

ABSTRACT
CbrA is a DivJ/PleC-like histidine kinase of DivK that is required for cell cycle progression and symbiosis in the alphaproteobacterium Sinorhizobium meliloti. Loss of cbrA results in increased levels of CtrA as well as its phosphorylation. While many of the known Caulobacter crescentus regulators of CtrA phosphorylation and proteolysis are phylogenetically conserved within S. meliloti, the latter lacks the PopA regulator that is required for CtrA degradation in C. crescentus. In order to investigate whether CtrA proteolysis occurs in S. meliloti, CtrA stability was assessed. During exponential growth, CtrA is unstable and therefore likely to be degraded in a cell cycle-regulated manner. Loss of cbrA significantly increases CtrA stability, but this phenotype is restored to that of the wild type by constitutive ectopic expression of a CpdR1 variant that cannot be phosphorylated (CpdR1D53A). Addition of CpdR1D53A fully suppresses cbrA mutant cell cycle defects, consistent with regulation of CtrA stability playing a key role in mediating proper cell cycle progression in S. meliloti. Importantly, the cbrA mutant symbiosis defect is also suppressed in the presence of CpdR1D53A. Thus, regulation of CtrA stability by CbrA and CpdR1 is associated with free-living cell cycle outcomes and symbiosis.
IMPORTANCE The cell cycle is a fundamental process required for bacterial growth, reproduction, and developmental differentiation. Our objective is to understand how a two-component signal transduction network directs cell cycle events during free-living growth and host colonization. The Sinorhizobium meliloti nitrogen-fixing symbiosis with plants is associated with novel cell cycle events. This study identifies a link between the regulated stability of an essential response regulator, free-living cell cycle progression, and symbiosis.
INTRODUCTION
Sinorhizobium meliloti is a member of the class Alphaproteobacteria that grows free-living in the soil or as a beneficial nitrogen-fixing symbiont in association with legumes in the genera Medicago, Melilotus, and Trigonella. As a free-living organism, S. meliloti undergoes an asymmetric cell division with once-and-only-once DNA replication per cell cycle (1, 2, 3). However, inside its host, this bacterium undergoes differentiation into a bacteroid that includes a novel cell cycle program of repeated DNA replication in the absence of cell division (endoreduplication) (3). The underlying molecular mechanisms that dictate cell cycle progression and differentiation in S. meliloti remain to be explored in detail and therefore represent a novel aspect of symbiont physiology. Known and putative S. meliloti cell cycle regulators are conserved among other members of the Alphaproteobacteria that also specialize in chronic colonization of eukaryotic hosts, in particular rhizobial species in the genera Agrobacterium, Bartonella, and Brucella (2, 4). Thus, a deeper understanding of S. meliloti cell cycle regulation will provide insight into processes that are broadly important to both cell cycle progression and host-microbe interaction (5, 6, 7, 8, 9, 10, 11, 12).
Caulobacter crescentus provides an intensively studied model for understanding the molecular mechanisms that underlie cell cycle control among alphaproteobacteria. A complex regulatory network, including a central two-component pathway (Fig. 1), plays a critical role in coordinating C. crescentus cell cycle progression and asymmetric daughter cell fate (13, 14, 15, 16). CtrA is an essential response regulator that contributes to these processes by regulating DNA replication initiation and methylation, as well as cell division and motility, by binding DNA in a phosphorylation-dependent manner. Since CtrA inhibits DNA replication initiation in G1 cells, its activity must be repressed in order to allow S phase to proceed and later restored to promote G2 events such as cell division. CtrA activity is then differentially inherited by the two daughter cells, being present in the small swarmer but not the large stalked cell, and in this way establishes their replicative asymmetry (17). The coordination of these diverse processes requires that CtrA activity be posttranslationally regulated in a temporal manner through protein-protein interaction with a transcription inhibitor, phosphorylation, degradation, and cellular localization (14, 18, 19, 20, 21, 22).
FIG 1.
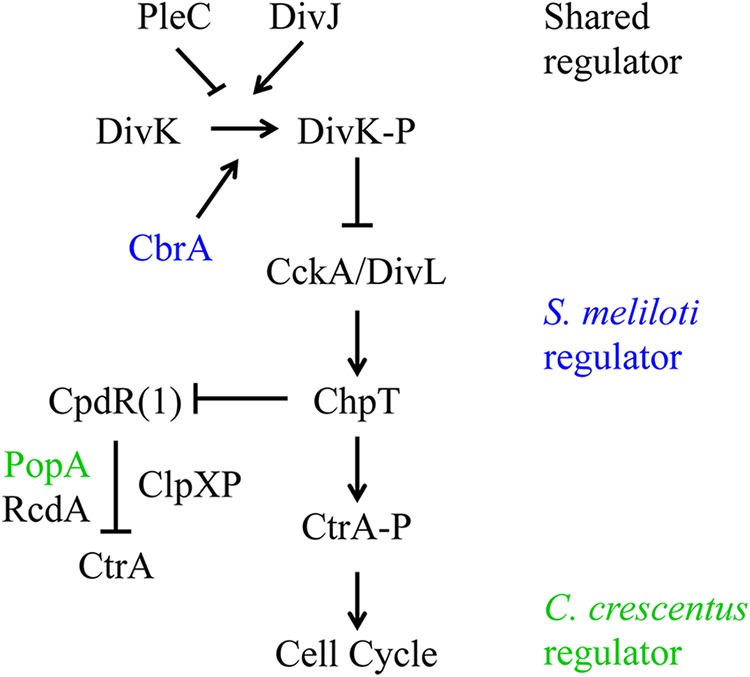
Two-component pathway model of CtrA cell cycle regulation. The S. meliloti genome encodes many of the two-component regulators of CtrA that have been identified in C. crescentus (black font). However, there are several points of divergence between these two distantly related alphaproteobacteria. S. meliloti contains an additional DivJ/PleC-like histidine kinase, CbrA, that promotes DivK phosphorylation (blue font). Additionally, S. meliloti lacks the c-di-GMP receptor PopA that is required to regulate CtrA proteolysis in C. crescentus (green font). DivK-P and CtrA-P, phosphorylated DivK and CtrA, respectively.
The DivK response regulator functions as an essential switch to indirectly regulate CtrA activity at the level of phosphorylation and degradation (23, 24) (Fig. 1). DivK is inactive when dephosphorylated by the PleC phosphatase and is active when phosphorylated by the DivJ kinase. Phosphorylated DivK inhibits the CckA hybrid histidine kinase (25); therefore, DivK dephosphorylation allows for CckA autophosphorylation and the subsequent transfer of this phosphate to the histidine phosphotransferase ChpT (26, 27). ChpT then transfers this phosphate to two substrates: CtrA and the response regulator CpdR (26, 27). Once CtrA is phosphorylated, it is competent to bind DNA and thereby regulate cell cycle events (28, 29, 30, 31). The second target of ChpT, CpdR, is required for ClpXP-mediated degradation of CtrA (32, 33); however, CpdR is inactivated by phosphorylation (34). In this manner, the CckA pathway simultaneously phosphorylates CtrA and protects it from proteolysis to stimulate its activity, or it dephosphorylates CtrA and promotes its degradation to repress its activity. Interestingly, phosphorylation and degradation are redundant mechanisms for regulating CtrA activity, although at least one is needed for cell viability (18).
Proteolysis of CtrA plays an important role in mediating cell cycle progression, and its recognition as a substrate of the ClpXP protease requires its terminal alanine-alanine dipeptide (35). CpdR-regulated proteolysis of CtrA also requires RcdA (36, 37) and the 3′,5′-cyclic diguanylic acid (c-di-GMP) receptor PopA (38, 39) (Fig. 1). RcdA and PopA form a complex that is activated by a CpdR-dependent increase in c-di-GMP levels and directly interacts with CtrA to recruit a complex containing CpdR and ClpXP to the old cell pole (36, 38, 39, 40, 41), although whether localization per se is critical to regulation of CtrA degradation remains unclear (37).
The two-component signaling pathway required for cell division and the establishment of asymmetric daughter cell fate in S. meliloti is just beginning to be characterized in molecular detail. However, S. meliloti has homologs of many C. crescentus regulators of CtrA activity (2, 4) (Fig. 1). For example, the DivJ/PleC homolog CbrA is necessary for proper cell cycle progression such that a null mutant displays aberrant morphologies indicative of cell division defects (9, 42). CbrA promotes DivK phosphorylation, and this is likely the reason for increased CtrA levels and phosphorylation in cbrA mutants (9, 10). DivJ has a similar function, and interestingly, divJ and cbrA null mutations are synthetically lethal (10). The gene encoding the PleC phosphatase is also essential in S. meliloti (43), suggesting that strict regulation of DivK phosphorylation is critical to growth and reproduction. Similarly, the presumed downstream target of DivK regulation, CtrA, is essential as in C. crescentus (44).
Although CtrA regulation at the level of phosphorylation has been shown in S. meliloti (10), it is unclear from bioinformatics analyses whether CtrA might also be regulated at the level of proteolysis. S. meliloti CtrA does have a C-terminal ETA motif (44), which is consistent with the known sequence requirements for ClpXP substrate recognition (45). S. meliloti also has CpdR and RcdA orthologs (4). RcdA function has not been examined yet, but CpdR1 is required for polar localization of ClpX and proper cell cycle progression (8), consistent with a role in CtrA cell cycle regulation. However, CpdR1 regulation of CtrA has not been tested directly, and more importantly, there is no PopA ortholog in S. meliloti (41) (Fig. 2), leaving regulation of CtrA at the level of proteolysis an open question.
FIG 2.

S. meliloti lacks a PopA ortholog. C. crescentus PopA was used to perform a reciprocal blastp search for orthologs. Sequences with a minimum of 21% identity were used to create a maximum likelihood phylogeny tree. If more than one ortholog was found in the same organism, the sequences were randomly labeled 1 and 2. C. crescentus PleD was included in the final sequence alignment and phylogenetic analysis for comparison. PopA is present in Caulobacterales and Rhodobacterales but absent from many alphaproteobacteria that engage in host-microbe interaction, including Bradyrhizobiaceae, Brucellaceae, and most members of the Rhizobiaceae family within the Rhizobiales order, as well as the Rickettsiales. Several species have a PopA ortholog that is more homologous to those present in a different order, and two PopA orthologs were identified outside the alphaproteobacterial division and are indicated by asterisks as follows. Woodsholea maritima (*) is a member of the Caulobacterales. Scytonema millei (**) and Mastigocoleus testarum (***) are from the division Cyanobacterium. Polymorphum gilvum (****) is an unclassified alphaproteobacterium with a PopA ortholog most closely related to those of the Rhodobacterales.
We therefore examined CtrA stability in S. meliloti and find that it is unstable during exponential growth. Previous studies observed increased levels of CtrA in cbrA mutants (9, 10). Here we show that the increased level of CtrA in a ΔcbrA mutant is due, in part, to a significant increase in its stability. We further show that constitutive ectopic expression of an unphosphorylatable version of CpdR1 (CpdR1D53A) restores wild-type CtrA instability to the ΔcbrA mutant. Thus, CtrA stability is regulated in a CbrA-dependent manner, and this stability is further influenced by CpdR1. Importantly, CpdR1D53A also suppresses ΔcbrA cell cycle and symbiosis defects, providing a strong link between the regulation of CtrA stability, cell cycle outcomes, and host colonization.
MATERIALS AND METHODS
Microbiological techniques.
Bacterial strains and plasmids are listed in Table 1. Sinorhizobium meliloti was grown at 30°C in LB/MC (LB supplemented with 2.5 mM calcium chloride and 2.5 mM magnesium sulfate), and Escherichia coli was grown at 37°C in LB. Strains were constructed through triparental mating as previously described (46). Exponential-phase cultures of S. meliloti were obtained by inoculating liquid medium with a single colony, then diluting the overnight culture to an optical density at 600 nm (OD600) of 0.1, and allowing the cells to grow to an OD600 of 0.6 to 0.8. LB/MC was supplemented with 0.02% calcofluor (fluorescent brighter 28; Sigma) and buffered with 10 mM HEPES (pH 7.4) to measure succinoglycan production. The following antibiotics were used at the specified concentration: streptomycin (500 μg/ml), neomycin (200 μg/ml), tetracycline (10 μg/ml), and chloramphenicol (20 μg/ml).
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant characteristic(s) | Resistance | Source or reference |
|---|---|---|---|
| S. meliloti strains | |||
| Rm1021 | Wild-type strain SU47 Smr | Sm | F. Ausubel |
| CSS6000 | Rm1021 ΔcbrA::cat | Cm | 46 |
| KBS1001 | Rm1021 carrying plasmid pcpdR1 | Tc | This study |
| KBS1002 | Rm1021 carrying plasmid pcpdR1D53A | Tc | This study |
| KBS1003 | CSS6000 carrying pcpdR1 | Tc | This study |
| KBS1004 | CSS6000 carrying pcpdR1D53A | Tc | This study |
| CSS6005 | Rm1021 carrying plasmid pLAFR2070 | Tc | This study |
| CSS6006 | Rm1021 carrying plasmid pLAFR1 | Tc | This study |
| CSS6002 | CSS6000 carrying pLAFR2070 | Cm Tc | 46 |
| CSS6003 | CSS6000 carrying pLAFR1 | Cm Tc | 46 |
| E. coli strains | |||
| MT616 | Strain MM294 carrying pRK600 Cmr | Cm | T. Finan |
| DH5α | endA1 hsdR17 supE44 thi-1 recA1 gyrA relA1 Δ(lacZYA-argG) | BRL Corp. | |
| BL21 | ompT hsdSB(rB− mB−) gal dcm (DE3) | Cm | Invitrogen |
| Plasmids | |||
| pLAFR1 | Low-copy-number vector | Tc | F. Ausubel |
| pLAFR2070 | cbrA+ complementation vector | Tc | 20 |
| pDEST17::ctrA | Gateway destination vector containing S. meliloti ctrA | Kn | 46 |
| pcpdR1 | pTH1227 cpdR1 | Tc | 33 |
| pcpdR1D53A | pTH1227 cpdR1D53A | Tc | 33 |
Bioinformatics identification of PopA and PleD homologs through reciprocal blastp analysis.
C. crescentus (NA1000) PopA (GenBank accession no. YP_002517291.1) was used as a query against the NCBI nonredundant database to perform a primary BLAST search for homologs using protein-protein BLAST (blastp). From the first 5,000 hits, candidate homologs with an E value of less than 0.001 and with at least 21% identity to C. crescentus PopA were selected; candidates with a partial species name were eliminated. These candidates were used as a query in a second blastp search. Hits from this second query were required to have an E value of less than 0.001 and to have the PopA sequence (GenBank accession no. YP_002517291.1) as their first hit of the query. If the query results had PopA as the first hit in this secondary blastp, the candidate was identified as a putative PopA ortholog, or PopA like (PAL). Forty sequences were obtained from the second blastp search. The PleD (GenBank accession no. YP_002517919.1) sequence was added to observe its divergence from PopA and PAL sequences, and the diguanylate cyclase from Pseudomonas aeruginosa (GenBank accession no. WP_034084052.1) was used as an outgroup to root the phylogenetic tree. Sequences were aligned using MUSCLE. This multiple-sequence alignment was used to infer evolutionary history using maximum likelihood phylogeny (InL) with 1,000 bootstraps based on the JTT matrix model (47). The tree with the highest log likelihood (−13,718.7871) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial trees for the heuristic search were obtained automatically by applying neighbor-joining and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model and then selecting the topology with superior log likelihood value. All positions containing gaps and missing data were eliminated. There were a total of 221 positions in the final data set. Evolutionary analyses were conducted using the publically available software MEGA version 5.10 (48).
S. meliloti CtrA purification and Western blot analysis.
CtrAHIS was purified and used as a positive control for Western blot analysis of cellular CtrA as described previously (9). Exponential-phase cultures were centrifuged at 4°C for 10 min at 5,000 × g. Cell pellets were resuspended in 2× Laemmli loading buffer and boiled for 5 min. The volume of lysate analyzed was normalized to OD600, subjected to 4 to 20% SDS-PAGE (Bio-Rad) with TRIS running buffer (250 mM Tris base, 1.92 M glycine, 1% SDS) at a constant 100 V for 100 min, and then transferred onto a low-fluorescence polyvinylidene difluoride (PVDF) membrane with Tris-glycine transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) at a constant 100 V for 1 h. The membrane was probed with C. crescentus anti-CtrA polyclonal antibodies (1:5,000 dilution in Li-Cor blocking buffer plus 0.2% Tween) for 16 h at 4°C and subsequently probed with donkey anti-rabbit IRDye 800 CW (1:20,000 dilution in Li-Cor blocking buffer plus 0.2% Tween and 0.1% SDS). Cross-reacting proteins were visualized with a Li-Cor Odyssey CLx infrared imaging system, and images were quantified with Image Study software. Subsequently, the membrane was stripped and reprobed with C. crescentus anti-ClpP polyclonal antibodies (1:25,000 dilution in Li-Cor blocking buffer plus 0.2% Tween) for 16 h at 4°C and subsequently probed with donkey anti-rabbit IRDye 800 CW (1:20,000 dilution in Li-Cor blocking buffer plus 0.2% Tween and 0.1% SDS). Cross-reacting proteins were visualized as described above for CtrA. Graphical results show the averages and standard deviations of a minimum of three biological replicates.
CtrA in vivo stability assay.
Exponential-phase cultures were divided and treated with either 20 μg chloramphenicol dissolved in 100% ethanol or 100% ethanol and then grown for 4 h. Aliquots were taken at 0, 30, 120, and 240 min. Samples were centrifuged at 4°C for 10 min at 5,000 × g. Cell pellets were resuspended in 2× Laemmli loading buffer, boiled for 5 min, and used for CtrA Western blot analysis as described above.
Motility assay.
Overnight cultures were diluted to an OD600 of 0.1 and grown on LB/MC with 0.22% agar. Measurement of the diameter of cell growth was taken after 6 days. Results are the averages of five biological replicates for each strain.
Microscopy and fluorescence flow cytometry.
Cell morphology was assessed with exponential cultures, and each strain was assayed using three biological replicates. Cell adhesion to a slide was performed as previously described (9). Cells were scored as the wild type when they showed the typical S. meliloti rod shape with an average size of 2 to 4 μm in length and 1 μm in width. Any cells that did not follow these criteria were considered non-wild type. All strains assessed for DNA content were grown in biological triplicates and prepared as previously described (9, 49). Cells were acquired on a modified Becton-Dickinson LSRII flow cytometer at the Massachusetts General Hospital Flow Cytometry Research Laboratory. Ten thousand cells were acquired for each replicate. The resulting data were analyzed as previously described using the FlowJo software package (9).
Symbiosis assay.
Symbiosis was assayed with 8 individual Medicago sativa plants per S. meliloti strain as previously described (9, 50). Nodule coloration and plant height (the length from the epicotyl stem to the apical node) was assessed 5 weeks postinoculation.
RESULTS
S. meliloti and closely related alphaproteobacteria lack a PopA ortholog.
CtrA is cell cycle regulated at the level of transcription, phosphorylation, and degradation in C. crescentus (16). In particular, CtrA degradation is one of the key factors contributing to cell cycle progression and the generation of asymmetric daughter cell fate (14, 15, 20). Consistent with the hypothesis that S. meliloti CtrA would also be regulated at the level of proteolysis, divJ and cbrA mutants with decreased DivK phosphorylation accumulate increased levels of CtrA and display cell cycle defects (9, 10). Moreover, the cpdR1 null mutant is unable to localize ClpX to the cell pole and also displays severe cell cycle defects, which can be complemented by C. crescentus CpdR (8). Orthologs of C. crescentus CpdR and RcdA have been identified in S. meliloti (4, 8), suggesting regulated ClpXP-dependent degradation of CtrA could be conserved. In contrast, recent analyses indicated the absence of a PopA ortholog in S. meliloti (41), and we were therefore interested in further corroborating this observation using a different bioinformatics approach.
A hallmark of PopA is that it can bind c-di-GMP but lacks catalytic activity due to the absence of a consensus GGDEF domain (38). A protein-protein BLAST (blastp) search of the S. meliloti genome was performed using C. crescentus PopA; however, the most closely related protein identified was a PleD ortholog containing consensus GGDEF and EAL domains. An extensive search for PopA orthologs was then performed using reciprocal blastp, and this revealed the limited presence of PopA orthologs primarily among members of the orders Caulobacterales and Rhodobacterales (Fig. 2 [PAL, PopA-like]). Although this analysis identified several PopA orthologs in addition to what has been described previously (41), particularly in the Rhodobacterales as well as the distantly related cyanobacteria Scytonema millei and Mastigocoleus testarum, this analysis was similarly unable to identify a PopA ortholog in S. meliloti. While CpdR and RcdA are present in Caulobacterales, they do not appear to be highly conserved among CtrA-containing Rhodobacterales (4), indicating either that PopA alone is sufficient for CtrA degradation or that PopA has an alternative function in these bacteria.
PopA orthologs appear to be absent from nearly all rhizobacterial families, including Bradyrhizobiaceae, Brucellaceae, and Rhizobiaceae, which have conserved the CtrA proteolysis regulators CpdR and RcdA (4). The one exception is the bradyrhizobial marine bacterium Rhodopseudomonas palustris, although its PopA ortholog is the most divergent within this protein family and retains similarity to PleD (Fig. 2). PopA also appears to be absent from the Rickettsiales; however, these bacteria also lack CpdR and RcdA (4), suggesting that regulated proteolysis of CtrA may not be conserved within this order of Alphaproteobacteria. Based on this analysis, if CtrA is regulated at the level of degradation in S. meliloti, then proteolysis occurs through a novel PopA-independent mechanism that may be common to a variety of host-associated Rhizobiales.
S. meliloti CtrA is unstable during exponential growth.
S. meliloti divJ and cbrA mutants with decreased DivK phosphorylation accumulate increased levels of CtrA and display cell cycle defects (9, 10). However, due to the absence of a PopA ortholog in S. meliloti, it was unclear whether the high levels of CtrA in these mutants could be the result of decreased proteolysis. To test for degradation, CtrA stability was measured in a protein shutoff assay by adding the translation inhibitor chloramphenicol to exponentially growing wild-type cells and assessing CtrA levels by Western blotting over the course of 4 h. CtrA levels begin to decrease at 30 min after the addition of chloramphenicol, and the amount of CtrA further decreases at each successive time point (Fig. 3A). ClpP levels were simultaneously assayed, but there was no change in protein level over the course of the experiment (Fig. 3A), as observed in C. crescentus (30). In order to quantitatively determine the relative amount of CtrA present over time, CtrA levels were normalized to ClpP as an internal loading control with the amount of CtrA at time t = 0 arbitrarily set at 1. The concentration of CtrA is approximately 60% of its starting levels at t = 30 min and at just under 30% of its starting levels at t = 240 min (Fig. 3C). Thus, S. meliloti CtrA is unstable during exponential growth despite the absence of a PopA ortholog.
FIG 3.

CtrA stability is regulated during exponential growth in a CbrA-dependent manner. (A and B) Western blots of whole-cell lysates were probed with anti-CtrA and anti-ClpP. Lane 1 contains purified S. meliloti CtrAHIS. In lanes 2 to 9, the levels of protein were assayed by Western blotting at different times (0, 30, 120, and 240 min) after treatment of cells with (+) and without (−) chloramphenicol (CHL). (C and D) CtrA signal in wild-type (WT) and ΔcbrA cells was first normalized to the ClpP loading control signal and then independently normalized to 1 using their value at 0 min. Graphical results are the averages for at least three biological replicates, and error bars represent the standard deviations. Solid line, no-chloramphenicol control; broken line, chloramphenicol treatment. The value that was significantly different from the value for the no-chloramphenicol control (P < 0.0001) is indicated by an asterisk.
S. meliloti CtrA stability is regulated in a CbrA-dependent manner.
CtrA levels are increased in a cbrA mutant that has decreased levels of phosphorylated DivK (9, 10). This observation is consistent with a model in which CbrA, through its effect on DivK, promotes the degradation of CtrA (Fig. 1). This was tested by measuring CtrA stability in a ΔcbrA null mutant as described above for the wild type. Western blot analysis shows that CtrA levels do not significantly decrease after treatment with chloramphenicol (Fig. 3B). In fact, CtrA levels in the ΔcbrA mutant are maintained at pretreatment levels for at least 240 min (Fig. 3B and D) in contrast to what is observed with the wild type (Fig. 3A and C). These results demonstrate that CtrA is normally unstable during exponential growth but is significantly stabilized in the ΔcbrA mutant. Based on C. crescentus, CbrA-dependent regulation of CtrA stability is most likely indirect and requires a CpdR response regulator ortholog whose activity is controlled by phosphorylation (Fig. 1).
CpdR1D53A restores wild-type CtrA stability to the ΔcbrA mutant.
In C. crescentus, CpdR plays an essential role in the directed proteolysis of CtrA by ClpXP (26, 33, 34, 40), such that in its unphosphorylated state CpdR promotes CtrA degradation (15). As CpdR can function in the place of S. meliloti CpdR1 (8), it is possible that a partially conserved mechanism for CtrA degradation exists in S. meliloti. We therefore predicted that the unphosphorylatable variant CpdR1D53A would function as a dominant-negative factor to promote proteolysis of CtrA. To test this, CpdR1 and CpdR1D53A were expressed from a constitutive promoter on a low-copy-number plasmid. We were unable to detect a significant change in CtrA levels in wild-type cells carrying plasmid pcpdR1 compared to those carrying plasmid pcpdR1D53A (Fig. 4A). Although somewhat surprising, this is consistent with the subtle cell cycle defects associated with plasmid pcpdR1D53A in the presence of native cpdR1 on the chromosome (8).
FIG 4.

CtrA instability is restored to the ΔcbrA mutant by constitutive ectopic expression of unphosphorylatable CpdR1D53A. Wild-type (WT) cells carrying plasmid pcpdR1 or pcpdR1D53A or ΔcbrA cells carrying plasmid pcpdR1 or pcpdR1D53A were studied. (A and C) CtrA levels during exponential growth were assayed by Western blotting in batch culture (A) or at different time points (0 and 240 min) after treatment of cells with (+) and without (−) chloramphenicol (CHL) (C). The leftmost lane of each Western blot contains purified S. meliloti CtrAHIS as a positive control for anti-CtrA. Each Western blot was probed with anti-CtrA and anti-ClpP. (B) For each strain indicated, CtrA signal from three biological replicates was normalized to the value for its internal ClpP loading control and then normalized to 1 using WT cells carrying pcpdR1 for comparison. The value that was significantly different (P < 0.0001) from the values for the other three strains is indicated by an asterisk. (D) For each strain indicated, CtrA signal from three biological replicates was normalized to its internal loading control, ClpP, and then independently normalized to 1 using its value at 0 min. The value that was significantly different (P < 0.0001) from the value for its no-chloramphenicol treatment control is indicated by an asterisk.
On the basis of C. crescentus, we predict that CpdR1 phosphorylation is regulated by CbrA indirectly through DivK and the CckA pathway (Fig. 1). This model further predicts that a ΔcbrA mutant would have aberrantly high levels of phosphorylated CpdR1 and therefore decreased proteolysis of CtrA. In this case, unphosphorylatable CpdR1D53A should function as a dominant extragenic suppressor of CtrA levels in the ΔcbrA mutant. Consistent with this hypothesis, lower levels of CtrA are present in ΔcbrA cells carrying plasmid pcpdR1D53A (ΔcbrA pcpdR1D53A cells) than in ΔcbrA cells carrying plasmid pcpdR1 (ΔcbrA pcpdR1 cells) and reflect a restoration to wild-type levels (Fig. 4A and B).
To test whether expression of CpdR1D53A in the ΔcbrA mutant restores CtrA instability, a protein shutoff assay was performed using chloramphenicol, and CtrA levels were measured by Western blotting at 0 and 240 min posttreatment. At 240 min, there is a significant decrease in CtrA levels in wild-type cells with either the pcpdR1 or pcpdR1D53A plasmid (Fig. 4C). There is also a significant decrease in CtrA levels in ΔcbrA pcpdR1D53A cells but not in ΔcbrA pcpdR1 cells (Fig. 4C). These results demonstrate that a modest input of constitutively active CpdR1D53A is able to suppress the ΔcbrA mutation with regard to CtrA levels by restoring a wild-type level of instability and support a model that regulation of CtrA degradation by CbrA occurs indirectly through downstream effects on CpdR1 activity (Fig. 1).
CpdR1D53A restores wild-type cell cycle progression to the ΔcbrA mutant.
CbrA contributes to cell cycle regulation such that the ΔcbrA mutant displays filamentous growth indicative of a cell division defect and is associated with cells containing an altered genome complement (9). CpdR1D53A is able to restore CtrA instability to wild-type levels in the ΔcbrA mutant and may therefore also restore wild-type cell cycle progression. As a preliminary test, we examined the ΔcbrA mutant calcofluor-bright phenotype that reflects overproduction of the exopolysaccharide EPS I (also referred to as succinoglycan) (9, 51). Wild-type cells with either the pcpdR1 or pcpdR1D53A plasmid produce normal levels of EPS I and are dim on calcofluor medium (Fig. 5A, strains 1 and 2). The ΔcbrA pcpdR1 cells retain their mutant calcofluor-bright phenotype, while the ΔcbrA pcpdR1D53A cells display a wild-type dim phenotype (Fig. 5A, strains 3 and 4). The dim phenotype of ΔcbrA mutant strain carrying plasmid pcpdR1D53A (ΔcbrA pcpdR1D53A strain) is indistinguishable from that of the ΔcbrA mutant strain complemented with pLAFR2070 (ΔcbrA pLAFR2070 complemented strain) and therefore reflects complete suppression of EPS I overproduction (Fig. 5A, strains 4 and 8). The ΔcbrA motility defect (Fig. 5B) is also fully suppressed by plasmid pcpdR1D53A but not by plasmid pcpdR1 (Fig. 5B).
FIG 5.

Free-living phenotypes of the ΔcbrA mutant are rescued by constitutive ectopic expression of unphosphorylatable CpdR1D53A. (A) Succinoglycan (EPS I) production was assessed on LB/MC medium supplemented with calcofluor. Strains are numbered on the plate as follows: 1, WT cells carrying plasmid pcpdR1; 2, WT cells carrying plasmid pcpdR1D53A; 3, ΔcbrA cells carrying pcpdR1; 4, ΔcbrA cells carrying pcpdR1D53A; 5, WT cells carrying plasmid pLAFR1; 6, WT cells carrying plasmid pLAFR2070; 7, ΔcbrA cells carrying pLAFR1; 8, ΔcbrA cells carrying pLAFR2070. (B) Motility was assessed using 0.2% LB/MC agar. The values that were significantly different (P < 0.0001) from the WT value are indicated by an asterisk.
As described previously (9), the ΔcbrA mutant displays an aberrant filamentous morphology (Fig. 6A, column 2) at an increased frequency compared to the wild type (Fig. 6B), and this phenotype is not altered by the addition of plasmid pcpdR1 (Fig. 6A and B). There is a modest but observable cell morphology defect associated with constitutive ectopic expression of CpdR1D53A but not CpdR1 in an otherwise wild-type background (Fig. 6A, column 2) such that 5% of cells are highly filamentous (Fig. 6B) (8). CpdR1D53A suppresses the ΔcbrA cellular filamentation defect (Fig. 6A) such that when plasmid pcpdR1D53A is combined with ΔcbrA cells, the rate of filamentation resembles that of plasmid pcpdR1D53A in wild-type cells (Fig. 6B). Similarly, combining plasmid pcpdR1D53A, but not plasmid pcpdR1, with ΔcbrA cells results in reversion to the bimodal 1N-2N DNA distribution of cells observed in unsynchronized wild-type populations (Fig. 6C and D). We therefore find that constitutive ectopic expression of CpdR1D53A, which restores wild-type levels of CtrA to the ΔcbrA mutant, is also able to fully suppress known ΔcbrA cell cycle defects during free-living growth.
FIG 6.

Cell cycle phenotypes of the ΔcbrA mutant are rescued by constitutive ectopic expression of unphosphorylatable CpdR1D53A. (A) Cellular morphology was observed and classified as either rod shaped (2 to 4 μm long and 1 μm wide; column 1) or aberrant (round or branched and filamentous; column 2). (B) The frequency of aberrant cell morphology in an exponentially growing population of cells was quantified. (C) Flow cytometry was performed to measure cellular genome content per cell. (D) The frequency of aberrant DNA content (either <1N or >2N) in an exponentially growing population of cells was quantified as the average percentage and its standard deviation.
CpdR1D53A restores wild-type symbiosis to the ΔcbrA mutant.
The cbrA mutant is unable to properly establish a symbiosis with Medicago sativa (51). The underlying reason for this defect has been unclear (52); however, we now predict that it may be due to high levels of CtrA and associated cell cycle defects (9). Since the ΔcbrA pcpdR1D53A mutant has wild-type levels of CtrA, we investigated whether it would be able to establish an effective symbiosis with M. sativa in order to test this prediction. Plants were inoculated with strains containing either pcpdR1 or pcpdR1D53A plasmid and were grown for 5 weeks. The percentage of pink nodules and total plant height were used as a set of metrics to assess the effectiveness of symbiosis. The extremely low percentage of pink nodules and decreased plant height observed with the ΔcbrA pcpdR1 strain shows that this strain fails to establish a functional symbiosis (Fig. 7), similar to the ΔcbrA strain alone (9). However, the ΔcbrA pcpdR1D53A strain shows a complete rescue of symbiosis to the level observed with the wild-type strain (Fig. 7). These results show that constitutively active CpdR1D53A is able to overcome the ΔcbrA symbiosis defect and are consistent with the model that this defect is due to misregulation of CtrA.
FIG 7.

The symbiosis defect of ΔcbrA is rescued by constitutive ectopic expression of unphosphorylatable CpdR1D53A. Symbiosis was assayed with Medicago sativa plants 5 weeks postinoculation. (A) Average percentage of pink nodules per total nodules per plant and its standard deviation. (B) Average plant height and its standard deviation. Values that were significantly different (P < 0.0001) from the values for WT cells carrying plasmid pcpdR1 or pcpdR1D53A are indicated by an asterisk. The value that was significantly different (P < 0.0007) from the value for ΔcbrA cells carrying plasmid pcpdR1D53A is indicated by a plus symbol.
DISCUSSION
A molecular understanding of the components that govern S. meliloti cell cycle progression during free-living growth is necessary to develop a model for how this program may be modified during host colonization. Several two-component histidine kinases function as part of the DivK pathway required for both free-living cell cycle progression and symbiosis. CbrA and DivJ function as kinases to phosphorylate DivK, while PleC functions as a DivK phosphatase (9, 10) (Fig. 1). While pleC is essential in S. meliloti (43), the individual loss of either divJ or cbrA is tolerated and leads to filamentation and ploidy defects during free-living growth as well as the loss of symbiosis (9, 10, 51). These phenotypic defects are correlated with increased levels and phosphorylation of the essential response regulator CtrA (9, 10, 44), suggesting that the DivK pathway serves to regulate the cell cycle by controlling CtrA activity (Fig. 1).
Since DivK-mediated regulation of CtrA at the level of phosphorylation appears to be conserved in S. meliloti (10), we examined whether CtrA regulation at the level of degradation is also conserved. CtrA degradation in C. crescentus is carried out by the ClpXP protease and requires CpdR, RcdA, and c-di-GMP-bound PopA for enhanced recognition of CtrA as a proteolytic substrate (40). Although S. meliloti has putative CpdR and RcdA orthologs (4, 8), there is no recognizable PopA ortholog, and it was therefore unclear whether CtrA would be regulated by proteolysis in S. meliloti (41) (Fig. 2).
We measured CtrA stability during exponential growth and found that CtrA is in fact unstable (Fig. 3A) despite the absence of PopA. Given that PopA serves as a c-di-GMP sensor (38), it is therefore unclear whether S. meliloti CtrA levels are responsive to fluctuations in this particular second messenger through an alternative c-di-GMP receptor or if this aspect of CtrA regulation is specific to C. crescentus and its more closely related class Alphaproteobacteria. We also measured CtrA stability in a ΔcbrA mutant and observed a significant stabilization of the protein (Fig. 3B), showing that CtrA stability is regulated in a CbrA-dependent manner.
CpdR1 is hypothesized to be a downstream target of CbrA regulation that is required to direct ClpXP-mediated degradation of CtrA in its unphosphorylated state (Fig. 1) (8). We tested this by examining whether addition of unphosphorylatable CpdR1D53A would impact CtrA levels. Consistent with this prediction, introduction of CpdR1D53A to the ΔcbrA mutant restores CtrA to wild-type levels (Fig. 4A and B) and leads to an increase in CtrA instability (Fig. 4C and D). Thus, CpdR1D53A behaves as a dominant factor that can overcome the loss of CbrA regulation. We were unable to directly test the epistatic relationship between cbrA and cpdR1 due to the severe growth defect of the ΔcpdR1 mutant (8); however, we favor a model in which CpdR1 functions downstream of CbrA as a target of DivK-mediated regulation of the CckA pathway (Fig. 1). Moreover, since CpdR1 influences ClpX localization (8), it is likely that unphosphorylated CpdR1 directly promotes ClpXP-mediated proteolysis of CtrA, although this remains to be tested directly.
Since CpdR1D53A restores wild-type CtrA levels to the ΔcbrA mutant, we examined whether it would also have an impact on the cell cycle. We find that CpdR1D53A is able to suppress ΔcbrA mutant free-living phenotypes (Fig. 5A and B). Importantly, the ΔcbrA mutant cell cycle defects of filamentous cell morphology (Fig. 6A and B) and aberrant DNA content are fully suppressed by the addition of CpdR1D53A (Fig. 6C and D). These observations provide a strong link between CbrA-dependent regulation of CtrA stability and free-living cell cycle outcomes. However, C. crescentus CpdR promotes the regulated degradation of multiple ClpXP targets, including the σ54-dependent response regulator TacA (53), the c-di-GMP phosphodiesterase PdeA (39, 54), and CtrA (33). It is therefore possible that CpdR1-dependent degradation of a target other than CtrA is responsible for CpdR1D53A suppression of ΔcbrA mutant phenotypes. Nevertheless, the simplest model is that a single event is responsible for CpdR1D53A suppression of all ΔcbrA phenotypes, including CtrA stability, which implies that regulated CtrA degradation is directly responsible for cell cycle outcomes (Fig. 1).
In C. crescentus, CtrA regulation is central to cell cycle progression and asymmetric cell division, and our results support a model in which this role is conserved in S. meliloti. Although it is possible that CtrA is not the only factor required for CbrA-dependent cell cycle regulation, it does remain highly likely that CtrA plays a role in cell cycle progression. CtrA is essential (44), so its presence is needed for cell viability, and the identification of putative CtrA binding sites points toward a role in promoting cell division (4, 55).
We also find that the addition of CpdR1D53A restores symbiosis to the ΔcbrA mutant (Fig. 7), which suggests that the regulation of CtrA stability may be critical to symbiosis between S. meliloti and M. sativa. While typical growth and cell division of S. meliloti has been suggested as the mechanism for host invasion (56), a novel cell cycle of bacterial endoreduplication has been observed during the intracellular colonization process of bacteroid formation (3). Given that CbrA is a regulator of CtrA and the cell cycle (9) and is absolutely required for symbiosis (51), it appears likely that regulation of the bacterial cell cycle plays an essential role in establishing the symbiosis. More specifically, it may be that endoreduplication requires that CtrA activity be repressed so that DNA replication can proceed in the absence of cell division or that bacterial exit from the cell cycle into a permanent nonreproductive G0 state involves CtrA regulation.
Several prior observations support this hypothesis that repression of CtrA is critical to symbiosis. We were unable to examine the CtrA phenotype of a ΔcpdR1 mutant due to its severe growth defect. However, based on our observation that CpdR1D53A decreases the stability of CtrA, it is likely that loss of cpdR1 results in increased levels of CtrA and this may be why the ΔcpdR1 mutant is able to infect host nodules but unable to properly differentiate into a bacteroid (8). More recently, it was shown that a nodule-specific cysteine-rich (NCR) peptide that is produced by the host within nodules and is able to induce free-living cells to differentiate into bacteroids ex planta is also able to alter the level of expression of putative CtrA gene targets (57). This is of broad significance given that CpdR1 and CtrA are conserved among rhizobial bacteria that engage in host-microbe interactions (4), particularly since regulation of the bacterial cell cycle is associated with the colonization process (3, 5).
ACKNOWLEDGMENTS
We extend our sincere thanks to Linda Huang (Department of Biology, University of Massachusetts, Boston) and her laboratory for use of their fluorescence microscope and microscopy expertise, Todd Riley and Trudy Gulick (Department of Biology, University of Massachusetts, Boston) for advice on bioinformatics methods for PopA/PleD phylogenic analysis, and members of the Gibson laboratory for many helpful discussions.
This work was supported by funding from various sources as follows. K.E.G. was supported by grants from the NIH (1 R15 GM099052-01) and NSF (IOS 1119866). P.C. was supported by grants from the NIH (GM111706 and GM084517). J.M. was supported by the NSF through a Research Experiences for Undergraduates (REU) grant to the University of Massachusetts, Boston (DBI1062748). K.B.S. was supported by the NIH through a U56 grant to the University of Massachusetts, Boston, (3U56CA1186305-05S1) and by the University of Massachusetts, Boston, through the Nancy Goranson Graduate Student Research Fund and the Sanofi Genzyme Doctoral Research Fellowship.
REFERENCES
- 1.Jacobs C, Hung D, Shapiro L. 2001. Dynamic localization of a cytoplasmic signal transduction response regulator controls morphogenesis during the Caulobacter cell cycle. Proc Natl Acad Sci U S A 98:4095–4100. doi: 10.1073/pnas.051609998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hallez R, Bellefontaine AF, Letesson JJ, De Bolle X. 2004. Morphological and functional asymmetry in alpha-proteobacteria. Trends Microbiol 12:361–365. doi: 10.1016/j.tim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Mergaert P, Uchiumi T, Alunni B, Evanno G, Cheron A, Catrice O, Mausset AE, Barloy-Hubler F, Galibert F, Kondorosi A, Kondorosi E. 2006. Eukaryotic control on bacterial cell cycle and differentiation in the Rhizobium-legume symbiosis. Proc Natl Acad Sci U S A 103:5230–5235. doi: 10.1073/pnas.0600912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brilli M, Fondi M, Fani R, Mengoni A, Ferri L, Bazzicalupo M, Biondi EG. 2010. The diversity and evolution of cell cycle regulation in alpha-proteobacteria: a comparative genomic analysis. BMC Syst Biol 4:52. doi: 10.1186/1752-0509-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deghelt M, Mullier C, Sternon JF, Francis N, Laloux G, Dotreppe D, Van der Henst C, Jacobs-Wagner C, Letesson JJ, De Bolle X. 2014. G1-arrested newborn cells are the predominant infectious form of the pathogen Brucella abortus. Nature Commun 5:4366. doi: 10.1038/ncomms5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng J, Sibley CD, Zaheer R, Finan TM. 2007. A Sinorhizobium meliloti minE mutant has an altered morphology and exhibits defects in legume symbiosis. Microbiology 153:375–387. doi: 10.1099/mic.0.2006/001362-0. [DOI] [PubMed] [Google Scholar]
- 7.Hallez R, Mignolet J, Van Mullem V, Wery M, Vandenhaute J, Letesson JJ, Jacobs-Wagner C, De Bolle X. 2007. The asymmetric distribution of the essential histidine kinase PdhS indicates a differentiation event in Brucella abortus. EMBO J 26:1444–1455. doi: 10.1038/sj.emboj.7601577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi H, De Nisco NJ, Chien P, Simmons LA, Walker GC. 2009. Sinorhizobium meliloti CpdR1 is critical for co-ordinating cell cycle progression and the symbiotic chronic infection. Mol Microbiol 73:586–600. doi: 10.1111/j.1365-2958.2009.06794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadowski C, Wilson D, Schallies K, Walker G, Gibson KE. 2013. The Sinorhizobium meliloti sensor histidine kinase CbrA contributes to free-living cell cycle regulation. Microbiology 159:1552–1563. doi: 10.1099/mic.0.067504-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pini F, Frage B, Ferri L, De Nisco NJ, Mohapatra SS, Taddei L, Fioravanti A, Dewitte F, Galardini M, Brilli M, Villeret V, Bazzicalupo M, Mengoni A, Walker GC, Becker A, Biondi EG. 2013. The DivJ, CbrA and PleC system controls DivK phosphorylation and symbiosis in Sinorhizobium meliloti. Mol Microbiol 90:54–71. doi: 10.1111/mmi.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van der Henst C, Beaufay F, Mignolet J, Didembourg C, Colinet J, Hallet B, Letesson JJ, De Bolle X. 2012. The histidine kinase PdhS controls cell cycle progression of the pathogenic alphaproteobacterium Brucella abortus. J Bacteriol 194:5305–5314. doi: 10.1128/JB.00699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Heindl JE, Fuqua C. 2013. Coordination of division and development influences complex multicellular behavior in Agrobacterium tumefaciens. PLoS One 8:e56682. doi: 10.1371/journal.pone.0056682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curtis PD, Brun YV. 2010. Getting in the loop: regulation of development in Caulobacter crescentus. Microbiol Mol Biol Rev 74:13–41. doi: 10.1128/MMBR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAdams HH, Shapiro L. 2009. System-level design of bacterial cell cycle control. FEBS Lett 583:3984–3991. doi: 10.1016/j.febslet.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsokos CG, Laub MT. 2012. Polarity and cell fate asymmetry in Caulobacter crescentus. Curr Opin Microbiol 15:744–750. doi: 10.1016/j.mib.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osteras M, Jenal U. 2000. Regulatory circuits in Caulobacter. Curr Opin Microbiol 3:171–176. doi: 10.1016/S1369-5274(00)00071-0. [DOI] [PubMed] [Google Scholar]
- 17.Chen YE, Tropini C, Jonas K, Tsokos CG, Huang KC, Laub MT. 2011. Spatial gradient of protein phosphorylation underlies replicative asymmetry in a bacterium. Proc Natl Acad Sci U S A 108:1052–1057. doi: 10.1073/pnas.1015397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domian IJ, Quon KC, Shapiro L. 1997. Cell type-specific phosphorylation and proteolysis of a transcriptional regulator controls the G1-to-S transition in a bacterial cell cycle. Cell 90:415–424. doi: 10.1016/S0092-8674(00)80502-4. [DOI] [PubMed] [Google Scholar]
- 19.Domian IJ, Reisenauer A, Shapiro L. 1999. Feedback control of a master bacterial cell-cycle regulator. Proc Natl Acad Sci U S A 96:6648–6653. doi: 10.1073/pnas.96.12.6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenal U. 2009. The role of proteolysis in the Caulobacter crescentus cell cycle and development. Res Microbiol 160:687–695. doi: 10.1016/j.resmic.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Gora KG, Cantin A, Wohlever M, Joshi KK, Perchuk BS, Chien P, Laub MT. 2013. Regulated proteolysis of a transcription factor complex is critical to cell cycle progression in Caulobacter crescentus. Mol Microbiol 87:1277–1289. doi: 10.1111/mmi.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gora KG, Tsokos CG, Chen YE, Srinivasan BS, Perchuk BS, Laub MT. 2010. A cell-type-specific protein-protein interaction modulates transcriptional activity of a master regulator in Caulobacter crescentus. Mol Cell 39:455–467. doi: 10.1016/j.molcel.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Ohta N, Newton A. 1998. An essential, multicomponent signal transduction pathway required for cell cycle regulation in Caulobacter. Proc Natl Acad Sci U S A 95:1443–1448. doi: 10.1073/pnas.95.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hung DY, Shapiro L. 2002. A signal transduction protein cues proteolytic events critical to Caulobacter cell cycle progression. Proc Natl Acad Sci U S A 99:13160–13165. doi: 10.1073/pnas.202495099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsokos CG, Perchuk BS, Laub MT. 2011. A dynamic complex of signaling proteins uses polar localization to regulate cell-fate asymmetry in Caulobacter crescentus. Dev Cell 20:329–341. doi: 10.1016/j.devcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biondi EG, Reisinger SJ, Skerker JM, Arif M, Perchuk BS, Ryan KR, Laub MT. 2006. Regulation of the bacterial cell cycle by an integrated genetic circuit. Nature 444:899–904. doi: 10.1038/nature05321. [DOI] [PubMed] [Google Scholar]
- 27.Chen YE, Tsokos CG, Biondi EG, Perchuk BS, Laub MT. 2009. Dynamics of two phosphorelays controlling cell cycle progression in Caulobacter crescentus. J Bacteriol 191:7417–7429. doi: 10.1128/JB.00992-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quon KC, Marczynski GT, Shapiro L. 1996. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell 84:83–93. doi: 10.1016/S0092-8674(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 29.Quon KC, Yang B, Domian IJ, Shapiro L, Marczynski GT. 1998. Negative control of bacterial DNA replication by a cell cycle regulatory protein that binds at the chromosome origin. Proc Natl Acad Sci U S A 95:120–125. doi: 10.1073/pnas.95.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laub MT, Chen SL, Shapiro L, McAdams HH. 2002. Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc Natl Acad Sci U S A 99:4632–4637. doi: 10.1073/pnas.062065699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laub MT, McAdams HH, Feldblyum T, Fraser CM, Shapiro L. 2000. Global analysis of the genetic network controlling a bacterial cell cycle. Science 290:2144–2148. doi: 10.1126/science.290.5499.2144. [DOI] [PubMed] [Google Scholar]
- 32.Jenal U, Fuchs T. 1998. An essential protease involved in bacterial cell-cycle control. EMBO J 17:5658–5669. doi: 10.1093/emboj/17.19.5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iniesta AA, McGrath PT, Reisenauer A, McAdams HH, Shapiro L. 2006. A phospho-signaling pathway controls the localization and activity of a protease complex critical for bacterial cell cycle progression. Proc Natl Acad Sci U S A 103:10935–10940. doi: 10.1073/pnas.0604554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iniesta AA, Shapiro L. 2008. A bacterial control circuit integrates polar localization and proteolysis of key regulatory proteins with a phospho-signaling cascade. Proc Natl Acad Sci U S A 105:16602–16607. doi: 10.1073/pnas.0808807105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan KR, Judd EM, Shapiro L. 2002. The CtrA response regulator essential for Caulobacter crescentus cell-cycle progression requires a bipartite degradation signal for temporally controlled proteolysis. J Mol Biol 324:443–455. doi: 10.1016/S0022-2836(02)01042-2. [DOI] [PubMed] [Google Scholar]
- 36.McGrath PT, Iniesta AA, Ryan KR, Shapiro L, McAdams HH. 2006. A dynamically localized protease complex and a polar specificity factor control a cell cycle master regulator. Cell 124:535–547. doi: 10.1016/j.cell.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 37.Taylor JA, Wilbur JD, Smith SC, Ryan KR. 2009. Mutations that alter RcdA surface residues decouple protein localization and CtrA proteolysis in Caulobacter crescentus. J Mol Biol 394:46–60. doi: 10.1016/j.jmb.2009.08.076. [DOI] [PubMed] [Google Scholar]
- 38.Duerig A, Abel S, Folcher M, Nicollier M, Schwede T, Amiot N, Giese B, Jenal U. 2009. Second messenger-mediated spatiotemporal control of protein degradation regulates bacterial cell cycle progression. Genes Dev 23:93–104. doi: 10.1101/gad.502409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abel S, Chien P, Wassmann P, Schirmer T, Kaever V, Laub MT, Baker TA, Jenal U. 2011. Regulatory cohesion of cell cycle and cell differentiation through interlinked phosphorylation and second messenger networks. Mol Cell 43:550–560. doi: 10.1016/j.molcel.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith SC, Joshi KK, Zik JJ, Trinh K, Kamajaya A, Chien P, Ryan KR. 2014. Cell cycle-dependent adaptor complex for ClpXP-mediated proteolysis directly integrates phosphorylation and second messenger signals. Proc Natl Acad Sci U S A 111:14229–14234. doi: 10.1073/pnas.1407862111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozaki S, Schalch-Moser A, Zumthor L, Manfredi P, Ebbensgaard A, Schirmer T, Jenal U. 2014. Activation and polar sequestration of PopA, a c-di-GMP effector protein involved in Caulobacter crescentus cell cycle control. Mol Microbiol 94:580–594. doi: 10.1111/mmi.12777. [DOI] [PubMed] [Google Scholar]
- 42.Wheeler RT, Shapiro L. 1999. Differential localization of two histidine kinases controlling bacterial cell differentiation. Mol Cell 4:683–694. doi: 10.1016/S1097-2765(00)80379-2. [DOI] [PubMed] [Google Scholar]
- 43.Fields AT, Navarrete CS, Zare AZ, Huang Z, Mostafavi M, Lewis JC, Rezaeihaghighi Y, Brezler BJ, Ray S, Rizzacasa AL, Barnett MJ, Long SR, Chen EJ, Chen JC. 2012. The conserved polarity factor podJ1 impacts multiple cell envelope-associated functions in Sinorhizobium meliloti. Mol Microbiol 84:892–920. doi: 10.1111/j.1365-2958.2012.08064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barnett MJ, Hung DY, Reisenauer A, Shapiro L, Long SR. 2001. A homolog of the CtrA cell cycle regulator is present and essential in Sinorhizobium meliloti. J Bacteriol 183:3204–3210. doi: 10.1128/JB.183.10.3204-3210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell 11:671–683. doi: 10.1016/S1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 46.Leigh JA, Signer ER, Walker GC. 1985. Exopolysaccharide-deficient mutants of Rhizobium meliloti that form ineffective nodules. Proc Natl Acad Sci U S A 82:6231–6235. doi: 10.1073/pnas.82.18.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282. [DOI] [PubMed] [Google Scholar]
- 48.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kahng LS, Shapiro L. 2001. The CcrM DNA methyltransferase of Agrobacterium tumefaciens is essential, and its activity is cell cycle regulated. J Bacteriol 183:3065–3075. doi: 10.1128/JB.183.10.3065-3075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ehrhardt DW, Atkinson EM, Long SR. 1992. Depolarization of alfalfa root hair membrane potential by Rhizobium meliloti Nod factors. Science 256:998–1000. doi: 10.1126/science.10744524. [DOI] [PubMed] [Google Scholar]
- 51.Gibson KE, Campbell GR, Lloret J, Walker GC. 2006. CbrA is a stationary-phase regulator of cell surface physiology and legume symbiosis in Sinorhizobium meliloti. J Bacteriol 188:4508–4521. doi: 10.1128/JB.01923-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibson KE, Barnett MJ, Toman CJ, Long SR, Walker GC. 2007. The symbiosis regulator CbrA modulates a complex regulatory network affecting the flagellar apparatus and cell envelope proteins. J Bacteriol 189:3591–3602. doi: 10.1128/JB.01834-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhat NH, Vass RH, Stoddard PR, Shin DK, Chien P. 2013. Identification of ClpP substrates in Caulobacter crescentus reveals a role for regulated proteolysis in bacterial development. Mol Microbiol 88:1083–1092. doi: 10.1111/mmi.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rood KL, Clark NE, Stoddard PR, Garman SC, Chien P. 2012. Adaptor-dependent degradation of a cell-cycle regulator uses a unique substrate architecture. Structure 20:1223–1232. doi: 10.1016/j.str.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Nisco NJ, Abo RP, Wu CM, Penterman J, Walker GC. 2014. Global analysis of cell cycle gene expression of the legume symbiont Sinorhizobium meliloti. Proc Natl Acad Sci U S A 111:3217–3224. doi: 10.1073/pnas.1400421111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gage DJ. 2002. Analysis of infection thread development using Gfp- and DsRed-expressing Sinorhizobium meliloti. J Bacteriol 184:7042–7046. doi: 10.1128/JB.184.24.7042-7046.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Penterman J, Abo RP, De Nisco NJ, Arnold MF, Longhi R, Zanda M, Walker GC. 2014. Host plant peptides elicit a transcriptional response to control the Sinorhizobium meliloti cell cycle during symbiosis. Proc Natl Acad Sci U S A 111:3561–3566. doi: 10.1073/pnas.1400450111. [DOI] [PMC free article] [PubMed] [Google Scholar]