

ABSTRACT
The opportunistic human pathogen Pseudomonas aeruginosa expresses numerous acute virulence factors in the initial phase of infection, and during long-term colonization it undergoes adaptations that optimize survival in the human host. Adaptive changes that often occur during chronic infection give rise to rugose small colony variants (RSCVs), which are hyper-biofilm-forming mutants that commonly possess mutations that increase production of the biofilm-promoting secondary messenger cyclic di-GMP (c-di-GMP). We show that RSCVs display a decreased production of acute virulence factors as a direct result of elevated c-di-GMP content. Overproduction of c-di-GMP causes a decrease in the transcription of virulence factor genes that are regulated by the global virulence regulator Vfr. The low level of Vfr-dependent transcription is caused by a low level of its coactivator, cyclic AMP (cAMP), which is decreased in response to a high level of c-di-GMP. Mutations that cause reversion of the RSCV phenotype concomitantly reactivate Vfr-cAMP signaling. Attempts to uncover the mechanism underlying the observed c-di-GMP-mediated lowering of cAMP content provided evidence that it is not caused by inhibition of adenylate cyclase production or activity and that it is not caused by activation of cAMP phosphodiesterase activity. In addition to the studies of the RSCVs, we present evidence that the deeper layers of wild-type P. aeruginosa biofilms have high c-di-GMP levels and low cAMP levels.
IMPORTANCE Our work suggests that cross talk between c-di-GMP and cAMP signaling pathways results in downregulation of acute virulence factors in P. aeruginosa biofilm infections. Knowledge about this cross-regulation adds to our understanding of virulence traits and immune evasion by P. aeruginosa in chronic infections and may provide new approaches to eradicate biofilm infections.
INTRODUCTION
Pseudomonas aeruginosa exhibiting a rugose small colony variant (RSCV) phenotype is frequently isolated from chronic infections (1–3). The RSCV phenotype promotes biofilm formation and provides advantages for the bacterium, such as increased tolerance toward antibiotics and immune responses of the host (4–7). As a consequence, the appearance of RSCVs in clinical samples from infected tissues is often associated with poor patient outcome (8, 9). The RSCV phenotype in P. aeruginosa is the result of increased production of exopolysaccharides and bacterial adhesins, which are central components of the biofilm matrix (10–15). The RSCV phenotype is most commonly caused by mutations that result in changed metabolism of the secondary messenger molecule cyclic di-GMP (c-di-GMP) (7, 16–18). c-di-GMP has been shown to positively regulate biofilm formation in various bacteria, including P. aeruginosa, at the transcriptional, translational, and posttranslational levels (19, 20). The synthesis of c-di-GMP in bacteria is accomplished by diguanylate cyclases (DGCs), whereas degradation of c-di-GMP is catalyzed by specific phosphodiesterases (PDEs) (19–21). The DGCs and PDEs frequently harbor sensory domains that enable translation of diverse environmental cues into specific c-di-GMP levels. In order to exert its effects, c-di-GMP binds to downstream effector molecules and modulates their function, thereby resulting in the production of adhesins and exopolymeric substances that promote biofilm formation (19, 20). In addition to its role as a positive regulator of biofilm formation, c-di-GMP has also been implicated in the regulation of virulence factors. In several pathogenic strains, including P. aeruginosa, Escherichia coli, Salmonella enterica, and Vibrio cholerae, high c-di-GMP levels have been shown to repress the production of acute virulence factors, whereas low levels enhance acute virulence (22–26).
In contrast to c-di-GMP, another second messenger, cyclic AMP (cAMP), has been shown to positively control the expression of acute virulence factors in P. aeruginosa (27–31). cAMP acts as a coactivator of the virulence factor regulator Vfr, and high levels of cAMP greatly increase the expression of numerous acute virulence factors, such as type II and III secretion systems and type IV pili (27, 30–33). Vfr has also been implicated in the regulation of quorum sensing and flagellum biosynthesis (34, 35). The production of cAMP in P. aeruginosa relies on two adenylate cyclases (ACs), CyaA and CyaB, whereas its degradation relies on the phosphodiesterase CpdA (27, 30, 31, 35). Decreased levels of cAMP in P. aeruginosa are associated with attenuated acute virulence (28).
In the present study, we provide evidence that the c-di-GMP and cAMP signaling pathways in P. aeruginosa are linked. We show that high levels of c-di-GMP in P. aeruginosa cause low levels of cAMP, resulting in low levels of acute virulence factor gene transcription.
MATERIALS AND METHODS
Strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 and S2, respectively, in the supplemental material. All strains were propagated in lysogeny broth (LB; containing per liter of ultrapure water 10.0 g tryptone, 5.0 g yeast extract, and 5.0 g NaCl). Alternatively, P. aeruginosa was grown in Vogel-Bonner minimal medium (VBMM), which was prepared as a 10× concentrate. This 10× VBMM contained per liter of ultrapure water 2.0 g MgSO4·7H2O, 20 g citric acid, 100 g K2HPO4, and 35 g NaNH4HPO4·4H2O and was adjusted to pH 7.0 and sterilized by filtration. The 10× VBMM solution was diluted 10-fold in sterile, ultrapure water as needed. Semisolid media were prepared by adding 15 g/liter Bacto agar to LB or 10 g/liter noble agar to VBMM. All measurements were conducted on cells grown in VBMM except for flow chamber experiments, which used fastidious anaerobe broth (FAB) medium with 0.3 mM glucose. Where appropriate, antibiotic selection was as follows: for E. coli, gentamicin (GEN) at 10 μg/ml, ampicillin (AMP) at 100 μg/ml, chloramphenicol (CHL) at 10 μg/ml, tetracycline (TET) at 10 μg/ml, and kanamycin (KAN) at 50 μg/ml; for P. aeruginosa, GEN at 100 μg/ml (30 μg/ml for mini-Tn7 mutants), TET at 100 μg/ml, and carbenicillin (CAR) at 300 μg/ml.
Congo red colony morphology assays and fluorescent imaging of macrocolonies.
Colony morphology was assessed on tryptone-Congo red (CR) plates (containing per liter of ultrapure water 10 g Bacto agar, 10 g tryptone, and 40 μg/ml Congo red). Overnight cultures were diluted in LB to an optical density at 600 nm (OD600) of 0.005, and 2-μl aliquots of these diluted cultures were spotted onto tryptone-CR agar. Colonies were allowed to grow for 4 days at room temperature. Pictures were taken using a Canon EOS 400D DSLR camera with a SteREO Lumar V12 fluorescent stereomicroscope (Zeiss). Heat maps of the fluorescent macrocolonies were generated using Fiji imaging software.
Crystal violet assay.
Overnight cultures in VBMM were diluted 1:100 in fresh VBMM, and 100-μl aliquots of the diluted cultures were transferred to the wells of a 96-well microtiter plate; plates were then incubated statically for 8 h at 37°C. Afterwards, the spent medium was removed and wells were washed twice with double-distilled water (ddH2O). Next, 125 μl of 0.1% crystal violet (CV) was added to each well and incubated for 15 min at room temperature. CV was removed and the wells were washed twice in ddH2O. The plates were allowed to dry before solubilizing the CV in 125 μl of 30% acetic acid. Optical densities of wells were read on a Victor plate reader (PerkinElmer) at 590 nm.
Standard molecular methods.
All basic molecular and microbiological techniques were executed according to standard protocols. Genomic DNA (gDNA) was purified using the DNeasy blood and tissue kit (Qiagen), plasmids were purified with the QIAprep Spin miniprep kit (Qiagen), and DNA was extracted from agarose gels with the QIAEX II gel extraction kit (Qiagen). Transformations of P. aeruginosa were carried out using established protocols for electroporation (36).
Allelic exchange vectors and mutant strain generation.
Mutant alleles and precise deletion mutations were created according to the method of Hmelo and colleagues (37). Briefly, mutant alleles were created by using two sets of PCR primers (see Table S1 in the supplemental material) which targeted the adjacent upstream and downstream regions of the chromosome flanking the gene of interest, to produce two PCR products. These products were subsequently joined via splicing by overlap extension (SOE) PCR to generate an in-frame deletion allele. Upstream forward and downstream reverse primers used to generate the deletion allele were tailed with either restriction sites or attB1 and attB2 sequences (see Table S1). SOE PCR products tailed with restriction sites were restricted and ligated into pEX18Gm by standard methods to produce allelic exchange vectors (pJJH55, pJJH57, and pJJH61) (see Table S2 in the supplemental material). In contrast, attB-tailed SOE PCR products were recombined with pDONRPEX18Gm by using BP Clonase II (Invitrogen) to produce allelic exchange vectors (pJJH206). Alternatively, attB-tailed SOE PCR products were first recombined with pDONR221 by using BP Clonase II and subsequently recombined with pEX18GmGW by using LR Clonase II to produce an allelic exchange vector (pJJH107). Plasmids with inserts were identified by PCR and sequenced using M13F(-21) and M13R primers (see Table S1).
To generate mutations in the P. aeruginosa chromosome, allelic exchange vectors were first transformed into the donor E. coli S17.1 strain and then introduced into P. aeruginosa via established protocols for biparental mating (37). Merodiploids were selected on VBMM containing 100 μg/ml GEN and subsequently streaked on no-salt LB agar containing 15% sucrose for counterselection. GEN-sensitive, sucrose-resistant strains with the mutant alleles were identified by PCR using the upstream forward and downstream reverse primers used to create the deletion allele (37).
Construction of vectors for overexpressing adenylate cyclases.
The cyaA (PA5272) and cyaB (PA3217) open reading frames (ORFs) were amplified using primer pairs cyaA-RBSII-NheI/cyaA-SacI and cyaB-up-EcoRI-RBSII/cyaB-dn-pstI, respectively (see Table S3 in the supplemental material). Here, the forward primers contained 5′ overhangs with a synthetic ribosomal binding site II (RBSII) sequence; additionally, all primers had 5′ overhangs with restriction sites (see Table S3). The PCR fragments produced with these primers were cloned into the arabinose-inducible shuttle vector pJN105 by restriction and ligation, producing the plasmids pJNcyaA and pJNcyaB, respectively.
Additionally, we constructed pUC18-miniTn7T-Gm vectors for making single-copy chromosomal insertions of arabinose-inducible genetic elements for cyaA and cyaB. This was accomplished by restricting pJNcyaA and pJNcyaB with KpnI/PstI and KpnI/SacI, respectively, and subsequently ligating the araC-PBAD-RBSII-cyaA and araC-PBAD-RBSII-cyaB fragments into pUC18-miniTn7T-Gm. This produced the vectors pHA104 and pHA59, respectively (see Table S2 in the supplemental material). These pHA104 and pHA59 mini-Tn7 vectors were subsequently introduced into the P. aeruginosa PAO1 chromosome by using the helper plasmid pTNS3 and established methods for electroporation, yielding the strains HA345 and HA346, respectively.
Construction of destination vectors pUCP22T2.1 and mini-CTX2T2.1-GW.
The Gateway compatible destination vectors pUCP22T2.1-GW and mini-CTXT2.1-GW were built for the purpose of creating transcriptional reporters and cloning divergently transcribed ORFs. These vectors were constructed with transcriptional terminators flanking both sides of the Gateway destination site. To build the pUCP22T2.1-GW plasmid, primers JJH876 and JJH878 were first used to clone the pUCP22 backbone (encompassing the origin of replication, bla, and aacC1 genes) from pMH487 (see Table S2 in the supplemental material). Next, primers JJH879 and JJH880 were used to clone the Gateway destination site (encompassing attR1 and attR2 sites, cat, and ccdB) as well as the flanking T4 and ToT1 terminators from pUC18-miniTn7T2.1-Gm-GW (see Table S2). Primers JJH876, JJH878, JJH879, and JJH880 were each synthesized with a 5′ overhang containing a HindIII site (see Table S3 in the supplemental material); thus, the resulting PCR products were digested with HindIII and ligated using standard procedures. The ligated plasmids were transformed into E. coli ccdB Survival 2 T1R (see Table S1 in the supplemental material) and selected on LB agar containing CHL, AMP, and GEN. Two colonies were streaked for isolation, and the resulting plasmid, pUCP22T2.1-GW, was purified and then fully sequenced by primer walking with Sanger sequencing (using primers JJH881, JJH894, JJH895, and JJH926 to JJH931 [see Table S3 in the supplemental material]).
To build the integration-proficient vector mini-CTX2T2.1-GW, primers JJH879 and JJH880 were first used to clone the Gateway destination site and the flanking T4 and ToT1 terminators from pUC18-miniTn7T2.1-Gm-GW (see Table S2 in the supplemental material). Subsequently, this PCR product was restricted with HindIII and ligated into the multiple-cloning site (MCS) of mini-CTX2 (see Table S2). Ligated plasmids were transformed into E. coli ccdB Survival 2 T1R (see Table S1 in the supplemental material) and selected on LB agar containing CHL and TET. Orientation of the insert was determined by Sanger sequencing using the primers JJH894 and JJH895 (see Table S3 in the supplemental material).
Construction of fluorescently labeled bacteria.
Multisite Gateway technology (Invitrogen) was used to create an integration-proficient mini-CTX2 vector in which a constitutively expressed, synthetic Ptrc promoter was fused to the mCherry ORF. To begin, the primers Ptrc-attB2-fwr and Ptrc-attB5-rev were used to amplify Ptrc from pMKB1::mCherry, and then the PCR product was recombined with pDONR221P2P5 by using BP Clonase II, generating the entry vector pHA50. Next, the primers mCherry-RBSII-attB5r-fwr and mCherry-attB1-rev were used to amplify the mCherry ORF from pMKB1::mCherry and place a synthetic RBSII adjacent to its start codon (see Table S1 in the supplemental material). Subsequently, BP Clonase II was used to recombine this PCR product with pDONR221P1P5r, generating the entry vector pHA46. Plasmids with inserts were identified by PCR and sequenced using M13F(-21) and M13R primers (see Table S1 in the supplemental material). Finally, pHA50 and pHA46 were recombined with mini-CTX2T2.1-GW by using LR Clonase II Plus, generating the Ptrc::mCherry fusion vector pHA51. The mCherry label from pHA51 was integrated into the P. aeruginosa PAO1 chromosome via an established electroporation protocol. The tetracycline resistance cassette was subsequently removed by using pFLP2 (38), creating the antibiotic-sensitive, mCherry-labeled strain P. aeruginosa HA147.
Reporter constructs.
Multisite Gateway technology was used to create transcriptional reporters for the P. aeruginosa PAO1 exoT and ptxR promoters as well as the E. coli K-12 lacP1 promoter. To begin, we used primers GFP(ASV)-RBSII-attB5r and GFP(ASV)-attB1 (see Table S3 in the supplemental material) to clone the short-half-life green fluorescent protein (GFP) gene from pUCP22Not::PcdrA-GFP(ASV) and flank it with a synthetic RBSII. Next, this PCR product was recombined with pDONR221P1P5r, creating the entry construct pHA60 (see Table S2 in the supplemental material). Plasmids with inserts were identified by PCR and sequenced using M13F(-21) and M13R primers.
Primer pairs attB2-PexoT-fwr/attB5-PexoT-rev, attB2-PptxR-fwr/attB5-PptxR-rev, and attB2-lacP1-fwr2/attB5-lacP1-rev2 were used to clone the exoT, ptxR, and lacP1 promoters, respectively. These PCR products were recombined with pDONR221P5P2 by using BP Clonase II to generate the entry vectors pHA36, pHA69, and pHA110, respectively (see Table S2 in the supplemental material). Plasmids with inserts were identified by PCR and sequenced using M13F(-21) and M13R primers. Finally, promoter-reporter fusions were generated through multisite recombination of pUCP22T2.1-GW and pHA60 with pHA36, pHA69, or pHA110 and by using LR Clonase II Plus, generating the GFP reporters pHA75, pHA76, and pHA67, respectively (see Table S2).
β-Galactosidase (LacZ) reporter measurements.
Overnight cultures of LacZ reporter strains were diluted 1:100 in VBMM. Cells were grown for an additional 8 h at 37°C and 200 rpm. Two 1-ml aliquots of culture were transferred into separate microcentrifuge tubes. Proteins were extracted from the first aliquot by using chloroform. LacZ activity in the aqueous phase was determined using the Tropix Galactostar chemiluminescent assay according to the manufacturer's directions (Invitrogen). The kit relies on the conversion of a chemiluminescent substrate for quantification of LacZ activity, and values are therefore given in relative light units (RLU). The second aliquot was spun at 13,000 × g for 5 min, and cell pellets were suspended in a 1:1 mixture of Laemmli buffer and Tris-Cl (pH 8.5). These samples were boiled for 15 min. The protein concentration was determined by using a Pierce 660-nm assay with ionic detergent compatibility reagent (IDCR; Thermo Scientific).
GFP reporter measurements.
Overnight cultures of GFP reporter strains were diluted 1:100 in VBMM. Cells were grown for an additional 8 h at 37°C and 200 rpm. Cultures were transferred to black, clear-bottom 96-well microtiter plates (Nunc). GFP fluorescence (excitation at 485 nm, emission at 535 nm) was measured on a Victor plate reader. Protein concentration was determined as described above for LacZ assays.
cAMP measurements.
Intracellular cAMP concentrations of P. aeruginosa were measured in an enzyme-linked immunosorbent assay (EIA; Cayman Chemical). Cells were grown in VBMM at 37°C to an OD450 of 1.5 (late exponential phase). Cells were collected (2 ml) by centrifugation at 13,000 × g for 2 min at 4°C and washed twice with 1 ml of cold phosphate-buffered saline (PBS). Nucleotides were extracted by suspending the cell pellets in 100 μl of 0.1 N HCl and incubating on ice for 10 min with occasional vortex mixing to lyse bacteria. Lysates were centrifuged at 13,000 × g for 5 min at 4°C to remove cellular material, and the supernatant was assayed for cAMP following the manufacturer's protocol for sample acetylation. Triplicate bacterial pellets for protein determinations were suspended in 100 μl of PBS and were lysed by three freeze-thaw cycles followed by centrifugation at 13,000 × g for 5 min to pellet unbroken cells. The total protein concentrations of the samples were determined by using the Pierce 660-nm reagent (Thermo Fisher Scientific). Assay values for cAMP were converted to intracellular concentrations based on the estimated cellular volume per milligram of protein (39).
Live cell imaging of planktonic bacteria.
Overnight cultures of GFP reporter strains were diluted 1:100 in VBMM supplemented with 100 μg/ml GEN. Cells were grown at 37°C at 200 rpm. Images were obtained at early exponential growth phase (OD450, 0.5) and at late exponential growth phase (OD450, 1.5). A 10-μl aliquot of each sample was spotted onto agarose pads (1% agarose, PBS) prepared on glass slides with a coverslip placed on top of the culture. Images were obtained using a LSM 710 confocal scanning laser microscope (CLSM; Zeiss) equipped with a Plan-Apochromat 63×/1.4 numerical aperture (NA) oil objective.
CLSM of flow chamber biofilms.
Flow chamber biofilms were grown in FAB containing 0.3 mM glucose and 15 μg/ml GEN. To begin, overnight cultures containing either the cAMP (pHA67) or c-di-GMP reporter [pCdrA-gfp(asv)] were grown in 5 ml LB with 100 μg/ml GEN. The flow chamber system was set up as previously described (40). Briefly, overnight cultures were diluted 1:100, grown to an OD600 of 1.0 in LB, then back-diluted again 1:1,000 in fresh medium. A 500-μl aliquot of this diluted culture was injected into the flow chamber system using a sterile syringe. Cells were allowed to attach for 1 h before flow was initiated at 1.75 rpm using a Watson Marlow 205S peristaltic pump. Images were obtained after 24 h and 72 h using the LSM 710 CLSM equipped with a Plan-Apochromat 63×/1.4 NA oil objective set for detection of GFP and mCherry.
CyaB protein expression and purification.
A plasmid (pQE30) containing a His-tagged version of the CyaB catalytic domain (amino acids 217 to 463) was overexpressed in E. coli XL1-Blue. An overnight culture was diluted to a start OD600 of 0.01 in LB medium at 37°C. Isopropyl-β-d-thiogalactopyranoside induction (1.0 mM) was started at an OD600 of 0.8, and the culture was placed at 18°C for 18 h (250 rpm). Cells were harvested by centrifugation (4,000 rpm, 10 min. Cell pellets were resuspended in lysis buffer solution (NPI), 1 mM imidazole, protease inhibitor cocktail. Cells were placed on ice for 15 min and lysed using sonication. Cellular debris was spun down for 30 min at 12,000 × g, and the soluble proteins were loaded onto Ni-nitrilotriacetic acid (NTA) columns. Purification was performed using a Qiagen Ni-NTA spin column kit according to the manufacturer's guidelines. Following the initial binding of the His-tagged proteins, the NPI buffer was replaced and cellular material was washed in HEPES buffer to avoid phosphate residues, which interfere with the EnzCheck assay (see below). SDS-PAGE was performed to confirm proper expression of the recombinant protein and purification steps.
Adenylate cyclase (AC) activity assays.
ATPase activity was assayed by measuring the production of inorganic phosphate (Pi) by using the Enzchek phosphate assay kit (Invitrogen). The reaction mixtures were set up as specified by the manufacturer, except that the MgCl2 concentration was increased to 10 mM. The total protein concentration per reaction mixture was 3.6 μM. Enzyme velocity was measured at a single concentration of substrate (ATP at 1 mM) with increasing concentrations of c-di-GMP (0.5 μM. 5 μM, 50 μM, 500 μM, and 1,000 μM). The enzyme had been incubated with the reaction mix and c-di-GMP for 30 min at room temperature before the addition of the ATP at 1 mM.
Statistical analysis.
Statistical significance was evaluated via a one-way or two-way analysis of variance (ANOVA).
RESULTS
P. aeruginosa RSCVs have decreased expression of acute virulence factor genes.
P. aeruginosa RSCVs with mutations in wspF (PA3703) or yfiR (PA1121) are frequently isolated from cystic fibrosis patients afflicted with persistent pulmonary biofilm infections (3, 7). Loss-of-function mutations in wspF or yfiR derepress the activities of the WspR and YfiN DGCs, respectively, increasing cellular c-di-GMP levels. Therefore, to begin investigating regulation of virulence factors in P. aeruginosa RSCVs, we employed two PAO1 strains with precise in-frame deletions in wspF (PA3703) and yfiR (PA1121) (7, 18).
The RSCV phenotype of both strains was verified by plating the cells on CR plates and by crystal violet staining of 8-hour-old biofilms grown in microtiter plates. Both the ΔwspF and ΔyfiR strains showed a wrinkly morphotype and increased CR binding, indicating increased production of matrix components compared to the wild-type PAO1 (see Fig. S1 in the supplemental material). Both strains overproduced biofilm compared to the wild type; however, unlike the CR binding assay, which showed markedly more CR binding to the ΔyfiR strain, the ΔwspF strain produced more biofilm in the plate assay (see Fig. S2 in the supplemental material). This was possibly due to pronounced aggregate formation by the ΔyfiR strain that may have prevented cells from coming in contact with the wall of the plate wells, which is necessary for attachment and biofilm formation.
Our labs recently developed and characterized a c-di-GMP bioreporter for gauging c-di-GMP levels in vivo (41). This bioreporter utilizes GFP as an indicator for transcription from the cdrA promoter, which is positively regulated by c-di-GMP through the transcriptional regulator FleQ (15, 17, 41). Both RSCVs showed a significant increase in PcdrA-gfp transcription, indicating high levels of c-di-GMP (Fig. 1). In particular, the ΔyfiR strain was found to have very high levels of c-di-GMP (more than a 20-fold increase above the wild type), in agreement with the high level of CR binding and aggregation.
FIG 1.
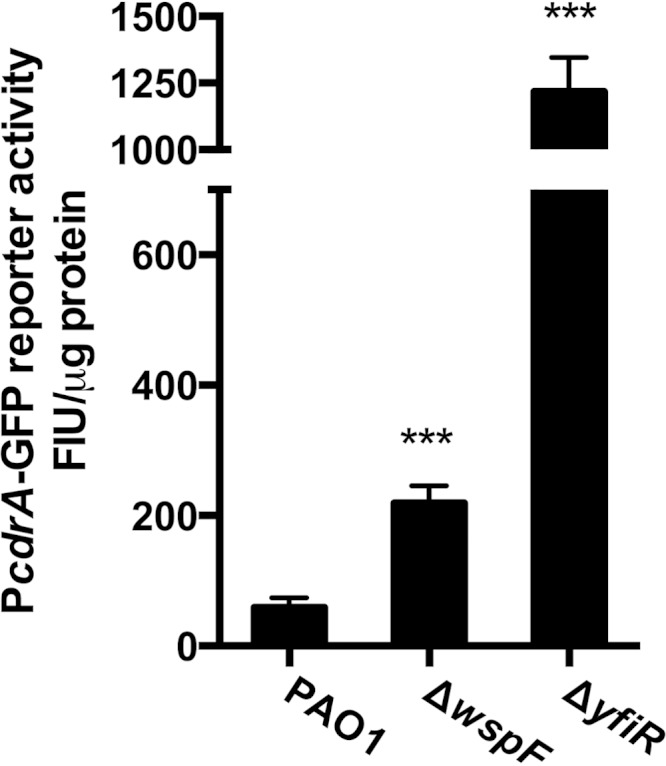
Increased c-di-GMP levels in RSCVs. c-di-GMP levels were estimated by using a transcriptional reporter [pUCp22Not::PcdrA-GFP(asv)] in wild-type PAO1 and the two RSCVs, ΔwspF and ΔyfiR. All measurements represent results from at least 2 biological experiments with at least 8 technical replicates. Fluorescence intensity units (FIU) from the reporter were normalized to the total protein content. Bars indicate standard deviations of the means. Statistical significance (P ≤ 0.001) is indicated with three asterisks.
Several studies have shown that c-di-GMP levels are inversely related to acute virulence in several species (22–26). In order to assess the expression of acute virulence factors in our two RSCV strains, we fused the promoter regions of the two virulence-associated genes exoT and ptxR to a short-half-life gfp gene. ExoT is a cytotoxic type III secretion system (T3SS) effector, whereas the ptxR gene encodes a transcription factor that promotes expression of exotoxin A (ExoA) and the LasB protease, which are released from bacteria by type II secretion. The plasmids pUCp22GW::PexoT-gfp(asv) and pUCp22GW::PptxR-gfp(asv) were introduced into the PAO1 wild type and ΔwspF and ΔyfiR mutant strains. We found that transcription from both the exoT and ptxR promoter was significantly decreased in the ΔwspF and ΔyfiR strains (Fig. 2). The exoT promoter activity was decreased more than 3-fold below wild-type levels in the ΔwspF and ΔyfiR strains, and a similar drop was observed for the ptxR promoter (Fig. 2).
FIG 2.

RSCVs show a decrease in transcription of acute virulence factors. Transcription levels of the exoT (black bars) and ptxR (white bars) promoters were measured using the fluorescence intensity from an unstable GFP reporter gene. exoT and ptxR transcription levels were measured in wild-type PAO1 and the two RSCVs, ΔwspF and ΔyfiR. Experiments were done in VBMM medium with a low calcium content. The data represent results of at least two biological experiments with 8 technical replicates each. Fluorescence intensity units (FIU) were normalized to the total protein content. Bars indicate standard deviations of the means. Statistical significance (P ≤ 0.001) is indicated with three asterisks.
P. aeruginosa RSCVs have decreased cellular cAMP levels.
The type II and type III secretion systems are both under positive regulation by the transcriptional regulator, Vfr. Vfr regulates multiple virulence factors in P. aeruginosa, many of which are essential for acute infection of eukaryotic cells (42). The transcriptional activity of Vfr is controlled by its coactivator, cAMP (27, 29). To test if cAMP levels were changed in our ΔwspF and ΔyfiR strains, we introduced a mini-CTX2::lacP1-lacZ construct into the ΦCTX site of the wild-type and RSCV strains. The Vfr-cAMP complex positively regulates the lacP1 promoter from E. coli in a dose-dependent manner in P. aeruginosa (30). As a control, we constructed a cAMP-negative P. aeruginosa mutant strain that also harbored the mini-CTX2::lacP1-lacZ construct. P. aeruginosa has two adenylate cyclases, CyaA (PA5272) and CyaB (PA3217). We created a ΔcyaA ΔcyaB double-knockout strain and introduced the mini-CTX2::lacP1-lacZ construct into the ΦCTX site of this cAMP-negative mutant. Utilizing the lacZ reporter assay, we assessed the cAMP levels in the wild-type, ΔwspF, ΔyfiR, and ΔcyaA ΔcyaB strains. The cAMP level in the ΔwspF strain was significantly reduced, and the reduction in cAMP levels was even greater in the ΔyfiR strain, which showed a reporter activity close to that of the cAMP-negative strain (Fig. 3A). In order to corroborate these findings, we also measured intracellular cAMP levels in the wild-type, ΔwspF, ΔyfiR, and ΔcyaA ΔcyaB strains via an EIA. The direct cAMP measurements using the EIA were in agreement with the reporter measurements (Fig. 3B).
FIG 3.

The RSCV phenotype is associated with severe repression in the cAMP-Vfr signaling cascade. (A) The transcriptional activity of the cAMP-Vfr complex was measured using the E. coli lacP1 promoter fused to lacZ. β-Galactosidase activity was assayed using a Tropix Galacto-Star kit. (B) Intracellular cAMP levels were measured in an EIA. The lacP1 transcriptional activity and cAMP levels were measured in the PAO1 wild type and the ΔwspF and ΔyfiR mutant strains. The cAMP-defective strain, ΔcyaA ΔcyaB, was included as a negative control. LacZ activity is reported in RLU (see Materials and Methods). Data sets represent results from at least two biological experiments with three technical replicates each. All measurements were standardized to the total protein content. Bars indicate standard deviations of the means. Statistical significance (P ≤ 0.001) is indicated with three asterisks.
P. aeruginosa shows an inverse relationship between cellular c-di-GMP and cAMP levels.
The data presented so far show that loss-of-function mutations in the wspF and yfiR genes in P. aeruginosa cause increased c-di-GMP levels and decreased transcription of acute virulence factors as well as decreased cAMP levels. We decided to directly test if changes in the c-di-GMP level per se would lead to changes in cAMP levels To avoid any pleitropic effects caused by extracellular matrix-mediated aggregation, we created a nonaggregating strain by deleting the pelF (PA3059) and pslD (PA2234) genes. The double-knockout strain ΔpelF ΔpslD was unable to aggregate in broth culture and unable to form biofilm in a standard crystal violet assay, even in the presence of high c-di-GMP levels (see Fig. S3 in the supplemental material). Subsequently, we introduced the cAMP reporter into the ΔpelF ΔpslD strain and then transformed this strain with plasmids harboring the gene for a DGC (PA1120/YfiN) or PDE (PA2133). Overexpression of the PA1120 DGC resulted in a significant decrease in cAMP levels, whereas overexpression of the PA2133 PDE resulted in a significant increase in cAMP levels (Fig. 4). These findings strongly indicated that the intracellular level of c-di-GMP affects the cAMP level in P. aeruginosa and thereby its virulence factor production.
FIG 4.

cAMP-Vfr signaling is repressed in response to c-di-GMP production. The direct effect of c-di-GMP production was assayed using an exopolysaccharide-negative strain (ΔpslD ΔpelF). The pBAD promoter was induced using 0.2% arabinose. The strains harbor a chromosomal insertion of a mini-CTX::lacP-lacZ construct. The data set represents results for at least two biological experiments with three technical replicates each. RLU were standardized to the total protein content. Bars indicate standard deviations of the means.
Reversion of the RSCV phenotype reactivates cAMP signaling.
The ΔyfiR RSCV frequently undergoes reversion to the ancestral smooth phenotype (18). Phenotypic reversion of ΔyfiR strains is commonly due to mutations in the yfiBNR operon that cause inactivation of the YfiN DGC (18). These mutations abolish c-di-GMP overproduction and restore the smooth colony phenotype. We hypothesized that mutations causing a reversion of the RSCV phenotype and lowering of c-di-GMP production would also restore cAMP levels. To test this hypothesis, we grew the ΔyfiR mutant strain with cyclic nucleotide reporters on agar plates for a prolonged time period and looked for phenotypic reversion. In order to visualize cAMP production in the colonies, we created a new GFP-based cAMP reporter. We tested this new reporter in P. aeruginosa mutant strains known to have either high or low cAMP levels, and we found no significant differences between the behaviors of our new GFP reporter and the previous lacZ-based reporter (see Fig. S4 in the supplemental material). When the ΔyfiR strain containing either the fluorescent cAMP or fluorescent c-di-GMP reporter had grown for 5 to 6 days on agar, mutants displaying a smooth colony morphology appeared in the periphery of the colonies (Fig. 5). The production of c-di-GMP was found to be highly upregulated in the central part of the colony, where the cells formed the characteristic wrinkled colony morphology. However, the reverted cells in the periphery of the colony that displayed the smooth colony phenotype showed little activity from the c-di-GMP reporter. The cAMP reporter, on the contrary, displayed little activity in the central parts of the colonies but appeared very bright in the reverted cells in the periphery of the colonies, indicating that cAMP signaling had been restored at the expense of increased c-di-GMP signaling (Fig. 5).
FIG 5.

Phenotypic reversion of RSCVs causes regain of cAMP-Vfr signaling. c-di-GMP signaling and Vfr-cAMP signaling were monitored for several days on CR agar plates by using fluorescent transcriptional reporters. Images were obtained after 4 days of growth at room temperature. Vfr-cAMP signaling was monitored for wild-type PAO1 (A), ΔyfiR (B), and a reverted ΔyfiR strain (C). c-di-GMP signaling was also monitored for wild-type PAO1 (D), ΔyfiR (E), and a reverted ΔyfiR strain (F). The calibration bar indicates fluorescence intensity units (FIU) for the biosensors.
The inner parts of P. aeruginosa wild-type biofilms have high c-di-GMP levels and low cAMP levels.
So far we have shown that RSCVs display a significant decrease in the Vfr-cAMP signaling pathway as a result of c-di-GMP overproduction and a concomitant lowering of cAMP levels. In contrast to the RSCVs, wild-type P. aeruginosa produces very little c-di-GMP when grown in a planktonic culture (43). However, substantial evidence suggests that biofilm-grown cells have greatly increased c-di-GMP levels (43). In light of the results presented here, it is therefore possible that cAMP levels are repressed in P. aeruginosa wild-type cells during the biofilm mode of growth. In agreement with previous studies, we saw little to no activity from the c-di-GMP reporter in P. aeruginosa wild-type cells during planktonic growth (Fig. 6A and B), whereas in a flow chamber-grown biofilm, we observed a significant increase in fluorescence from the reporter, especially along the bottom of the biofilm and in the central part of the microcolonies (Fig. 6C). The opposite was found to be the case for the cAMP reporter, which showed high and increasing activity during planktonic growth of P. aeruginosa wild-type cells (Fig. 6D and E) and little activity during biofilm growth except in the outer layer of cells, where it was highly expressed (Fig. 6F).
FIG 6.

P. aeruginosa biofilms shows heterogeneity in cAMP and c-di-GMP signaling. cAMP and c-di-GMP signaling levels were monitored by the use of GFP fluorescent reporters during planktonic growth and in biofilms grown in a continuous flow system. The activity of each signaling system was monitored using fluorescent transcriptional reporters. Images of c-di-GMP-producing cells were obtained from a mid-log-phase (A) and a late-log/early-stationary-phase (B) planktonic culture and from a 72-h-old biofilm (C). Images of cAMP-producing cells were also obtained from a mid-log-phase (D) and a late-log-/early-stationary-phase (E) planktonic culture and from a 72-h-old biofilm (F). Bar, 5 μM (planktonic cultures) or 10 μm (flow chambers). The calibration bar indicates fluorescence intensity units (FIU) for the biosensors.
c-di-GMP-dependent repression of cAMP does not occur through the Chp chemosensory system.
We next attempted to gain insight into the mechanistic basis underlying the observed inverse relationship between c-di-GMP and cAMP levels in P. aeruginosa. The Chp chemosensory system has been shown to regulate cAMP metabolism in P. aeruginosa by controlling the activation of CyaB (30, 31). To date, it is the only known system that directly regulates the production of cAMP in P. aeruginosa. A recent study by Düvel et al. identified ChpA as a possible c-di-GMP binding protein in a surface plasmon resonance screen (44). ChpA is a histidine kinase and the central component of the Chp chemosensory system. In the light of this work, we decided to test if c-di-GMP-mediated repression of cAMP acts through the Chp chemosensory system. We hypothesized that if c-di-GMP inhibits CyaB activity through the Chp chemosensory system, a ΔchpA (PA0413) mutant strain would be unresponsive to changes in the c-di-GMP level. We therefore deleted the chpA gene from P. aeruginosa and introduced the PA1120 DGC in the deletion mutant. Subsequently, we monitored cAMP levels in this strain by using our lacP-lacZ cAMP reporter. In accordance with previous reports, deletion of chpA caused a drop in cAMP levels (Fig. 7) (30). However, similar to what was observed in the wild-type strain, the induction of the PA1120 DGC also caused a lowering of cAMP in the ΔchpA mutant strain (Fig. 7). Based on these results, we conclude that c-di-GMP does not cause repression of cAMP levels through interference with the Chp chemosensory system in P. aeruginosa.
FIG 7.
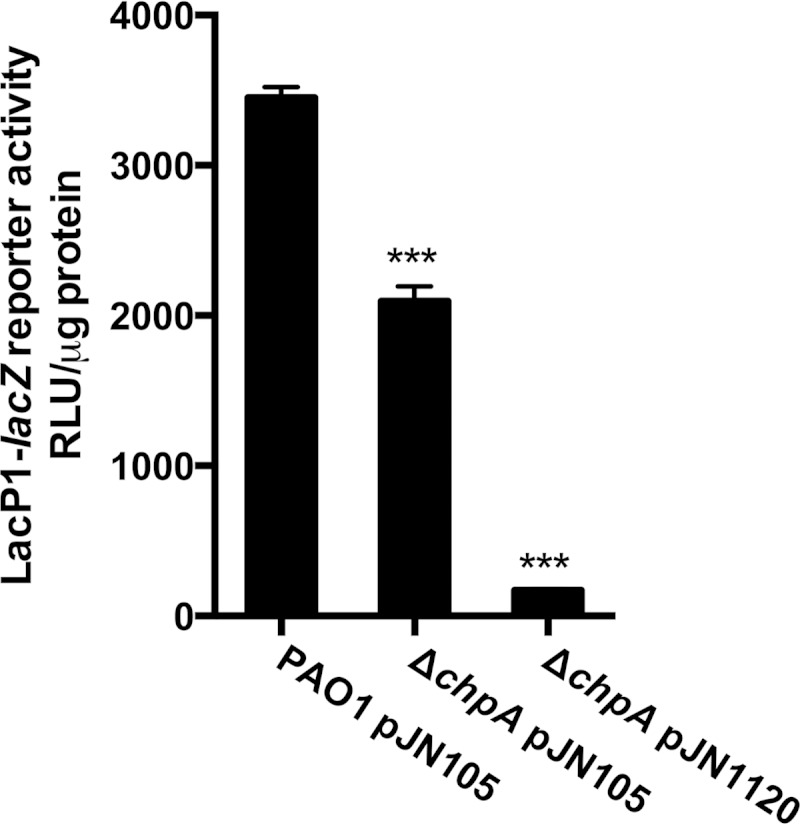
The Chp chemosensory system is not involved in c-di-GMP inhibition of cAMP-Vfr signaling. The transcriptional activity of the cAMP-Vfr reporter was measured in a ΔchpA mutant strain during PA1120 DGC expression (pJN1120). Wild-type PAO1 and ΔchpA strain containing the empty vector (pJN105) were included for comparison. All strains contained the mini-CTX::lacP1-lacZ construct inserted on the chromosome. Data sets represent results from at least two biological experiments with three technical replicates each. All measurements were standardized to the total protein content. Bars indicate standard deviations of the means. Statistical significance (P ≤ 0.001) is indicated with three asterisks.
c-di-GMP-dependent lowering of cAMP levels is not AC specific.
Subsequently, we sought to investigate if c-di-GMP-mediated repression of cAMP levels occurs through inhibition of a specific AC (either CyaA or CyaB). To this end, we created deletion mutations in each of the ACs. The deletion of the cyaA or cyaB gene, respectively, resulted in different changes in overall cAMP levels or Vfr-cAMP transcriptional activity (Fig. 8). The ΔcyaA strain showed only minor changes from wild-type PAO1 in both intracellular levels of cAMP and Vfr-cAMP transcription (Fig. 8). The ΔcyaB mutant strain, however, displayed a much larger drop in intracellular cAMP levels and was strongly impaired in Vfr-cAMP transcription. This is consistent with previous reports, which ascribed the CyaB AC as the major contributor to both overall cAMP levels in the cell and the primary contributor of cAMP for the Vfr-cAMP signaling cascade (27, 30). We introduced the arabinose-inducible PA1120 DGC in both the ΔcyaA and ΔcyaB strain in order to test if c-di-GMP-dependent inhibition of Vfr-cAMP signaling was specific either to the CyaA or CyaB AC. In both cases we found that DGC activation caused a further reduction in cAMP-Vfr transcription, compared to what was observed from the AC deletion alone (Fig. 8). These results suggest that c-di-GMP can repress Vfr-cAMP transcription regardless of which AC is responsible for producing the cAMP.
FIG 8.

cAMP levels produced by both the ACs are affected by DGC expression. Intracellular levels of cAMP in ΔcyaA and ΔcyaB mutant strains were measured using an EIA (A). The cAMP levels were measured during expression of PA1120 DGC (pJN1120) or with the vector control (pJN105). Vfr-cAMP transcriptional activity was measured in the same strain by using the mini-CTX::lacP-lacZ transcriptional reporter (B). Data sets represent results of at least two biological experiments with three technical replicates each. All measurements were standardized to the total protein content. Bars indicate standard deviations of the means. Statistical significance (P ≤ 0.001) is indicated with three asterisks.
Transcription and translation of the AC genes are not affected by c-di-GMP.
We next sought to investigate if c-di-GMP affects expression of the ACs at the level of transcription or translation of the AC genes in P. aeruginosa. We cloned the cyaA and cyaB genes so that they were controlled by the arabinose-inducible pBAD promoter and the ribosome binding site RBSII, instead of their native promoter and ribosome binding site. The pUC18miniTn7::pBAD-RBSII::cyaA and pUC18miniTn7::pBAD-RBSII::cyaB plasmids were then transformed into a P. aeruginosa ΔcyaA ΔcyaB strain in which we had introduced the pJN1120 plasmid, allowing overexpression of the PA1120 DGC. Overexpression of the PA1120 DGC resulted in lowering of the cAMP content in both the strain containing pUC18miniTn7::pBAD-RBSII::cyaA and pUC18miniTn7::pBAD-RBSII::cyaB (Fig. 9). From these results, we inferred that c-di-GMP does not have an effect on transcription or translation upstream of the start codon for either AC genes in P. aeruginosa. Because the cyaA and cyaB structural genes are very different, it is unlikely that c-di-GMP affects their transcription or translation via mechanisms acting downstream of their start codons.
FIG 9.

Transcription and translation of adenylate cyclases are not affected by c-di-GMP. The effects of c-di-GMP on Vfr-cAMP signaling through inhibition of AC transcription and translation were tested. The native AC genes (cyaA and cyaB) were deleted and were subsequently reintroduced individually under the control of the pBAD promoter with RBSII. Vfr-cAMP transcriptional activity was measured via LacZ measurement using the attCTX::lacP1-lacZ fusion. Data sets represent at results of least two biological experiments with three technical replicates each. All measurements were standardized to the total protein content. Bars indicate standard deviations of the means. Statistical significance (P ≤ 0.001) is indicated with three asterisks.
c-di-GMP does not interfere with AC cyclase activity.
Another possible mechanism for c-di-GMP-mediated lowering of cAMP levels is through allosteric inhibition of the ACs. A recent study showed that c-di-GMP could act as a competitive inhibitor for certain enzymes capable of catabolizing ATP (45). The cyclase activity of ACs and the biosynthesis of cAMP also require ATP. We tested this hypothesis on a recombinant version of the catalytic domain of CyaB (46). The enzymatic activity of the CyaB enzyme was tested in the presence of 0.5 to 1,000 μM c-di-GMP. However, we did not observe any effect on cyclase activity even at the highest concentration of c-di-GMP (1,000 μM), which is about 25 to 50 times higher than c-di-GMP levels found in RSCV cells (41) (Fig. 10).
FIG 10.

Adenylate cyclase activity is not inhibited by c-di-GMP. Vmax activity of CyaB-His was measured in an EnzCheck pyrophosphatase assay. The cyclase activity of CyaB was assayed with different concentrations of c-di-GMP, ranging from 0.5 μM to 1 mM. ATP (1 mM) substrate was used. The assay was performed with at least two replicates.
c-di-GMP-mediated lowering of cAMP does not occur through CpdA.
Since we did not obtain evidence for an effect of c-di-GMP on the production or activity of the ACs, we next sought to investigate if c-di-GMP might affect the degradation of cAMP. To date, the only known phosphodiesterase capable of degrading cAMP in P. aeruginosa is CpdA. One explanation for the observed decrease in cAMP levels under high-c-di-GMP conditions could be that CpdA expression is upregulated in response to c-di-GMP. This would in turn lead to an increased degradation of cAMP and a decrease in Vfr-cAMP transcription. To test this hypothesis, we created an in-frame deletion of the cpdA gene (PA4969) and introduced the mini-CTX::lacP1-lacZ reporter. Consistent with previous studies, the deletion of the cpdA gene caused the cells to accumulate cAMP (30, 31, 35). The ΔcpdA mutant was found to have cAMP levels more than 20-fold higher than wild-type levels (see Fig. S5 in the supplemental material). Similarly, cAMP-Vfr transcription was also highly upregulated in the absence of cpdA (Fig. 11). However, overexpression of the PA1120 DGC in the ΔcpdA mutant resulted in cAMP-Vfr transcriptional activity, which was significantly lower than the wild-type level. The results thus indicate that c-di-GMP-dependent inhibition of Vfr-cAMP signaling does not occur through increased CpdA-mediated cAMP degradation.
FIG 11.
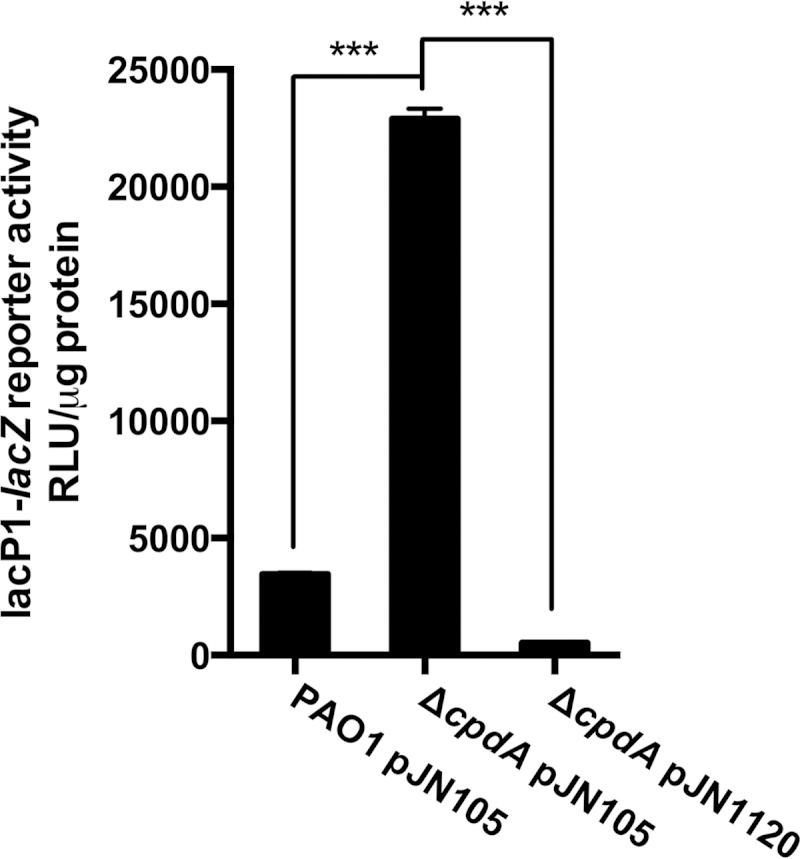
Deletion of the cpdA gene does not affect c-di-GMP inhibition of cAMP-Vfr signaling. The transcriptional activity of the cAMP-Vfr complex was measured in the ΔcpdA mutant strain during PA1120 DGC expression (pJN1120). Wild-type PAO1 and the ΔcpdA strain containing the empty vector (pJN105) were included for comparison. All strains contained the lacP1-lacZ construct allowing quantification of cAMP-Vfr transcriptional activity via LacZ measurements. Data sets represent results of at least two biological experiments with three technical replicates each. All measurements were standardized to the total protein content. Bars indicate standard deviations of the means. Statistical significance (P ≤ 0.001) is indicated with three asterisks.
DISCUSSION
In the present study, we have provided evidence that high levels of c-di-GMP in P. aeruginosa cause low levels of cAMP, resulting in low levels of acute virulence factor gene transcription. Our study suggests that c-di-GMP plays a more direct role in the repression of acute virulence than what has previously been appreciated. This coordination between c-di-GMP and cAMP signaling in P. aeruginosa may play an important role in the transition between the virulent planktonic lifestyle and the persistent biofilm lifestyle. Our work suggests that high levels of c-di-GMP not only increase the biofilm-forming capabilities of P. aeruginosa but also directly cause downregulation of several acute virulence factors. The acute virulence factors of bacteria comprise components that may be targeted by the host immune system. Downregulation of the expression of acute virulence factors during chronic infection may dampen immune responses and enable the bacteria to evade the immune system.
Type III secretion is considered an archetypical acute virulence factor in P. aeruginosa. A number of previous studies, both in in vitro systems and in longitudinal studies of sputum from cystic fibrosis patients, have reported that the type III secretion system is downregulated in P. aeruginosa biofilms (3, 47), although a study indicating that type III secretion is active in biofilms has also been reported (48). Recently it was reported that the translocon apparatus of the type III secretion system, but not the effectors themselves, is required for cell-associated aggregation of P. aeruginosa on the surface of polarized epithelial cells (49). The emerging consensus, which is in agreement with the findings reported here, appears to be that type III secretion may be required for the early phase of P. aeruginosa surface attachment and biofilm growth but is downregulated in the later stages of biofilm development.
Unlike type III secretion, type VI secretion has previously been linked to biofilm formation and chronic infection (50). Moscoso et al. provided evidence that the RetS/GacS/LadS system acts in coordination with c-di-GMP signaling in a switch between type III and type VI secretion (25). Deletion of the retS gene (PA4856) resulted in increased levels of c-di-GMP, and the ΔretS strain displayed an RSCV phenotype. It was further shown that the ΔretS strain had decreased expression of the type III secretion system, while type VI secretion was upregulated. Through overexpression of DGC's and PDE's Moscoso et al. provided evidence that the switch between type III and type VI secretion occurs as a direct result of changes in c-di-GMP metabolism. It was subsequently demonstrated that the SadC DGC is a central link between the RetS/GacS/LadS system and c-di-GMP signaling in P. aeruginosa (51). In the present study, we have added to these findings by demonstrating that c-di-GMP metabolism may change the expression of virulence factors through interference with the Vfr-cAMP signaling system. Through modulation of intracellular cAMP levels, the c-di-GMP level may affect the expression of several virulence factors under the transcriptional control of Vfr in P. aeruginosa. These studies are part of an increasing amount of evidence, for P. aeruginosa and also other bacteria, that high c-di-GMP levels repress virulence while low levels promote it (22–26).
The results from our flow chamber experiments indicated that during biofilm formation the bacteria display heterogeneity in their expression of the c-di-GMP and cAMP signaling systems. While the outer layers of the biofilm showed significant expression of our cAMP reporter, the inner part displayed low expression of the cAMP reporter and high expression of the c-di-GMP reporter. We hypothesize that high c-di-GMP production in the inner part of the biofilm serves to maintain the structural components of the biofilm, while cells residing in the outer layers remain more motile and possibly more virulent. This model is supported by a study that showed that P. aeruginosa bacteria from a biofilm increased the expression of the type III secretion system in response to predation (52). Those authors showed that upon coming in contact with the biofilm, the predatory amoeba Acanthamoeba castellanii was rapidly attacked and surrounded by highly virulent and motile bacteria (52).
The CRP-like proteins, Clp in Xanthomonas axonopodis pv. citri and Bcam1349 in Burkholderia cenocepacia, have been shown to be allosterically inhibited or activated, respectively, by c-di-GMP (53, 54). However, several studies have shown that Vfr is incapable of binding c-di-GMP (29, 55, 56). In addition, microarray analysis has provided evidence that c-di-GMP does not regulate vfr at the transcriptional level (17).
The mechanistic basis underlying the observed c-di-GMP-mediated lowering of cAMP in P. aeruginosa remains undiscovered. We have provided evidence that it is not caused by inhibition of AC production or activity, and it is not caused by activation of CpdA activity. Monds et al. demonstrated that diadenosine tetraphosphate (Ap4A) metabolism impacts biofilm formation by Pseudomonas fluorescens via modulation of c-di-GMP-dependent pathways, and they hypothesized that changes in the substrate (GTP) concentration mediated by altered flux through nucleotide biosynthetic pathways may be a significant point of regulation for c-di-GMP biosynthesis and regulation of biofilm formation (57). Similarly, we suggest that changes in the c-di-GMP level may affect the flux through the nucleotide biosynthetic pathways, resulting in changes in the level of substrate (ATP) for cAMP production. However, the average cellular concentration of ATP is expected to be more than 2 orders higher than the average cellular concentration of c-di-GMP (58, 59), so this scenario would involve compartmentalization. Further work is needed to confirm or refute this hypothesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Danish Council for Independent Research (DFF-1323-00177; to T.T.-N.), the Canadian Institutes of Health Research and Natural Sciences and Engineering Research Council (435631; to J.J.H.), and the American National Institute for Allergy and Infectious Diseases (2R01AI077628-05A1; to M.R.P.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00193-15.
REFERENCES
- 1.Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW. 2001. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis 183:444–452. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 2.Haussler S. 2003. Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J Med Microbiol 52:295–301. doi: 10.1099/jmm.0.05069-0. [DOI] [PubMed] [Google Scholar]
- 3.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drenkard E, Ausubel FM. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740–743. doi: 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- 5.Mishra M, Byrd MS, Sergeant S, Azad AK, Parsek MR, McPhail L, Schlesinger LS, Wozniak DJ. 2012. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell Microbiol 14:95–106. doi: 10.1111/j.1462-5822.2011.01704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, Wozniak DJ, Harwood CS, Parsek MR. 2009. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malone JG, Jaeger T, Spangler C, Ritz D, Spang A, Arrieumerlou C, Kaever V, Landmann R, Jenal U. 2010. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog 6:e1000804. doi: 10.1371/journal.ppat.1000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayer-Hamblett N, Ramsey BW, Kulasekara HD, Wolter DJ, Houston LS, Pope CE, Kulasekara BR, Armbruster CR, Burns JL, Retsch-Bogart G, Rosenfeld M, Gibson RL, Miller SI, Khan U, Hoffman LR. 2014. Pseudomonas aeruginosa phenotypes associated with eradication failure in children with cystic fibrosis. Clin Infect Dis 59:624–631. doi: 10.1093/cid/ciu385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayer-Hamblett N, Rosenfeld M, Gibson RL, Ramsey BW, Kulasekara HD, Retsch-Bogart GZ, Morgan W, Wolter DJ, Pope CE, Houston LS, Kulasekara BR, Khan U, Burns JL, Miller SI, Hoffman LR. 2014. Pseudomonas aeruginosa in vitro phenotypes distinguish cystic fibrosis infection stages and outcomes. Am J Respir Crit Care Med 190:289–297. doi: 10.1164/rccm.201404-0681OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman L, Kolter R. 2004. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol 186:4457–4465. doi: 10.1128/JB.186.14.4457-4465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman L, Kolter R. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol 51:675–690. doi: 10.1046/j.1365-2958.2003.03877.x. [DOI] [PubMed] [Google Scholar]
- 12.Jackson KD, Starkey M, Kremer S, Parsek MR, Wozniak DJ. 2004. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J Bacteriol 186:4466–4475. doi: 10.1128/JB.186.14.4466-4475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasseur P, Vallet-Gely I, Soscia C, Genin S, Filloux A. 2005. The pel genes of the Pseudomonas aeruginosa PAK strain are involved at early and late stages of biofilm formation. Microbiology 151:985–997. doi: 10.1099/mic.0.27410-0. [DOI] [PubMed] [Google Scholar]
- 14.Wozniak DJ, Wyckoff TJ, Starkey M, Keyser R, Azadi P, O'Toole GA, Parsek MR. 2003. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci U S A 100:7907–7912. doi: 10.1073/pnas.1231792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. 2010. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol 75:827–842. doi: 10.1111/j.1365-2958.2009.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D'Argenio DA, Calfee MW, Rainey PB, Pesci EC. 2002. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol 184:6481–6489. doi: 10.1128/JB.184.23.6481-6489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hickman JW, Tifrea DF, Harwood CS. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A 102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malone JG, Jaeger T, Manfredi P, Dotsch A, Blanka A, Bos R, Cornelis GR, Haussler S, Jenal U. 2012. The YfiBNR signal transduction mechanism reveals novel targets for the evolution of persistent Pseudomonas aeruginosa in cystic fibrosis airways. PLoS Pathog 8:e1002760. doi: 10.1371/journal.ppat.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fazli M, Almblad H, Rybtke ML, Givskov M, Eberl L, Tolker-Nielsen T. 2014. Regulation of biofilm formation in Pseudomonas and Burkholderia species. Environ Microbiol 16:1961–1981. doi: 10.1111/1462-2920.12448. [DOI] [PubMed] [Google Scholar]
- 20.Hengge R. 2009. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 21.Simm R, Morr M, Kader A, Nimtz M, Romling U. 2004. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol Microbiol 53:1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- 22.Chua SL, Liu Y, Yam JK, Chen Y, Vejborg RM, Tan BG, Kjelleberg S, Tolker-Nielsen T, Givskov M, Yang L. 2014. Dispersed cells represent a distinct stage in the transition from bacterial biofilm to planktonic lifestyles. Nat Commun 5:4462. doi: 10.1038/ncomms5462. [DOI] [PubMed] [Google Scholar]
- 23.Kuchma SL, Connolly JP, O'Toole GA. 2005. A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J Bacteriol 187:1441–1454. doi: 10.1128/JB.187.4.1441-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, Lee DG, Neely AN, Hyodo M, Hayakawa Y, Ausubel FM, Lory S. 2006. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A 103:2839–2844. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moscoso JA, Mikkelsen H, Heeb S, Williams P, Filloux A. 2011. The Pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ Microbiol 13:3128–3138. doi: 10.1111/j.1462-2920.2011.02595.x. [DOI] [PubMed] [Google Scholar]
- 26.Tischler AD, Camilli A. 2005. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun 73:5873–5882. doi: 10.1128/IAI.73.9.5873-5882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolfgang MC, Lee VT, Gilmore ME, Lory S. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell 4:253–263. doi: 10.1016/S1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 28.Smith RS, Wolfgang MC, Lory S. 2004. An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect Immun 72:1677–1684. doi: 10.1128/IAI.72.3.1677-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuchs EL, Brutinel ED, Jones AK, Fulcher NB, Urbanowski ML, Yahr TL, Wolfgang MC. 2010. The Pseudomonas aeruginosa Vfr regulator controls global virulence factor expression through cyclic AMP-dependent and -independent mechanisms. J Bacteriol 192:3553–3564. doi: 10.1128/JB.00363-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fulcher NB, Holliday PM, Klem E, Cann MJ, Wolfgang MC. 2010. The Pseudomonas aeruginosa Chp chemosensory system regulates intracellular cAMP levels by modulating adenylate cyclase activity. Mol Microbiol 76:889–904. doi: 10.1111/j.1365-2958.2010.07135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inclan YF, Huseby MJ, Engel JN. 2011. FimL regulates cAMP synthesis in Pseudomonas aeruginosa. PLoS One 6:e15867. doi: 10.1371/journal.pone.0015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrell E, Carty NL, Colmer-Hamood JA, Hamood AN, West SE. 2008. Regulation of Pseudomonas aeruginosa ptxR by Vfr. Microbiology 154:431–439. doi: 10.1099/mic.0.2007/011577-0. [DOI] [PubMed] [Google Scholar]
- 33.Whitchurch CB, Beatson SA, Comolli JC, Jakobsen T, Sargent JL, Bertrand JJ, West J, Klausen M, Waite LL, Kang PJ, Tolker-Nielsen T, Mattick JS, Engel JN. 2005. Pseudomonas aeruginosa fimL regulates multiple virulence functions by intersecting with Vfr-modulated pathways. Mol Microbiol 55:1357–1378. doi: 10.1111/j.1365-2958.2005.04479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dasgupta N, Ferrell EP, Kanack KJ, West SE, Ramphal R. 2002. fleQ, the gene encoding the major flagellar regulator of Pseudomonas aeruginosa, is σ70 dependent and is downregulated by Vfr, a homolog of Escherichia coli cyclic AMP receptor protein. J Bacteriol 184:5240–5250. doi: 10.1128/JB.184.19.5240-5250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuchs EL, Brutinel ED, Klem ER, Fehr AR, Yahr TL, Wolfgang MC. 2010. In vitro and in vivo characterization of the Pseudomonas aeruginosa cyclic AMP (cAMP) phosphodiesterase CpdA, required for cAMP homeostasis and virulence factor regulation. J Bacteriol 192:2779–2790. doi: 10.1128/JB.00168-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi KH, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Tseng BS, Zhang W, Harrison JJ, Quach TP, Song JL, Penterman J, Singh PK, Chopp DL, Packman AI, Parsek MR. 2013. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ Microbiol 15:2865–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomal located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 39.D'Souza-Ault MR, Smith LT, Smith GM. 1993. Roles of N-acetylglutaminylglutamine amide and glycine betaine in adaptation of Pseudomonas aeruginosa to osmotic stress. Appl Environ Microbiol 59:473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tolker-Nielsen T, Sternberg C. 2014. Methods for studying biofilm formation: flow cells and confocal laser scanning microscopy. Methods Mol Biol 1149:615–629. doi: 10.1007/978-1-4939-0473-0_47. [DOI] [PubMed] [Google Scholar]
- 41.Rybtke MT, Borlee BR, Murakami K, Irie Y, Hentzer M, Nielsen TE, Givskov M, Parsek MR, Tolker-Nielsen T. 2012. Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosa. Appl Environ Microbiol 78:5060–5069. doi: 10.1128/AEM.00414-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holder IA, Neely AN, Frank DW. 2001. Type III secretion/intoxication system important in virulence of Pseudomonas aeruginosa infections in burns. Burns 27:129–130. doi: 10.1016/S0305-4179(00)00142-X. [DOI] [PubMed] [Google Scholar]
- 43.Roy AB, Petrova OE, Sauer K. 2012. The phosphodiesterase DipA (PA5017) is essential for Pseudomonas aeruginosa biofilm dispersion. J Bacteriol 194:2904–2915. doi: 10.1128/JB.05346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duvel J, Bertinetti D, Moller S, Schwede F, Morr M, Wissing J, Radamm L, Zimmermann B, Genieser HG, Jansch L, Herberg FW, Haussler S. 2012. A chemical proteomics approach to identify c-di-GMP binding proteins in Pseudomonas aeruginosa. J Microbiol Methods 88:229–236. doi: 10.1016/j.mimet.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 45.Baraquet C, Harwood CS. 2013. Cyclic diguanosine monophosphate represses bacterial flagella synthesis by interacting with the Walker A motif of the enhancer-binding protein FleQ. Proc Natl Acad Sci U S A 110:18478–18483. doi: 10.1073/pnas.1318972110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topal H, Fulcher NB, Bitterman J, Salazar E, Buck J, Levin LR, Cann MJ, Wolfgang MC, Steegborn C. 2012. Crystal structure and regulation mechanisms of the CyaB adenylyl cyclase from the human pathogen Pseudomonas aeruginosa. J Mol Biol 416:271–286. doi: 10.1016/j.jmb.2011.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Speert DP, Farmer SW, Campbell ME, Musser JM, Selander RK, Kuo S. 1990. Conversion of Pseudomonas aeruginosa to the phenotype characteristic of strains from patients with cystic fibrosis. J Clin Microbiol 28:188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mikkelsen H, Bond NJ, Skindersoe ME, Givskov M, Lilley KS, Welch M. 2009. Biofilms and type III secretion are not mutually exclusive in Pseudomonas aeruginosa. Microbiology 155:687–698. doi: 10.1099/mic.0.025551-0. [DOI] [PubMed] [Google Scholar]
- 49.Tran CS, Rangel SM, Almblad H, Kierbel A, Givskov M, Tolker-Nielsen T, Hauser AR, Engel JN. 2014. The Pseudomonas aeruginosa type III translocon is required for biofilm formation at the epithelial barrier. PLoS Pathog 10:e1004479. doi: 10.1371/journal.ppat.1004479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, Walz T, Joachimiak A, Mekalanos JJ. 2006. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moscoso JA, Jaeger T, Valentini M, Hui K, Jenal U, Filloux A. 2014. The diguanylate cyclase SadC is a central player in Gac/Rsm-mediated biofilm formation in Pseudomonas aeruginosa. J Bacteriol 196:4081–4088. doi: 10.1128/JB.01850-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matz C, Moreno AM, Alhede M, Manefield M, Hauser AR, Givskov M, Kjelleberg S. 2008. Pseudomonas aeruginosa uses type III secretion system to kill biofilm-associated amoebae. ISME J 2:843–852. doi: 10.1038/ismej.2008.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fazli M, O'Connell A, Nilsson M, Niehaus K, Dow JM, Givskov M, Ryan RP, Tolker-Nielsen T. 2011. The CRP/FNR family protein Bcam1349 is a c-di-GMP effector that regulates biofilm formation in the respiratory pathogen Burkholderia cenocepacia. Mol Microbiol 82:327–341. doi: 10.1111/j.1365-2958.2011.07814.x. [DOI] [PubMed] [Google Scholar]
- 54.Leduc JL, Roberts GP. 2009. Cyclic di-GMP allosterically inhibits the CRP-like protein (Clp) of Xanthomonas axonopodis pv. citri. J Bacteriol 191:7121–7122. doi: 10.1128/JB.00845-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cordes TJ, Worzalla GA, Ginster AM, Forest KT. 2011. Crystal structure of the Pseudomonas aeruginosa virulence factor regulator. J Bacteriol 193:4069–4074. doi: 10.1128/JB.00666-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serate J, Roberts GP, Berg O, Youn H. 2011. Ligand responses of Vfr, the virulence factor regulator from Pseudomonas aeruginosa. J Bacteriol 193:4859–4868. doi: 10.1128/JB.00352-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monds RD, Newell PD, Wagner JC, Schwartzman JA, Lu W, Rabinowitz JD, O'Toole GA. 2010. Di-adenosine tetraphosphate (Ap4A) metabolism impacts biofilm formation by Pseudomonas fluorescens via modulation of c-di-GMP-dependent pathways. J Bacteriol 192:3011–3023. doi: 10.1128/JB.01571-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bennett BD, et al. 2009. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol 5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Irie Y, Parsek MR. 2014. LC/MS/MS-based quantitative assay for the secondary messenger molecule, c-di-GMP. Methods Mol Biol 1149:271–279. doi: 10.1007/978-1-4939-0473-0_22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.