and the gene-specific sequence, which were linked by a second round of PCR. The fusion fragments were introduced into plasmid pHGM01 by using Gateway BP Clonase II enzyme mixture (Invitrogen) according to the manufacturer's instructions. The resultant plasmid was maintained in E. coli strain WM3064 and transferred to S. oneidensis by conjugation. Integration of the deletion constructs into the chromosome was selected by resistance to gentamicin and confirmed by PCR. Verified transconjugants were grown in LB broth in the absence of NaCl and plated on LB supplemented with 10% sucrose. Gentamicin-sensitive and sucrose-resistant colonies were screened by PCR for deletion of the target gene. To facilitate the growth of mutants, thiourea and/or catalase (from bovine liver; Sigma) were added to plates at the final resolution step for genes critical for survival through ROS. Mutants were verified by sequencing the mutated regions.
The promoterless plasmid pHG101 was used in genetic complementation of mutants as described previously (32). For inducible gene expression, the gene of interest was generated by PCR and introduced into the inducible plasmid pHGE-Ptac under the control of the promoter Ptac (33). After sequencing verification, the resulting vectors were transferred into the relevant strains via conjugation.
β-Galactosidase activity assay.
The β-galactosidase activity assay was performed to determine gene expression using the integrative lacZ reporter pHGEI01 (34). In brief, the sequence of ∼400 bp upstream from the gene of interest was amplified and placed in front of the full-length E. coli lacZ gene. The resulting vector was maintained in E. coli WM3064 and transferred to S. oneidensis after verification by sequencing. Cells of the log-phase culture (optical density at 600 nm [OD600] of ∼0.4) were harvested by centrifugation, washed with PBS, treated with lysis buffer (0.25 M Tris-HCl, 0.5% Triton X-100, pH 7.5) for 30 min, and subjected to the o-nitrophenyl-β-d-galactopyranoside (ONPG)-based assay as described previously (35). β-Galactosidase activity was determined by monitoring the color development at 420 nm using a Synergy 2 multimode microplate reader (M200 Pro; Tecan), and the results are presented as Miller units.
H2O2 assay.
To measure the H2O2 concentrations in samples without exogenous iron species, the ferrous ion oxidation-xylenol orange (FOX) method was used (36). Mid-log-phase cells in liquid medium or cells grown overnight on plates were collected, washed twice in 50 mM NaHPO4 buffer (pH 7.0), and resuspended in the same buffer to an OD600 of 0.1. H2O2 was added to a final concentration of 0.5 mM, and the cells were incubated at 30°C. After 5 min, cells were filtered out and the elutions were assayed for the remaining H2O2. To assay the H2O2 concentrations in samples containing iron species, the leuco crystal violet (LCV) method was used (37). In brief, reagents were added for a total volume of 2 ml in the following order: 50 μM H2O2, compounds tested at the required concentrations, 100 mM KH2PO4 buffer (pH 4.0), 41 mM LCV dissolved with HCl, and 4 mg/50 ml horseradish peroxidase containing 1.5 mM sodium azide. The mixture was kept in the dark at room temperature for 30 min, to stabilize absorbance. Aliquots of 200 μl of the mixture were transferred into a 96-well plate, and the absorbance of crystal violet cation (CV+) was measured using a Tecan M200 pro microplate reader.
Viability assay.
Cells of the log-phase cultures (OD600 of ∼0.4) were collected by centrifugation and adjusted to 109 cell/ml, which was set as the undiluted (dilution factor 0) level. Tenfold serial dilutions were prepared with fresh LB. Five microliters of each dilution was spotted onto LB plates, on which additives (metal ions, catalase, thiourea, etc.) were added at the concentrations indicated in the figures. For a 15- by 100-mm plate containing approximately 25 ml of medium, metal ions and thiourea sterilized by filtration were added before pouring, while 2,000 units of catalase was spread evenly over the surface after solidification. The plates were incubated for 24 h or longer in the dark before being read. All experiments were repeated at least three times.
Quantification of intracellular total iron.
Quantification of total iron was performed as described previously (38). Cells grown overnight on LB plates were collected, washed with phosphate-buffered saline (PBS), and adjusted to similar densities (OD600 of ∼0.6). Aliquots of 50 ml were mixed with 5 ml of 50 mM NaOH, sonicated on ice using 60 3-s bursts with a 4-s cooling period between each burst, and centrifuged at 5,000 × g for 10 min. The cell lysates (100 μl) were then mixed with 100 μl 10 mM HCl and 100 μl iron-releasing reagent (1.4 M HCl and 4.5% [wt/vol] KMnO4 [1:1]) and treated at 60°C for 2 h. After cooling, the iron detection reagents (6.5 mM ferrozine, 6.5 mM neocuproine, 2.5 M ammonium acetate, and 1 M ascorbic acid in water) were added. The absorbance of samples was measured at 550 nm 30 min later. The standard curve was constructed using 0 to ∼300 μM ferric chloride.
Calcein assay to measure intracellular free iron.
Quantification of intracellular free iron was performed as described previously (39). S. oneidensis strains grown overnight on LB plates with or without 2 mM iron ions were collected by scraping cells with a metal loop, washing with PBS, and adjusting them to the same OD600. Aliquots of cells were mixed with 5 μM calcein-AM (acetomethoxy derivate of calcein) for 30 min at 37°C. At the same time, other aliquots were treated with the iron chelator dipyridyl for 10 min prior to treatment with calcein-AM for comparison. Cells were then washed with PBS to remove the extracellular calcein-AM and monitored for signal changes using a Tecan M200 pro microplate reader (excitation at 485 nm and emission at 535 nm).
RESULTS
Plating defect of ΔoxyR mutant is alleviated on minimal medium agar plates.
Previously, we showed that the plating defect of the S. oneidensis oxyR mutant can best be rescued by adding either thiourea or iron species into LB medium (29). As shown by the results in Fig. 1A, visible growth of the wild-type culture following 105× dilution (dilution factor 5, ∼5 × 103 cell/ml) was observed, whereas the ΔoxyR strain could show growth only when it was undiluted (dilution factor 0, ∼5 × 108 cell/ml). In the presence of thiourea at 5 and 10 mM, the viability was almost recovered to the wild-type level. Thiourea has been used routinely as a scavenger of hydroxyl radicals, but it was recently suggested that the ability of the chemical to help combat ROS should be attributed at least partially to its inhibitory effects on growth and/or metabolic rates (40). Hence, we reasoned that a medium allowing metabolism to run more slowly than in LB should alleviate the plating defect. To test this, we plated ΔoxyR cells on MS minimal medium agar plates (Fig. 1B). Indeed, the ability for colony development of the mutant was just one degree less robust than that of the wild type. However, the ΔoxyR colonies were still significantly smaller (see Fig. S1A in the supplemental material), indicating that the MS medium helps correct the plating but not the slow growth phenotype. To further explore the idea that the increased viability was associated with the reduced growth and/or metabolic rate, we assessed the effect of sucrose on colony formation of the ΔoxyR strain on LB plates. Sucrose, as an osmotic agent, hinders growth significantly at high concentrations (see Fig. S1B), a scenario observed previously (41). However, the viability defect was not evidently rescued by the addition of sucrose (see Fig. S1C). These data imply that the suppression of the plating defect of the ΔoxyR strain on MS plates may not be mainly attributable to the reduced metabolic rate and that there must be an unknown factor(s) responsible for the plating defect on LB plates.
FIG 1.
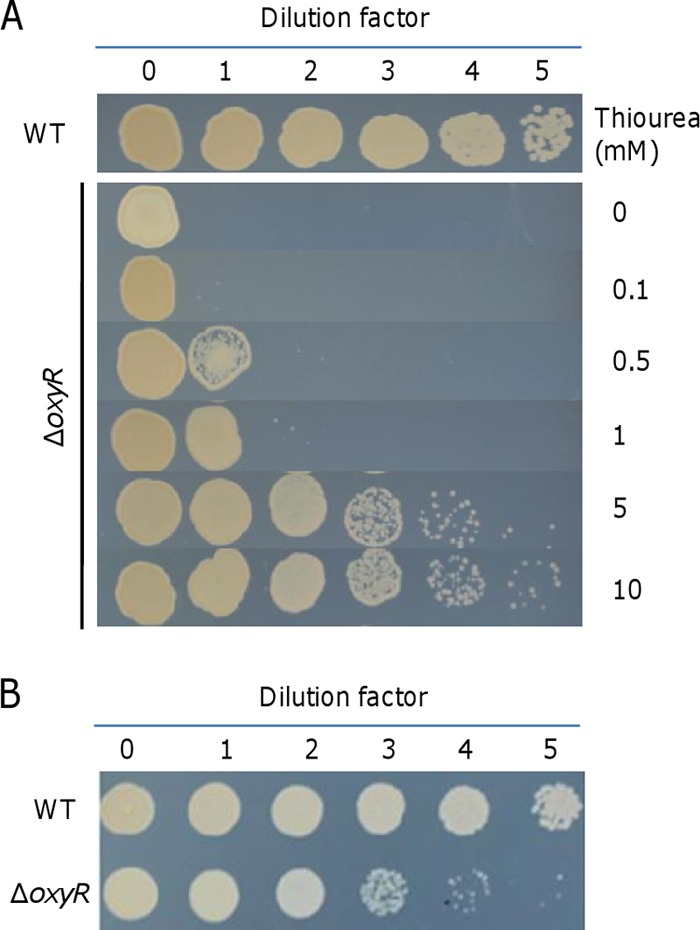
Plating defect of the oxyR mutant. The mutant was validated by genetic complementation in our previous study (29). Cultures at mid-log phase were collected, adjusted to 109 cell/ml (dilution factor 0), and then serially diluted, and 5 μl of each dilution was dropped onto agar plates as indicated. The results shown are from after growth of the wild type plated at the lowest cell density (104 cell/ml, dilution factor 5) became evident. Experiments were performed at least three times, and representative results are presented. (A) Effects of thiourea on the plating defect on LB plates. (B) Results on MS plates.
Catalase suppresses the plating defect of the ΔoxyR mutant.
The finding that the ΔoxyR strain has a plating defect on LB but not on MS plates prompted us to screen for agents that prevent survival/growth of the strain on LB plates. Although loss of OxyR derepresses the expression of the katB gene, conferring an enhanced H2O2 removal capacity on cells (29), we examined the effect of exogenous catalase (2,000 U [the same concentration was used in all experiments where exogenous catalase was added]) on the viability and growth of ΔoxyR cells on LB plates. As shown by the results in Fig. 2A, the plating defect was fully rescued, implying that H2O2 was present in the medium. We therefore reasoned that a katB mutant would be affected with respect to survival and growth on LB plates. Indeed, the loss of the katB gene greatly impaired viability; no growth was observed with dilutions of 100× or higher. As expected, the plating defect was not observed on MS plates (data not shown). We then performed an H2O2 assay to confirm the presence of the oxidant. In LB broth (8 h after autoclaving), the concentrations of H2O2 were approximately 5.2 ± 0.6 μM (mean ± standard deviation), and they were fairly stable with time (Fig. 2B). When catalase (2,000 U) was added, the H2O2 levels were reduced to the detection limit of the method. Similarly, H2O2 in the supernatants of ΔkatB cultures remained at relatively high levels, about 4 ± 0.5 μM at both the exponential (OD600 of ∼0.3) and stationary (OD600 of ∼1.2) phase. In contrast, the levels of H2O2 in the supernatants of cultures of the other strains tested decreased significantly. The wild-type cultures at exponential and stationary phases had values of 1 ± 0.2 and 3.8 ± 0.4 μM, respectively. Because of derepressed production of KatB, the ΔoxyR strain restricted the H2O2 levels by no more than 1.3 ± 0.3 μM. In the case of the MS medium, the levels of H2O2 were below 1 μM under all test conditions. These data imply a possibility that the plating defect is due at least in part to H2O2 generated in LB without the involvement of living cells. Moreover, the plating defect of the ΔoxyR strain was observed on LB plates lacking either tryptone or yeast extract (see Fig. S2 in the supplemental material). While this result indicates that both ingredients may be involved in H2O2 generation, tryptone alone evidently results in a defect more severe than is seen with yeast extract alone, suggesting that peptides and amino acids are the main source of abiotic H2O2 generation. It is worth mentioning that the slow-growth phenotype of the ΔoxyR strain remained the same in the presence of catalase (see Fig. S1A), implying that these two defects may result from functional impairment of different sets of proteins.
FIG 2.

LB contains H2O2. (A) Effects of catalase (2,000 U) on the plating defect of indicated strains on LB plates. Experiments were performed at least three times, and similar results were obtained each time. (B) H2O2 assay. H2O2 concentrations in indicated media (No cell) and in cultures at the indicated cell densities (OD600 of 0.3 and 1.2) were determined. The data reported represent the mean results ± standard deviations (SD) (n = 4).
Cells of the ΔoxyR strain on LB plates die quickly.
To address the severe plating defect of the ΔoxyR strain, the catalase rescue assay was performed. We dropped wild-type and ΔoxyR cultures at 1,000× and 10× dilutions onto LB plates such that the wild-type but not the ΔoxyR strain could proceed to colony development (Fig. 3A). Catalase was then applied to the ΔoxyR culture drops at different times. We found that cells could form visible colonies only when the application was carried out immediately (0 h). These results show that diluted ΔoxyR cells not promptly protected by the catalase treatment simply die out.
FIG 3.
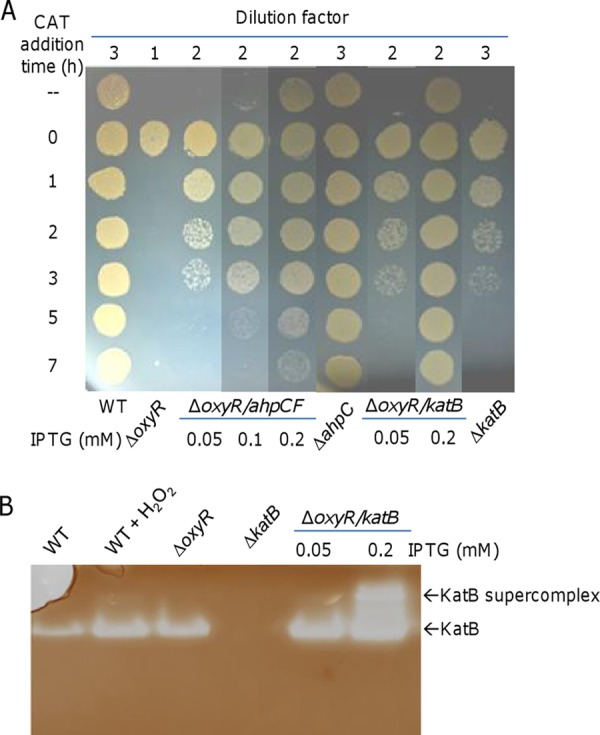
Cells of the ΔoxyR strain on LB plates die quickly. Experiments were performed at least three times, and similar results were obtained. (A) Effects of catalase on the plating defect of indicated strains on LB plates. Cultures were serially diluted, and the indicated dilution of each strain was dropped on LB plates. Catalase (2,000 U) was applied to the drops at the indicated times. Production of AhpCF and KatB at various levels was induced by IPTG. (B) Catalase staining analysis. Cells were collected just before and 30 min after the addition of 0.2 mM H2O2. Proteins from the indicated cell lysates were separated by native PAGE and stained for catalase activity. Overproduction of KatB induced by 0.2 mM IPTG resulted in KatB aggregates, forming the supercomplex.
In view of the protective effect of catalase on the viability of the ΔoxyR strain, we proposed that the plating defect was likely due to its impaired defense system. In addition to KatB, Ahp, a complex formed by the two subunits AhpC and AhpF, is another major H2O2 scavenger in S. oneidensis (29, 30). Because the transcription of ahp relies on OxyR activation, the production of Ahp is extremely low in the ΔoxyR strain (29). To test whether Ahp plays a role in the reduced viability conferred by the oxyR mutation, we placed the ahpCF genes under the control of the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible Ptac promoter within pHGE-Ptac (33). AhpCF produced at various levels by IPTG induction significantly improved the viability of the ΔoxyR strain (Fig. 3A). The ability of the mutant to achieve enhanced viability appeared proportional to the level of IPTG; with IPTG at 0.2 mM, cultures of 100× dilution were able to grow in the absence of catalase. However, the impaired viability of the ΔoxyR strain was apparently not due to reduced Ahp production alone, as an ahpCF deletion mutant exhibited normal survival and growth on LB plates (Fig. 3A).
Because of the derepression in the ΔoxyR strain, KatB is produced at a substantially elevated level compared to its level in the wild-type strain under noninducible conditions (Fig. 3B). Given that overproduction of Ahp could improve the viability of the ΔoxyR strain, we wondered whether increasing KatB production further would have a similar effect. Consistent with its reduced viability (Fig. 2A), the ΔkatB strain at 1,000× dilution survived and grew only when exogenous catalase was applied onto culture drops within 3 h (Fig. 3A). By using the same vector, the level of KatB production was controlled by IPTG. While the inducer at 0.05 mM did not show any noticeable effect on the viability of the ΔoxyR strain, the defect was completely corrected with 0.2 mM IPTG (Fig. 3A). To confirm overproduction of KatB, we performed catalase staining (Fig. 3B). Besides the fact that the expression pattern of KatB was in excellent agreement with the results of our previous study (29), we found that the levels of KatB increased drastically in the presence of IPTG at 0.2 mM, leading to the formation of a KatB supercomplex. These data collectively indicate that the ΔoxyR strain was vulnerable to H2O2 generated extracellularly and that this vulnerability could be rescued either by the removal of the oxidant with exogenous catalase or by enhancement of the intracellular scavenging ability.
The plating defect of the oxyR mutant is not caused by iron starvation.
One of the most striking findings about the S. oneidensis oxyR mutant was that the plating defect could be corrected by adding iron species into LB plates (29). To assess whether this phenomenon is specific to iron, we tested the ΔoxyR strain with other metals. As shown by the results presented in Fig. 4A, in contrast to the results for Fe2+, Zn2+, Mg2+, and K+ hardly improved the viability of the ΔoxyR and ΔkatB strains. Mn2+, however, appeared as effective as Fe2+ in elevating the viability of the katB mutant. While an effect of Mn2+ was evident in the oxyR mutant, it could not fully correct the defect in this strain. Compared to Fe2+, Fe3+ displayed a significantly reduced ability to correct the defect, to a level similar to that of Mn2+ (Fig. 4). These results imply a possibility that the ΔoxyR strain suffers from an iron shortage. However, the odds are low given that the defect is not observed on low-MS medium, which contains 3.6 μM Fe3+, about 5 times lower than the ∼17 μM iron concentration in LB (42). This is confirmed by the finding that levels of total iron in the wild-type and ΔoxyR strains were comparable under all test conditions (see Fig. S3 in the supplemental material).
FIG 4.

Plating defect of the oxyR mutant is not caused by iron starvation. Effects of indicated metal ions (Fe2+, FeSO4; Mn2+, MnCl2; Zn2+, ZnSO4; Mg2+, MgCl2; K+, KCl; and Fe3+, FeCl3) on the plating defect of indicated strains on LB plates. For each metal ion, five different concentrations (2× increases) were tested, and the results for the concentrations that had the most significant effect are shown.
One explanation for this dilemma may be that the levels of free Fe2+ are low in the ΔoxyR strain. Similar to KatB, the iron storage protein Dps is produced at substantially increased levels upon the loss of OxyR (29). As a result, more free Fe2+ is sequestered by Dps, triggering a requirement for iron. To test this notion, we evaluated the intracellular free-iron content by measuring the expression of the ryhB gene, whose product is a small RNA responding to free Fe2+ to regulate the expression of genes involved in iron metabolism (43, 44). The results demonstrated that the ryhB promoter activities of the wild-type and ΔoxyR strains were not significantly different under the same conditions, suggesting that their free-iron levels are comparable (see Fig. S3A in the supplemental material). Similar results were obtained for the ΔkatB strain (see Fig. S3A). To further confirm this, we measured the intracellular levels of free iron in cells grown on LB plates with or without iron supplement by using the calcein assay (39). Consistent with the results for the ryhB promoter assay, the same pattern of response was displayed in all wild-type and mutant strains, including the ΔoxyR and ΔkatB strains (see Fig. S3B). These data collectively indicate that the intracellular iron content, either total or free, was not significantly affected by the oxyR mutation.
Iron species decompose H2O2 directly.
It is clear that the oxyR mutation has little influence on intracellular iron levels, ruling out the possibility that the addition of exogenous iron functions mainly to rescue an iron shortage. Alternatively, iron may scavenge H2O2 without involving cells, given that both ferrous and ferric ions were reported to react with H2O2 many years ago (6). To investigate this, the effects of Fe2+ and Fe3+ on H2O2 decomposition were examined using leuco crystal violet (LCV) reagent, which can reliably quantify H2O2 in the presence of iron species (37). In the presence of H2O2 and horseradish peroxidase, LCV forms a crystal violet cation, CV+, which absorbs at 590 nm and remains stable for days (37). As shown by the results in Fig. 5, in the presence of 50 μM Fe2+, only 28% of the initially added H2O2 (200 μM) remained 30 min after the addition (the same treatment time below unless otherwise noted). When Fe2+ was added to final concentrations of 150 and 200 μM, the levels of H2O2 remaining were reduced to ∼15 and less than 1 μM, respectively. For the ferric ion, the addition had no significant effect on H2O2 degradation when its concentration was no more than 600 μM (Fig. 5). However, degradation of H2O2 became evident with Fe3+ at 800 μM or higher. These results indicate that both ferrous and ferric ions are able to react with H2O2 but that Fe2+ is substantially more effective than Fe3+. We then examined whether Mn2+ and thiourea have similar effects given that both suppress the plating defect of the ΔoxyR strain to some extent. Interestingly, although thiourea at low concentrations (as shown by the results at 100 μM) displayed an ability to remove H2O2, it was less effective than iron species, leaving a significant amount of the oxidant in the medium even when the agent was added at levels greater than 1 mM. In contrast, degradation of H2O2 was not observed with Mn2+ at levels up to 2 mM. These results, in line with those showing that catalase is the most effective agent to suppress the plating defect of the ΔoxyR strain, suggest that direct H2O2 removal is likely the major mechanism involved. In parallel, other strategies, including at least one that is exploited by Mn2+, must exist.
FIG 5.
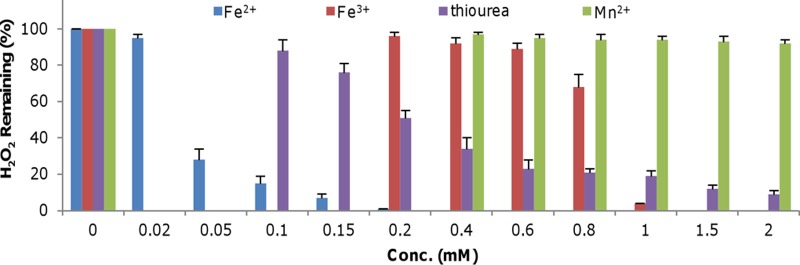
Iron species decompose H2O2 directly. LB broth containing 200 μM H2O2 was mixed with the indicated chemicals at various concentrations. After 30 min of incubation, the amount of H2O2 remaining was determined using the LCV method. The data were normalized to the initial concentration. The concentrations of the chemicals tested in the assay were as follows: Fe2+, 0.02 to 0.2 mM; Fe3+, 0.2 to 1 mM; thiourea, 0.1 to 2 mM; and Mn2+, 0.4 to 2 mM. The data reported represent the mean results ± SD (n = 4).
Iron ions facilitate degradation of H2O2 by living cells.
Although exogenous iron can scavenge H2O2 in cultures directly, the possibility that it also influences the ability of S. oneidensis cells to decompose the oxidant could not be excluded. To test this, we examined the capacities of relevant strains to decompose H2O2 when grown on LB plates in the absence and presence of exogenous iron. Cells grown on plates for 18 h were collected, washed, suspended to similar OD values, and subjected to the H2O2 degradation assay. Compared to cells on LB plates, the wild-type cells grown with exogenous Fe2+ and Fe3+ displayed faster H2O2 degradation (Fig. 6A). However, the enhanced capacities for H2O2 removal induced by the two iron ions were similar, in contrast with their substantially different abilities to remove H2O2 in cell-free environments. Interestingly, the increased H2O2 removal capacities are, at least in part, independent of KatB, as cells of the katB mutant grown with Fe2+ and Fe3+ also degraded H2O2 more rapidly than those grown without (Fig. 6A). To determine whether the increased H2O2-degrading capacities depend on the living cells, we performed the H2O2 assay with the same samples which were disrupted by sonication. All samples displayed reduced capacities for degrading H2O2 compared to the capacity of the untreated control (Fig. 6A). As catalases are H2O2-scavenging enzymes that can function independently of living cells (8), the results suggest that the increased H2O2-degrading capacity in cells grown with iron species is likely due to noncatalase enzymes, such as peroxidase. The experiment was then repeated with the addition of thiourea and Mn2+. While thiourea did not show any influence on the H2O2-degrading capacity, Mn2+ induced an increase in the ability to decompose the oxidant to levels similar to those observed with Fe2+ and Fe3+ (see Fig. S4 in the supplemental material).
FIG 6.

Iron ions facilitate degradation of H2O2 by living cells. (A) Cells of the indicated strains grown overnight on LB plates with or without the indicated additives were collected with a metal loop, washed, and suspended to an OD600 of ∼0.1. Five minutes after the addition of 0.5 mM H2O2, the remaining H2O2 was determined. Con, initial concentration; −, LB without additive; LC, living cell; DC, disrupted cell. Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars indicate SD (n = 4). (B) Effects of indicated mutations on the plating defect on LB plates without or with 2 mM Fe2+. (C) Expression of ahp and katB in the indicated strains under indicated conditions. Cells grown on the LB plates without or with 2 mM Fe2+ for 16 h were collected, washed, and resuspended in PBS. Aliquots were treated with 200 μM H2O2 for 10 min, and ahp and katB promoter activities assayed using an integrative lacZ reporter.
Like the ΔkatB strain, the ΔoxyR strain also displayed elevated H2O2 removal capacities in the presence of Fe2+ and Fe3+, although the effects were much more modest than the effects on the wild type (Fig. 6A). This observation implies that the auxiliary H2O2-scavenging enzymes are unlikely to be tightly regulated by OxyR. Unfortunately, this could not be tested directly because we failed to remove the katB gene from the ΔoxyR strain. As alternatives, we examined the contributions of Ahp, KatG1, and KatG2 to the increased H2O2 removal capacities given that these proteins are a major peroxidase and additional catalases, respectively (29). ΔkatB ΔkatG1 ΔkatG2 (ΔCAT), and ΔkatB Δahp mutants were generated and characterized with respect to viability when grown with Fe2+ or without (Fig. 6B). Consistent with a previous finding (29), KatG1 and KatG2 were dispensable. However, the removal of ahp from the ΔkatB strain further reduced viability and, more importantly, the impaired viability could only be partially rescued by exogenous Fe2+. This result implies that Ahp may be responsible for the difference in the H2O2-degrading capacities of living and disrupted cells shown by the results in Fig. 6A. To test this, we assayed the H2O2 removal capacities of the ΔkatB Δahp strain grown with exogenous Fe2+ and Fe3+. As expected, the double mutant exhibited a further-reduced H2O2-degrading capacity but it was still able to decompose H2O2 (Fig. 6A), showing that additional systems exist for H2O2 removal. Additionally, we assessed the contributions to viability of CcpA and Ohr, two peroxidases that have been implicated in H2O2 scavenging in previous reports (30, 45). However, the viabilities of the ΔkatB ΔccpA and ΔkatB Δohr strains were indistinguishable from that of the ΔkatB strain under both conditions (Fig. 6B).
Furthermore, we determined whether exogenous Fe2+ affects the expression of these genes given that both katB and ahp contribute to viability. By using an integrative lacZ reporter system (34), we found that, in the wild-type strain, both genes were induced by the addition of Fe2+ into the medium (Fig. 6C). In the ΔoxyR strain, the katB gene was no longer responsive but the ahp gene displayed enhanced expression, albeit less substantial than its enhancement in the wild type, indicating that katB may be solely controlled by OxyR, while ahp is subject to regulation by an additional factor(s). Overall, these results indicate that KatB and Ahp are the predominant forces for H2O2 removal and that S. oneidensis is also equipped with OxyR-independent ROS-scavenging enzymes.
DISCUSSION
OxyR is now regarded as a pleiotropic regulator, and its loss causes multiple defects in various biological processes (1, 12). One of these is the viability deficiency. Since in most bacteria, OxyR functions as an activator, at least for H2O2-scavenging genes, the defect is attributed to reduced ability to scavenge endogenous H2O2, as most if not all of the enzymes carrying out the task require OxyR for expression. In S. oneidensis, OxyR acts as a repressor of the katB gene such that the production level of KatB in the oxyR mutant is comparable to the level in the wild type under H2O2-induced conditions, leading to rapid H2O2 degradation (29). We were surprised when we found that the S. oneidensis oxyR mutant also carries the viability defect. The purpose of this study was to identify the factors that account for this phenotype of the oxyR mutant.
The S. oneidensis oxyR mutant is not vulnerable to endogenous H2O2, as aerobic growth at various rates appears irrelevant to the plating defect. This notion is supported by the finding that the katB gene could not be removed from the oxyR mutant, given its predominance in limiting the accumulation of endogenous H2O2 (29). In contrast, the oxyR mutant cells suffer severe damage from the stress imposed by ambient H2O2, based on the unparalleled efficacy of exogenous catalase in suppressing the defect and its enzymatic specificity. Although abiotic generation of H2O2, up to 20 μM, in sterile laboratory medium has been mentioned in a previous review (3), such a finding remains unreported in research articles (personal communication with J. A. Imlay). Here, we showed that the amounts of H2O2 generated in LB are sufficient to differentiate mutants with impaired ability to respond to oxidative stress from the wild type, whereas the defined MS medium does not generate H2O2 to detectable levels.
Based upon the revelation of exogenous H2O2 as the major factor for the plating defect, we suggest a model to explain what we have observed in this study (Fig. 7). In diluted cultures, cells on plates are likely separated from one another and, thus, have to cope with H2O2 produced abiotically in the medium individually. The oxyR mutant cells carry pleiotropic defects, and one of the consequences is that they are vulnerable to extracellular H2O2. By inhibiting cellular components that are essential for survival and/or growth, ambient H2O2 rapidly kills these oxyR mutant cells (Fig. 7A). In contrast, in cultures with high cell densities, the cells exist in close proximity, resulting in patches of cells within which some cells are sequestered from a lethal dose of H2O2 and can proceed to replicate (Fig. 7B). In addition, H2O2 inhibition and killing are conceivably alleviated with higher starting cell numbers, as there is less H2O2 per cell and so less oxidative damage. In the case of exogenously added catalase, growth is allowed simply because the catalase effectively and efficiently removes H2O2. While this mechanism is applicable for presumably any agents that are able to scavenge H2O2, such as thiourea tested in this study, the degrees of efficacy can be critically different depending on the chemical features of the agents. Alternatively, the plating defect can be suppressed by enhancing the intracellular H2O2-scavenging ability of cells (Fig. 7C). When extracellular H2O2 is present, an H2O2 gradient is formed across the cytoplasmic membrane; the internal concentration is approximately an order of magnitude lower than the external concentration (46). In overabundance, KatB or Ahp confers upon cells a substantially elevated capacity to decompose H2O2, leading to low internal H2O2 concentrations. A direct consequence is that the flux of H2O2 into the cell is accelerated, promptly lowering the concentration of H2O2 in the proximity to a level permissive for survival and growth. It is worth mentioning that the oxyR mutation in S. oneidensis does not significantly affect growth in liquid LB, a scenario also found in some other bacteria (22, 23). A possible explanation is that some of the ΔoxyR cells lyse after inoculation, releasing catalase, which decomposes H2O2, into the liquid medium. On the plates, catalase from dead cells could not make such a contribution.
FIG 7.
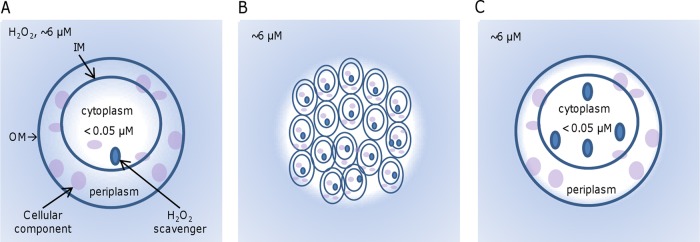
Model illustrating the mechanism underlying the plating defect of the oxyR mutant. Cells face H2O2 at ∼6 μM (blue background) generated abiotically in LB but have intracellular levels lower than 0.05 μM (white background). An H2O2 gradient forms from extracellular space to the cytoplasm membrane because of the H2O2 influx. (A) The oxyR mutant cells, whose H2O2 scavengers are able to keep intracellular H2O2 levels below 0.05 μM, carry pleiotropic defects and are vulnerable to extracellular H2O2. OM, outer membrane; IM, inner membrane. (B) A high cell density allows the oxyR mutant cells to form aggregates. As a result, cells within aggregates are not accessible to extracellular H2O2, and thus, they survive and are able to grow. (C) Increased H2O2-degrading capacity promptly removes intracellular H2O2 and speeds up the H2O2 influx, reducing the concentration of H2O2 in the proximity and facilitating survival and growth.
This logic implies that many cellular components outside the S. oneidensis cytoplasm are vulnerable to H2O2. In support of this idea, many predicted targets of H2O2, including mononuclear iron proteins, Fe-S cluster-containing enzymes, proteins carrying active cysteine residues, and lipids (1, 47–50), are extensively localized outside the cytoplasm. This is particularly true with S. oneidensis, as the bacterium expresses more than 40 c-type cytochromes and many Fe-S cluster-containing proteins that reside exclusively and largely in the membranes and the periplasm, respectively. Conceivably, some of these proteins may carry out a function essential for growth or viability, and thus, the cell would stop growing and die if they were inactivated by ROS.
Among the metal ions tested, Fe2+ was the most effective in H2O2 removal. The addition of this iron species would be expected to reduce peroxide to hydroxyl radicals, as it does inside the cell. As such, the damage caused by hydroxyl would be limited to LB constituents or the exopolymeric substances on the surface of the cell. We are not certain of the mechanism underlying the direct decomposition of H2O2 by Fe3+ in the abiotic system used in this study. Compared to that of Fe2+, the efficiency was considerably lower. However, the impacts of Fe3+ and Fe2+ on the plating defect of the oxyR mutant were similar. One explanation is that S. oneidensis, which is renowned for metal reduction, is able to reduce Fe3+ to Fe2+ under aerobic conditions (51). It is also possible that LB also contains some superoxide, which can reduce Fe3+ to Fe2+ through the first step of the Harber-Weiss reaction (52). The subsequent second step is the Fenton reaction, which converts H2O2 to hydroxyl radicals. The presence of superoxide, generated biotically, abiotically, or both, may also offer an explanation for the plating defect and the effect of Mn2+, which can largely rescue the plating defect of the oxyR and katB mutants, although it is responsible for a marginal amount of decomposition of H2O2. Mn2+, established as the oxidative stress reliever, can scavenge superoxide either by cofactoring superoxide dismutase or functioning abiotically (53). Additionally, it is proposed that Mn2+ acts as an oxidant-resistant substitute for iron in the active site of many nonheme iron enzymes, which are vulnerable during oxidative stress (54). The S. oneidensis genome encodes additional catalases and a large number of peroxidases, some of which remain uncharacterized, implying the presence of OxyR-independent H2O2-scavenging enzymes. Our finding that katB-, katB ahp-, and oxyR-deficient cells grown in the presence of iron and manganese species had increased H2O2-scavenging capacities supports this notion. In E. coli, excess copper is found to be able to help protect cells from H2O2 damage by enhancing the activity of hydroperoxidase I (KatG), hydroperoxidase II, and superoxide dismutase, which is dependent on neither OxyR nor SoxS (55). However, it is clear that the additional help from proteins beyond the OxyR regulon is minor. Notably, the results presented here exclude the involvement of Ohr or CcpA. This is somewhat unexpected because previous work has shown (i) that the organic peroxide (OP)-responding regulator OhrR is also responsive to H2O2 in S. oneidensis and has a larger regulon than those studied (30) and (ii) that CcpA is a periplasmic cytochrome c peroxidase capable of removing H2O2 (45). In contrast, the increased H2O2-scavenging capacities are due to some of the OxyR regulon members which can be activated by a different regulator in the absence of OxyR. Our data indicate that Ahp is such a member. Based on the fact that its production increases in the presence of both Fe2+ and Mn2+, it is conceivable that the unknown regulator may be involved in the response to heavy metals. Efforts to test these notions are under way.
The work presented here also raises unanswered questions about the slow growth of the oxyR mutant. In bacteria in which OxyR functions solely as an activator, this universal phenotype resulting from the oxyR mutation has been conveniently attributed to the reduced H2O2-scavenging capacity (20–23). Our data suggest that this may not be the case, at least in the context of S. oneidensis. Although exogenous catalase perfectly suppresses the plating defect, the oxyR mutant still grows significantly more slowly than the wild type, indicating that this growth defect does not appear to be associated with H2O2-scavenging capacity. Addressing the underlying mechanisms for this represents an important challenge for future work.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Natural Science Foundation of China (grants 31270097 and 41476105), the Major State Basic Research Development Program (973 Program, grant 2010CB833803), and the Doctoral Fund of the Ministry of Education of China (grant 20130101110142).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00154-15.
REFERENCES
- 1.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews SC, Robinson AK, Rodríguez-Quiñones F. 2003. Bacterial iron homeostasis. FEMS Microbiol Rev 27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 3.Imlay JA. 2008. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korshunov S, Imlay JA. 2010. Two sources of endogenous hydrogen peroxide in Escherichia coli. Mol Microbiol 75:1389–1401. doi: 10.1111/j.1365-2958.2010.07059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravindra Kumar S, Imlay JA. 2013. How Escherichia coli tolerates profuse hydrogen peroxide formation by a catabolic pathway. J Bacteriol 195:4569–4579. doi: 10.1128/JB.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barb WG, Baxendale JH, George P, Hargrave KR. 1949. Reactions of ferrous and ferric ions with hydrogen peroxide. Nature 163:692–694. doi: 10.1038/163692a0. [DOI] [Google Scholar]
- 7.Seaver LC, Imlay JA. 2004. Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J Biol Chem 279:48742–48750. doi: 10.1074/jbc.M408754200. [DOI] [PubMed] [Google Scholar]
- 8.Mishra S, Imlay J. 2012. Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch Biochem Biophys 525:145–160. doi: 10.1016/j.abb.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horst SA, Jaeger T, Denkel LA, Rouf SF, Rhen M, Bange F-C. 2010. Thiol peroxidase protects Salmonella enterica from hydrogen peroxide stress in vitro and facilitates intracellular growth. J Bacteriol 192:2929–2932. doi: 10.1128/JB.01652-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liochev SI, Fridovich I. 1992. Fumarase C, the stable fumarase of Escherichia coli, is controlled by the SoxRS regulon. Proc Natl Acad Sci U S A 89:5892–5896. doi: 10.1073/pnas.89.13.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang S, Imlay JA. 2010. Hydrogen peroxide inactivates the Escherichia coli Isc iron-sulphur assembly system, and OxyR induces the Suf system to compensate. Mol Microbiol 78:1448–1467. doi: 10.1111/j.1365-2958.2010.07418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubbs JM, Mongkolsuk S. 2012. Peroxide-sensing transcriptional regulators in bacteria. J Bacteriol 194:5495–5503. doi: 10.1128/JB.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storz G, Tartaglia LA, Ames BN. 1990. Transcriptional regulator of oxidative stress-inducible genes: direct activation by oxidation. Science 248:189–194. doi: 10.1126/science.2183352. [DOI] [PubMed] [Google Scholar]
- 14.Storz G, Imlay JA. 1999. Oxidative stress. Curr Opin Microbiol 2:188–194. doi: 10.1016/S1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- 15.Christman MF, Morgan RW, Jacobson FS, Ames BN. 1985. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell 41:753–762. doi: 10.1016/S0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- 16.Christman MF, Storz G, Ames BN. 1989. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc Natl Acad Sci U S A 86:3484–3488. doi: 10.1073/pnas.86.10.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maciver I, Hansen EJ. 1996. Lack of expression of the global regulator OxyR in Haemophilus influenzae has a profound effect on growth phenotype. Infect Immun 64:4618–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ. 2000. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J Bacteriol 182:4533–4544. doi: 10.1128/JB.182.16.4533-4544.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamet A, Kiss E, Batut J, Puppo A, Hérouart D. 2005. The katA catalase gene is regulated by OxyR in both free-living and symbiotic Sinorhizobium meliloti. J Bacteriol 187:376–381. doi: 10.1128/JB.187.1.376-381.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hennequin C, Forestier C. 2009. OxyR, a LysR-type regulator involved in Klebsiella pneumoniae mucosal and abiotic colonization. Infect Immun 77:5449–5457. doi: 10.1128/IAI.00837-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mongkolsuk S, Sukchawalit R, Loprasert S, Praituan W, Upaichit A. 1998. Construction and physiological analysis of a Xanthomonas mutant to examine the role of the oxyR gene in oxidant-induced protection against peroxide killing. J Bacteriol 180:3988–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.González-Flecha B, Demple B. 1997. Transcriptional regulation of the Escherichia coli oxyR gene as a function of cell growth. J Bacteriol 179:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hahn J-S, Oh S-Y, Roe J-H. 2002. Role of OxyR as a peroxide-sensing positive regulator in Streptomyces coelicolor A3(2). J Bacteriol 184:5214–5222. doi: 10.1128/JB.184.19.5214-5222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tseng H-J, McEwan AG, Apicella MA, Jennings MP. 2003. OxyR acts as a repressor of catalase expression in Neisseria gonorrhoeae. Infect Immun 71:550–556. doi: 10.1128/IAI.71.1.550-556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seib KL, Wu H-J, Srikhanta YN, Edwards JL, Falsetta ML, Hamilton AJ, Maguire TL, Grimmond SM, Apicella MA, McEwan AG, Jennings MP. 2007. Characterization of the OxyR regulon of Neisseria gonorrhoeae. Mol Microbiol 63:54–68. doi: 10.1111/j.1365-2958.2006.05478.x. [DOI] [PubMed] [Google Scholar]
- 26.Ieva R, Roncarati D, Metruccio MME, Seib KL, Scarlato V, Delany I. 2008. OxyR tightly regulates catalase expression in Neisseria meningitidis through both repression and activation mechanisms. Mol Microbiol 70:1152–1165. doi: 10.1111/j.1365-2958.2008.06468.x. [DOI] [PubMed] [Google Scholar]
- 27.Fredrickson JK, Romine MF, Beliaev AS, Auchtung JM, Driscoll ME, Gardner TS, Nealson KH, Osterman AL, Pinchuk G, Reed JL, Rodionov DA, Rodrigues JLM, Saffarini DA, Serres MH, Spormann AM, Zhulin IB, Tiedje JM. 2008. Towards environmental systems biology of Shewanella. Nat Rev Microbiol 6:592–603. doi: 10.1038/nrmicro1947. [DOI] [PubMed] [Google Scholar]
- 28.Ghosal D, Omelchenko MV, Gaidamakova EK, Matrosova VY, Vasilenko A, Venkateswaran A, Zhai M, Kostandarithes HM, Brim H, Makarova KS, Wackett LP, Fredrickson JK, Daly MJ. 2005. How radiation kills cells: survival of Deinococcus radiodurans and Shewanella oneidensis under oxidative stress. FEMS Microbiol Rev 29:361–375. doi: 10.1016/j.fmrre.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, Dong Y, Luo Q, Li N, Wu G, Gao H. 2014. Protection from oxidative stress relies mainly on derepression of OxyR-dependent KatB and Dps in Shewanella oneidensis. J Bacteriol 196:445–458. doi: 10.1128/JB.01077-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li N, Luo Q, Jiang Y, Wu G, Gao H. 2014. Managing oxidative stresses in Shewanella oneidensis: intertwined roles of the OxyR and OhrR regulons. Environ Microbiol 16:1821–1834. doi: 10.1111/1462-2920.12418. [DOI] [PubMed] [Google Scholar]
- 31.Jin M, Jiang Y, Sun L, Yin J, Fu H, Wu G, Gao H. 2013. Unique organizational and functional features of the cytochrome c maturation system in Shewanella oneidensis. PLoS One 8:e75610. doi: 10.1371/journal.pone.0075610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu L, Wang J, Tang P, Chen H, Gao H. 2011. Genetic and molecular characterization of flagellar assembly in Shewanella oneidensis. PLoS One 6:e21479. doi: 10.1371/journal.pone.0021479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Q, Dong Y, Chen H, Gao H. 2013. Mislocalization of Rieske protein PetA predominantly accounts for the aerobic growth defect of tat mutants in Shewanella oneidensis. PLoS One 8:e62064. doi: 10.1371/journal.pone.0062064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu H, Jin M, Ju L, Mao Y, Gao H. 2014. Evidence for function overlapping of CymA and the cytochrome bc1 complex in the Shewanella oneidensis nitrate and nitrite respiration. Environ Microbiol 16:3181–3195. doi: 10.1111/1462-2920.12457. [DOI] [PubMed] [Google Scholar]
- 35.Shi M, Wu L, Xia Y, Chen H, Luo Q, Sun L, Gao H. 2013. Exoprotein production correlates with morphotype changes of nonmotile Shewanella oneidensis mutants. J Bacteriol 195:1463–1474. doi: 10.1128/JB.02187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolff SP. 1994. Ferrous ion oxidation in presence of ferric iron indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol 233:182–189. [Google Scholar]
- 37.Cohn CA, Pak A, Strongin D, Schoonen MA. 2005. Quantifying hydrogen peroxide in iron-containing solutions using leuco crystal violet. Geochem Trans 6:47. doi: 10.1186/1467-4866-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. 2004. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem 331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 39.Deb S, Johnson EE, Robalinho-Teixeira RL, Wessling-Resnick M. 2009. Modulation of intracellular iron levels by oxidative stress implicates a novel role for iron in signal transduction. Biometals 22:855–862. doi: 10.1007/s10534-009-9214-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Imlay JA. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan J, Wei B, Shi M, Gao H. 2011. Functional assessment of EnvZ/OmpR two-component system in Shewanella oneidensis. PLoS One 6:e23701. doi: 10.1371/journal.pone.0023701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abdul-Tehrani H, Hudson AJ, Chang Y-S, Timms AR, Hawkins C, Williams JM, Harrison PM, Guest JR, Andrews SC. 1999. Ferritin mutants of Escherichia coli are iron deficient and growth impaired, and fur mutants are iron deficient. J Bacteriol 181:1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massé E, Gottesman S. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci U S A 99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y, Harris D, Luo F, Xiong W, Joachimiak M, Wu L, Dehal P, Jacobsen J, Yang Z, Palumbo A, Arkin A, Zhou J. 2009. Snapshot of iron response in Shewanella oneidensis by gene network reconstruction. BMC Genomics 10:131. doi: 10.1186/1471-2164-10-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schütz B, Seidel J, Sturm G, Einsle O, Gescher J. 2011. Investigation of the electron transport chain to and the catalytic activity of the diheme cytochrome c peroxidase CcpA of Shewanella oneidensis. Appl Environ Microbiol 77:6172–6180. doi: 10.1128/AEM.00606-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seaver LC, Imlay JA. 2001. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol 183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imlay J, Chin S, Linn S. 1988. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 48.Wallace MA, Liou LL, Martins J, Clement MHS, Bailey S, Longo VD, Valentine JS, Gralla EB. 2004. Superoxide inhibits 4Fe-4S cluster enzymes involved in amino acid biosynthesis: cross-compartment protection by CuZn-superoxide dismutase. J Biol Chem 279:32055–32062. doi: 10.1074/jbc.M403590200. [DOI] [PubMed] [Google Scholar]
- 49.Anjem A, Imlay JA. 2012. Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J Biol Chem 287:15544–15556. doi: 10.1074/jbc.M111.330365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sobota JM, Gu MZ, Imlay JA. 2014. Intracellular hydrogen peroxide and superoxide poison 3-deoxy-d-arabinoheptulosonate 7-phosphate synthase, the first committed enzyme in the aromatic biosynthetic pathway of Escherichia coli. J Bacteriol 196:1980–1991. doi: 10.1128/JB.01573-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan J, Chen Y, Zhou G, Chen H, Gao H. 2013. Investigation of roles of divalent cations in Shewanella oneidensis pellicle formation reveals unique impacts of insoluble iron. Biochim Biophys Acta 1830:5248–5257. doi: 10.1016/j.bbagen.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 52.Koppenol WH. 2001. The Haber-Weiss cycle: 70 years later. Redox Rep 6:229–234. doi: 10.1179/135100001101536373. [DOI] [PubMed] [Google Scholar]
- 53.Barnese K, Gralla EB, Valentine JS, Cabelli DE. 2012. Biologically relevant mechanism for catalytic superoxide removal by simple manganese compounds. Proc Natl Acad Sci U S A 109:6892–6897. doi: 10.1073/pnas.1203051109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imlay JA. 2014. The mismetallation of enzymes during oxidative stress. J Biol Chem 289:28121–28128. doi: 10.1074/jbc.R114.588814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Macomber L, Rensing C, Imlay JA. 2007. Intracellular copper does not catalyze the formation of oxidative DNA damage in Escherichia coli. J Bacteriol 189:1616–1626. doi: 10.1128/JB.01357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fu H, Chen H, Wang J, Zhou G, Zhang H, Zhang L, Gao H. 2013. Crp-dependent cytochrome bd oxidase confers nitrite resistance to Shewanella oneidensis. Environ Microbiol 15:2198–2212. doi: 10.1111/1462-2920.12091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.