

Abstract
The efficacy of two highly specific IκB-α kinase β (IKK-β) inhibitors in reducing the enhanced basal activation of the NF-κB pathway in dystrophic muscle was assessed by determining the effects of these inhibitors in increasing the expression of cytosolic IκB-α and reducing the enhanced expression of nuclear p65 in adult mdx costal diaphragm preparations. In vivo and in vitro treatment with BMS-345541 was ineffective at altering these variables when administered at concentrations that were highly effective in models of acute inflammation. PHA-408 increased cytosolic IκB-α and reduced nuclear p65 at a concentration in vitro (20 μM) that was 500 fold higher than the IC50 for inhibiting purified activity. Long term daily oral administration of PHA-408 increased cytosolic IκB-α but did not influence nuclear p65. Long term intraperitoneal administration of PHA-408 reduced nuclear p65 by approximately 50%. In comparison to their potent effects in models of acute inflammation, these results indicate a reduced efficacy of the specific IKKβ inhibitors in ameliorating the enhanced basal activation of the NF-κB pathway in dystrophic muscle, and suggest that the therapeutic potential of IKK-β inhibitors in treating muscular dystrophy would be enhanced by simultaneous treatment with agents which more directly interfere with NF-κB transactivation.
Keywords: Duchenne muscular dystrophy, mdx mouse, inflammation, NF-κB signaling, p65, IκB-α, BMS-345541, PHA-408, IκB-α kinase
Introduction
Skeletal muscle from mice lacking dystrophin (mdx mouse; [1]) and from patients with Duchenne muscular dystrophy is characterized by elevated NF-κB dependent signaling that is associated with elevations in the basal activity of the enzyme IκB-α kinase (IKK; [2,3]). IKK phosphorylates the cytosolic inhibitory protein IκB-α and initiates its ubiquitination for subsequent proteasomal degradation. In the unphosphorylated state, IκB-α bound to the NF-κB p65-p50 dimer shields a nuclear localization sequence on p65, thereby promoting the retention of the transcription factor in the cytosol. Following the proteasomal degradation of cytosolic IκB-α, the nuclear localization sequence on p65 is uncovered, and the steady state level of the NF-κB dimer in the nucleus is increased [4]. Subsequent transactivation of NF-κB dependent genes by p65 leads to increases in a large variety of cytokines, inflammatory mediators, and pro-survival proteins [5,6].
Several reports indicate that long term treatment with agents that inhibit the NF-κB pathway have beneficial effects on dystrophic (mdx) muscle structure and function. The first such agent to be examined was pyrrolidine dithiocarbamate (PDTC), an inhibitor of the ubiquitination of cytosolic IκB-α [7]. Daily treatment with PDTC improved the resting membrane potential and survival of striated muscle fibers in the highly dystrophic mdx triangularis sterni (TS) muscle [8], increased forelimb grip strength and forward pulling tension in mdx mice [9,10], reduced necrosis and increased muscle regeneration in the mdx biceps muscle [9], and increased individual fiber growth and fiber density in the mdx TS muscle [11]. Similar effects were observed following daily treatment with ursodeoxycholic acid (UDCA; [10,11]), which suppresses p65 transactivation following its interaction with the glucocorticoid receptor [12]. Neither of these agents influenced collagen expression or fibrosis in the mdx mouse [13]. Several other agents which inhibit the NF-κB pathway in various systems have also been shown to be useful in ameliorating the dystrophic phenotype in the mdx mouse model for Duchenne and Becker muscular dystrophy. These agents include the IKK inhibitory peptide (NEMO binding domain peptide; [14]) and several relatively non-specific agents which inhibit the NF-κB pathway by a variety of mechanisms [15-18].
The central role of the IKK enzyme in controlling the nuclear localization of NF-κB, along with the elevation in nuclear p65 expression and nuclear activation in several cancer cell lines [19-21], has fueled efforts to identify compounds which specifically and potently inhibit IKK in vitro and in vivo. IKK is composed of 3 subunits; IKK-α, IKK-β, and IKK-γ (NEMO). The IKK-β (IKK-2) subunit is the primary catalytically active subunit and is activated by TNF-α and IL-1β via phosphorylation at two Ser residues (Ser177 and Ser181; [22]). Although both IKK-α and IKK-β can phosphorylate IκB-α, knockout studies indicate the continued presence of TNF-α (or IL-1β) -mediated IκB-α phosphorylation in IKK-α knockouts [23] and essentially no IκB-α phosphorylation following cytokine exposure in IKK-β knockouts [24]. This primacy of the IKK-β subunit in phosphorylating IκB-α has led to the development of IKK inhibitors that are specific to blocking the catalytic activity of the IKK-β subunit.
One of the first specific IKK-β inhibitors to be developed was BMS-345541, which inhibited IκB-α phosphorylation by IKK-β in the range of 0.1 to 10 μM, and inhibited nearly 100% of lipopolysaccharide (LPS) stimulated cytokine production in THP-1 cells at 25 μM [25]. Perioral administration of BMS-345541 at concentrations of 10 to 100 mg/kg reduced by 50 to 100% the increase in serum TNF-α induced by intra-peritoneal LPS injection. A tight binding inhibitor with a 2 hour half-life of IKK-β binding, PHA-408, inhibited IκB-α phosphorylation by IKK-β at concentrations of 0.1 to 1 μM and inhibited LPS induced IκB-α phosphorylation in peripheral blood mononuclear cells (PBMCs) by approximately 100% at concentrations of 1 to 10 μM [26]. Oral administration of PHA-408 at 30 mg/kg/3 days inhibited 80% of IKK-β activity and paw swelling induced by streptococcal cell wall injection [26]. The purpose of the present studies was to determine the efficacy of BMS-345541 and PHA-408 in influencing the relevant endpoints of NF-κB signaling in adult mdx muscle; namely in increasing the basal cytosolic expression of IκB-α and in reducing the nuclear expression of p65. These endpoints represent particularly robust outcome measures for assessing the therapeutic efficacy of NF-κB inhibitors and have been used previously to identify effective inhibition of the pathway in the mdx signaling environment [2,3,8-10,14].
Significantly enhanced NF-κB signaling in the costal diaphragm is present as early as 15 days postnatal [2] and throughout adulthood in the mdx mouse [27]. Furthermore, the level of nuclear p65 activation in the adult mdx costal diaphragm is increased approximately 5 fold above the levels observed in the nondystrophic costal diaphragm, representing substantially larger proportional increases than those observed in either the mdx gastrocnemius or triangularis sterni muscles [27]. The mdx costal diaphragm muscle also exhibits a more severe dystrophic phenotype than the mdx limb musculature [13,28]. These considerations indicate that an examination of NF-κB signaling in the mdx costal diaphragm represents a particularly robust method for identifying inhibitors that could be useful in pre-clinical and clinical investigations.
Inhibitors of the NF-κB pathway have been shown in this laboratory to enhance cytosolic IκB-α and reduce nuclear p65 activation in the mdx costal diaphragm, and have positive effects on muscle structure and function following a 1 month in vivo exposure to young adult (1 month old) mdx mice [8,10,11]. These inhibitors have also been shown to be effective in older mdx mice [8,10,11] and following brief (3 hours) exposure in freshly isolated adult mdx costal diaphragms [29]. In the present experiments, we examine the efficacy of the IKK-β inhibitors under the same general conditions as those used in previous experiments; following in vivo exposure to young adult and mature mdx mice and following direct in vitro exposure to freshly isolated adult mdx costal diaphragms. The results indicate a reduced efficacy of the IKK-β inhibitors in the dystrophic signaling environment in comparison to their efficacy in models of acute inflammation [25,26]. Therefore, we believe that these agents would be most effective in dystrophic muscle when used in conjunction with additional treatments to reduce p65 expression and/or transactivation.
Materials and methods
Drug administration
The IKK-β inhibitors used in this study were administered in vivo and in vitro using protocols that were previously found effective in acute studies involving TNF-α or IL-1β activation of the NF-κB pathway [25,26]. Since dystrophic muscle exhibits elevated nuclear activation of NF-κB and increased basal activation of the NF-κB pathway [2,14,27], the inhibitors were administered directly without pretreatment with exogenous TNF-α or IL-1β, and the influence of the treatment on the nuclear expression of p65 and the cytosolic expression of IκB-α was determined. The mdx mouse model for Duchenne muscular dystrophy [1] was used exclusively in these studies. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the Kirksville College Osteopathic Medicine.
To examine the efficacy of BMS-345541, an acute in vivo protocol was initially used. BMS-345541 was administered in vivo by oral gavage at a dose of 100 mg/kg in H2O [25]. Age-matched control adult mdx mice received the water vehicle. The mice were euthanized and costal hemi-diaphragms were isolated at 7 hours after administration. A second acute in vivo protocol was used in which mdx mice received a single intraperitoneal (ip) injection at 100 mg/kg in filtered (0.2 mm) HEPES Ringer solution (147.5 mM NaCl, 5 mM KCl, 2 mM CaCl2, 11 mM glucose, 5 mM HEPES, pH 7.35). Vehicle injected (HEPES Ringer) and experimental mice were euthanized and the costal hemi-diaphragms were isolated 5 hours after the injection. To examine the efficacy of BMS-345541 in vitro, freshly isolated, adult mdx costal diaphragms were pinned and stretched to approximate resting length on Sylgard coated plastic Petri dishes. Age- and gender-matched costal hemidiaphragms were exposed to either vehicle (0.1% DMSO) or BMS-345541 in HEPES Ringer (0.1% DMSO) at concentrations of 1, 10, or 50 μM for 6 hours at room temperature. At the end of the treatment period, the costal hemidiaphragms were flash frozen for subsequent biochemical analyses.
PHA-408 was initially administered by oral gavage to mdx mice at a dose of 50 mg/kg in a vehicle of H2O containing 25% Tween 20 and 5% methyl cellulose [26]. Control mice received vehicle. This dose was used for a perod of 30 consecutive days before the mice were euthanized and costal diaphragm samples were obtained. The effect of an acute dose of 100 mg/kg by oral gavage was also examined by euthanizing the mice and obtaining costal diaphragm samples 5 hours after the treatment. In addition, the drug was administered ip at 0.8 mg/kg/day in a HEPES buffered isotonic saline (pH 7.3) to 1 month old mdx mice for 30 consecutive days (2 doses per day) with the corresponding control mice receiving vehicle injections. To examine the efficacy of PHA-408 in vitro, freshly isolated, adult, age- and gender-matched mdx costal diaphragms were pinned at approximate resting length and exposed to vehicle (0.1% DMSO) or 1, 10 or 20 μM PHA-408 in HEPES Ringer for 6 hours. At the end of the treatment, the muscles were flash frozen for subsequent analyses. As a further examination of the efficacy of PHA-408 in vitro, cultured mdx myotubes were exposed to 20 μM PHA-408 for 52 hours and the distribution of p65 in the cytosol and nucleus was determined using confocal immunofluorescence.
Cytosolic and nuclear extracts
Cytosolic and nuclear extracts were obtained as described in Singh et al [27]. Isolated costal diaphragms from vehicle and drug treated preparations were homogenized by Dounce homogenizer at 1 mg wet weight per 18 μl of low salt lysis buffer (LSLB: 10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 1.0 mg/ml benzamidine, 0.5 mM phenylmethylsulfonyl fluoride, 4.0 ml/ml protease inhibitor cocktail Sigma # 8340, 10 μl/ml phosphatase inhibitor Sigma # P2850, pH 7.9). The homogenized muscle was then maintained on dry ice for at least 10 min, and subjected to two cycles of freeze-thaw lysis using dry ice (5 minute freeze, thaw at room temperature) before centrifugation (13,000 rpm, 20 seconds, 4°C) and removal of the supernatant (cytosolic extract).
The remaining pellets were then washed twice with LSLB (50 μl) to remove any cytosolic contaminants, and re-suspended in ice-cold high-salt lysis buffer (HSLB: 20 mM HEPES (pH 7.9), 420 mM NaCl, 1 mM EDTA, 1 mM EGTA, 25% glycerol, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 1.0 mg/ml benzamidine, 4 μl/ml protease inhibitor cocktail, 10 μl/ml phosphate inhibitor; 4 ml/mg wet weight; 30 min incubation). The suspension was vigorously mixed (5 seconds, vortex) 6 times at 5 minute intervals and centrifuged (13,000 rpm, 6 min, 4°C) to obtain the nuclear extract. Both the nuclear and cytosolic extracts were stored at -80°C. Protein concentration was determined using the Bradford assay (Bio-Rad 500-0006) in a standard 96 well plate.
Western blot
Western blots were obtained as described in Singh et al [27]. Extracts (20 to 40 mg total protein) were separated by SDS PAGE under reducing conditions and transferred to a PVDF membrane (BioRad). Blots were incubated in rabbit p65 (Cell Signaling Technology (CST) #3034S), rabbit IκB-α (CST #9242), and/or rabbit phospho-IκB-α (ser 32; CST #2859) at 1:1000 or 1:500 dilution with 5% bovine serum albumin (BSA) in Tris buffered saline containing Tween 20 (TBST) overnight at 4°C. To assess loading, the blots were stripped and exposed to rabbit glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (CST #14C10; 1:20000; 1 hr, room temperature; cytosolic extracts) or rabbit Oct 1 antibody (Abcam #15112; nuclear extracts). The secondary antibody was HRP conjugated anti-rabbit IgG (Jackson Laboratory #111-035-003) administered in 5% BSA in TBST for 1 hour at room temperature. Immunoblot detection was performed by the ECL detection method (Amersham, UK).
Densitometric analyses
Image J was used to determine the density of p65 and IκB-α on Western blots relative to the corresponding density of GAPDH (cytosolic extracts; p65/GAPDH, IκB-α/GAPDH) or Oct1 (nuclear extracts; p65/Oct1). The proportion of phosphorylated IκB-α was also determined (phospho-IκB-α/IκB-α). Statistical significance (p < 0.05) was determined by one tailed t-tests using Sigmaplot (V 11, 12, 12.5) statistical software.
Primary cultures
Primary cultures of mdx (C57Bl10ScSn-mdx) myotubes were obtained using a previously described protocol [30]. Satellite cells from 14 to 17 day mdx embryos were dissociated using standard procedures in a medium containing 4 mg/ml collagenase Type A, 0.5 mg/ml trypsin, and 1 mg/ml deoxyribonuclease in a HEPES (25 mM HEPES, pH 7.3) buffered saline solution (137 mM NaCl, 0.7 mM Na2HPO4) maintained at room temperature. The cells were plated (560,000 cells/ml, 1.5 ml) in DMEM-HAMS F12 (Sigma) supplemented with 10% fetal bovine serum (Sigma), 100 units penicillin, and 100 mg streptomycin (Sigma) on laminin coated (Sigma # L2020; 2 mg/cm2) thin glass coverslips placed within 35 mm plastic Petri dishes (Fisher Scientific). The cells were maintained at 37°C in a humidified CO2 environment (NuAire Model NU-4950 incubator) and culture media was replaced every other day. At 5 to 7 days after plating, myoblasts reach confluence and fuse to form myotubes, which usually begin to twitch spontaneously at 7 days. Fibroblasts as well as myoblasts were allowed to proliferate within the laminin matrix. The laminin substrate and associated bed of fibroblasts provides a stable matrix for the twitching myotubes which have been maintained for as long as 30 days in culture [30,31]. Myotubes were exposed to 20 μM PHA-408 for 52 hours beginning at culture day 7 after plating. Vehicle-treated (0.1% DMSO) myotubes served as controls.
Immunofluorescent staining for p65
After removing the culture media, which contained 20 μM PHA-408 or vehicle (0.1% DMSO), the cells were washed 3 times in phosphate buffered saline (PBS) and subsequently fixed in 4% paraformaldehyde in PBS (15 minutes). The myotubes were then exposed to 0.1% Triton X-100 for five minutes, followed by three 5 minute washes with PBS. The samples were exposed to 1.5 ml blocking buffer (1.275 mL dd H2O, 150 uL 10X PBS, 4.5 uL Triton X-100, 75 uL goat serum; Sigma-Aldrich, G9023) for one hour. The blocking buffer was then removed and the myotubes incubated in 0.5 ml primary antibody dilution buffer containing rabbit anti-p65 (number 3987S, Cell Signaling Technology; 1:500) at 4°C overnight with gentle rocking. Control experiments were also performed in which the primary antibody was exposed to Jurkat nuclear extract (Active Motif, # 36110) prior to addition to the fixed myotubes. The cells were subsequently washed 3 times in PBS (5 minute wash). Secondary antibody dilution buffer containing goat anti-rabbit IgG conjugated with Alexa Fluor 645 (4414, Cell Signaling Technology; 1:250 dilution) was then added for one hour. After removing the secondary antibody, the dishes were washed in PBS (3 times, 5 minute wash) before being examined in PBS using a Leica DMI660B confocal microscope. Sequential scanning was used to capture both confocal and fluorescent images.
Results
In vitro exposure to IKKβ inhibitors
Adult freshly isolated costal diaphragms were exposed in vitro to various concentrations of BMS-345541. Examples of Western blots obtained from costal diaphragms exposed to vehicle (0.1% DMSO) or 50 μM BMS-345541 are shown in Figure 1A. Densitometric measurements indicated that exposure to 50 μM BMS-345541 had no effect on nuclear p65 or cytosolic IκB-α expression (Figure 1B). An examination of the effects of exposure to 1, 10, and 50 μM BMS-345541 revealed no significant effect of treatment on the expression of cytosolic IκB-α, the phosphorylation of cytosolic IκB-α, or the expression of nuclear p65 (Table 1).
Figure 1.
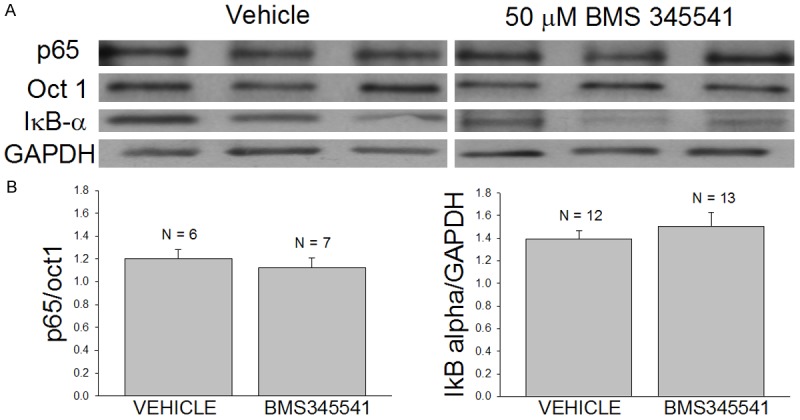
Exposure to 50 μM BMS-345541 does not influence the expression of nuclear p65 or cytosolic IκB-α in freshly isolated mdx costal diaphragms. A. Western blots of the nuclear expression of p65 and Oct 1 and the cytosolic expression of IκB-α and GAPDH. Freshly isolated mdx costal diaphragms were exposed to either vehicle (0.1% DMSO) or 50 μM BMS-345541 for 6 hours at room temperature before being flash frozen for subsequent analyses of the expression of p65 and IκB-α in nuclear and cytosolic fractions of the muscle, respectively. B. Densitometry results from nuclear (left - p65/oct1) and cytosolic fractions (right - IκB-alpha/GAPDH) obtained from the vehicle-treated and drug-treated (50 μM BMS-345541) preparations. Note that BMS-345541 produced a slight increase in cytosolic IκB-α and reduction in nuclear p65 that did not reach statistical significance.
Table 1.
The effect of BMS-345541 on the expression and phosphorylation of cytosolic IκB-α and expression of nuclear p65
| Treatment | Cytosolic IκB-α/GAPDH | Proportion of Cytosolic Phosphorylated IκB-α | Nuclear p65/Oct1 |
|---|---|---|---|
| Vehicle | 0.92 ± 0.09, N = 7 | 0.55 ± 0.07, N = 4 | 0.26 ± 0.09, N = 7 |
| 1 uM BMS345541 | 0.74 ± 0.11, N = 7 | 0.68 ± 0.06, N = 4 | 0.46 ± 0.11, N = 7 |
| Vehicle | 1.765 ± 0.47, N = 4 | 0.35 ± 0.9, N = 4 | 0.39 ± 0.05, N = 20 |
| 10 uM BMS345541 | 1.16 ± 0.20, N = 4 | 0.48 ± 0.08, N = 4 | 0.47 ± 0.08, N = 20 |
| Vehicle | 1.39 ± 0.08, N = 12 | --------- | 1.20 ± 0.08, N = 6 |
| 50 uM BMS345541 | 1.1 ± 0.12, N = 13 | --------- | 1.12 ± 0.08, N = 7 |
Shown are the means ± SEM and N values for densitometric results obtained from the appropriate cell extracts of hemidiaphragm exposed to either vehicle or the indicated concentrations of BMS-345541. N represents the number of costal hemidiaphragms used to obtain the densitometric measurements from Western blots of the appropriate protein, using GAPDH as a loading control for cytosolic extracts and Oct 1 as a loading control for nuclear extracts. Separate vehicle controls were used for each concentration of BMS-345541 analyzed. BMS-345541 did not significantly influence cytosolic IκB-α expression, the proportion of phosphorylated IκB-α in the cytosol, or the nuclear expression of p65 at any concentration (1, 10, 50 μM).
Similarly, exposure to PHA-408 at concentrations of 1 and 10 μM did not influence the cytosolic expression of IκB-α nor the nuclear expression of p65 (Figure 2A1, 2A2, 2B1, 2B2). However, exposure to the drug at 20 μM significantly increased the cytosolic expression of IκB-α (p < 0.001; Figure 2A3) and reduced the nuclear expression of p65 (p < 0.01; Figure 2B3). Consistent with this observation, cultured mdx myotubes exposed to 20 μM PHA-408 for 52 hours exhibited reduced nuclear p65 immunofluorescence compared to myotubes exposed to vehicle alone (0.1% DMSO; Figure 3).
Figure 2.
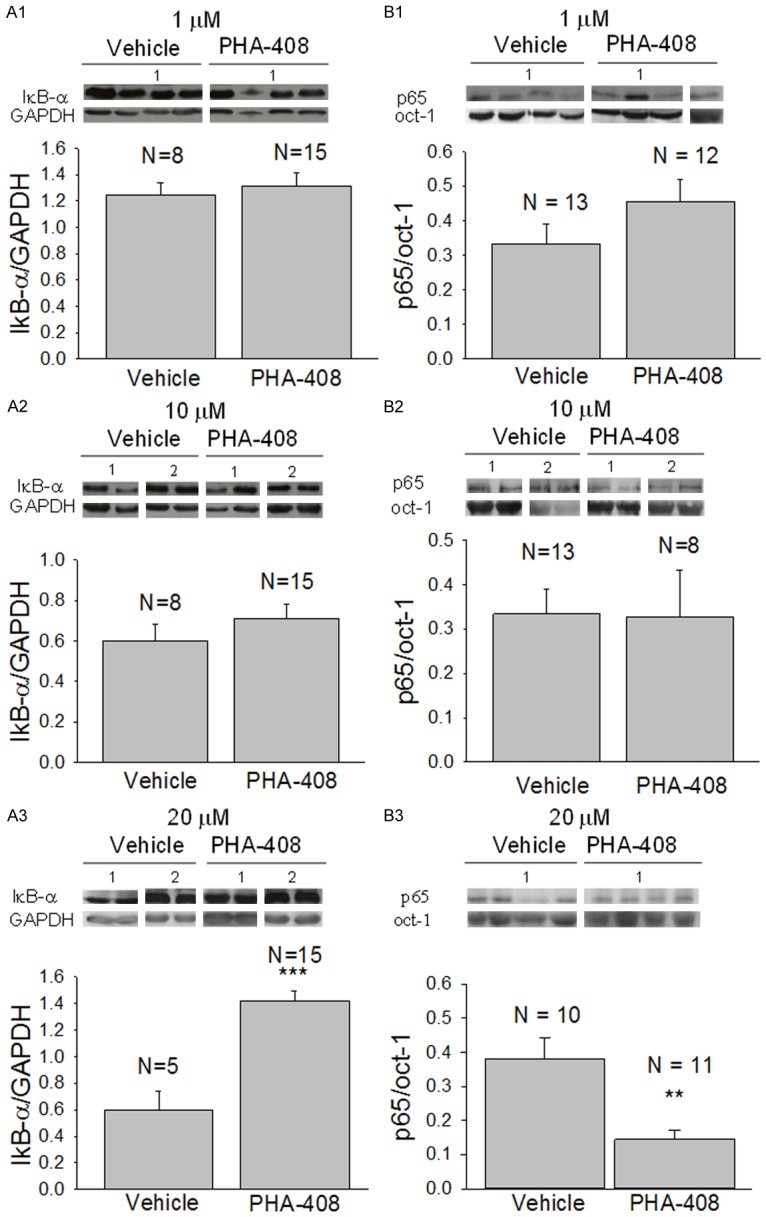
The effect of PHA-408 on the cytosolic expression of IκB-α and the nuclear expression of p65. Freshly isolated mdx costal diaphragms were exposed to either vehicle (0.1% DMSO) or PHA-408 for 6 hours at room temperature before being flash frozen for subsequent analyses of the expression of p65 and IκB-α in nuclear and cytosolic fractions of the muscle, respectively. Insets show Western blots for IκB-α, GAPDH, p65, and Oct-1. Numbers indicate corresponding blots for vehicle and treated preparations. Note that in B1 an additional blot was used to complete the analyses of p65 expression in treated preparations. Bar graphs show densitometric results (+SEM) using the appropriate loading controls. N is the number of isolated mdx costal hemi-diaphragms. ** and *** indicate p < 0.01 or 0.001, respectively. A1-A3 show the effects of a 6 hour exposure to 1, 10, or 20 μM PHA-408, respectively, on the cytosolic expression of IκB-α. B1-B3 show the corresponding effects on the nuclear levels of p65. Note that a concentration of 20 μM PHA-408 significantly increased the cytosolic expression of IκB-α (A3; p < 0.001) and significantly reduced nuclear p65 (B3; p < 0.01).
Figure 3.

The effect of exposure to 20 μM PHA-408 on the relative nuclear expression of p65 in cultured mdx myotubes. Confocal immunofluorescent images of p65 indicate the relative distribution of the transcription factor in the nuclei (arrows) and cytosol of cultured myotubes. Inset indicates magnification of each image. A1-A3 are representative images of myotubes exposed to vehicle (0.1 % DMSO). B1-B3 represent images from myotubes exposed to 20 μM PHA-408 in culture media for 52 hours. Note that the nuclei in the myotubes treated with PHA-408 are more distinct due to a substantial decrease in the relative nuclear intensity of p65 immunofluorescent staining (B1-B3).
In vivo exposure to IKKβ inhibitors
BMS-345541 or a water vehicle was administered by oral gavage at a dose of 100 mg/kg to age-matched adult mdx mice and the mice were euthanized at 7 hours after the injection. The costal diaphragms from the vehicle and drug treated mdx mice were isolated and flash frozen for subsequent biochemical analyses. Although BMS-345541 produced a 17% reduction in the nuclear expression of p65, this effect did not reach statistical significance (p = 0.24; Figure 4). Similarly, ip administration of the same dose of BMS-345541 produced a 22% reduction in nuclear p65 (5 hours post-injection) that did not reach statistical significance (p = 0.224; Figure 5).
Figure 4.
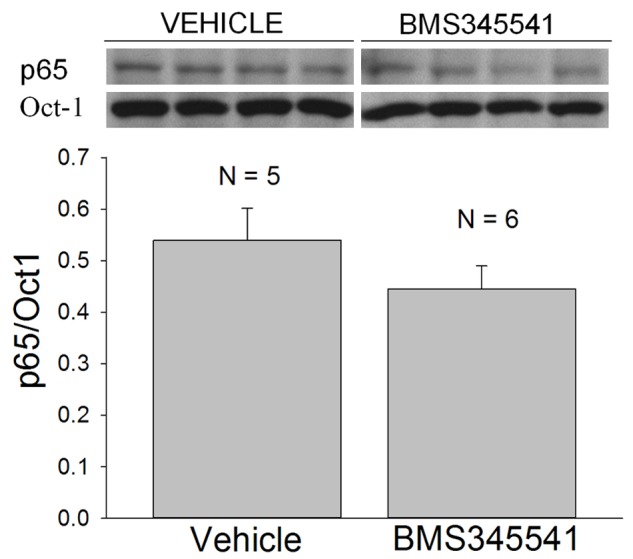
The acute effects of the oral administration of BMS-345541 to adult mdx mice. Inset shows Western blots of p65 and Oct-1, which were obtained from the nuclear extracts of mdx costal hemi-diaphragms isolated 7 hours following drug (100 mg/kg) or vehicle (water) administration. N is the number of mice and costal hemi-diaphragms. The densitometric results (bar graph) for p65/Oct-1 did not reach statistical significance.
Figure 5.
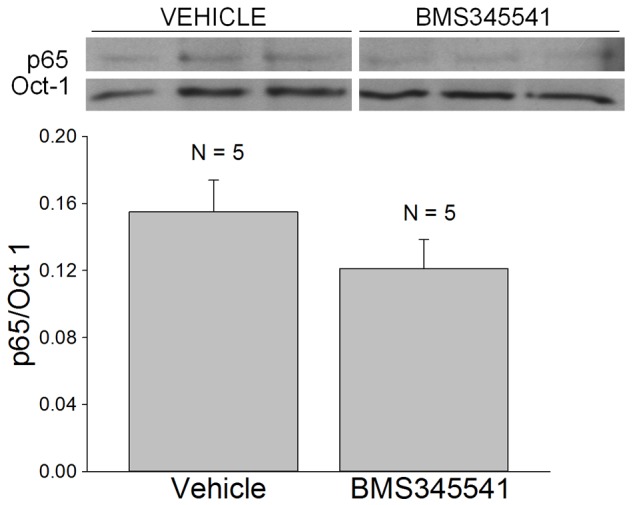
The acute effects of BMS-345541 administered intraperitoneally to adult mdx mice. Inset shows Western blots of p65 and Oct-1 which were obtained from the nuclear extracts of costal hemi-diaphragms isolated 5 hours after drug (100 mg/kg in HEPES Ringer) or vehicle (HEPES Ringer) injection. N is the number of mice and costal hemi-diaphragms. The densitometric results (p65/Oct-1) did not reveal a significant effect of drug administration on nuclear p65 expression.
PHA-408 was initially administered daily by oral gavage (50 mg/kg) for 30 consecutive days beginning at 1 month of age. Nuclear extracts from the drug-treated mdx mice showed no change in nuclear p65 expression relative to the vehicle-treated controls (p = 0.425; Figure 6A, 6B). Cytosolic extracts from the drug-treated mdx mice, however, showed a slight increase in the expression of cytosolic IκB-α which was significant (p = 0.049; Figure 6A, 6C). To further examine the potential efficacy of PHA-408 administered orally to mdx mice, the effect of a single oral administration of 100 mg/kg PHA-408 on the expression of nuclear p65 was determined. The results clearly showed no difference in the nuclear expression of p65 at 5 hours after the injection (vehicle: p65/Oct1 = 0.12 ± 0.01 SEM, N = 6; PHA-408: p65/Oct1 = 0.12 ± 0.02 SEM, N = 6).
Figure 6.
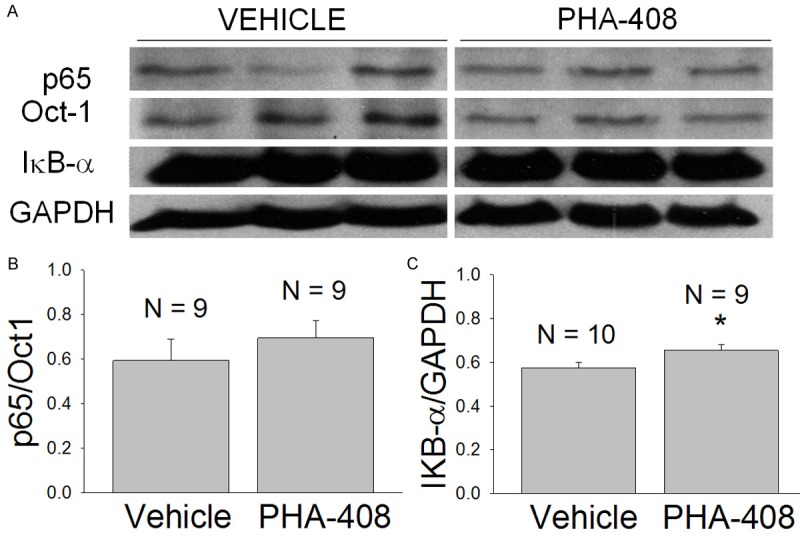
The effects of long term (30 day) oral administration of PHA-408 to young adult (1 month old) mdx mice. Daily administration of either vehicle (H2O containing 25% Tween 20 and 5% methyl cellulose) or PHA-408 (50 mg/kg in vehicle) was continued for 30 days prior to muscle isolation. N refers to the number of mice and costal hemi-diaphragms. A. Western blots for p65, Oct-1, IκB-α, and GAPDH for vehicle and drug-treated preparations. B. Densitometric measurement of nuclear p65 expression (p65/Oct-1). C. Densitometric measurement of cytosolic IκB-α expression (IκB-α/GAPDH). The 14% increase in cytosolic IκB-α expression in the drug-treated group was statistically significant (*-p < 0.05).
A second long term study examining the action of ip administration of PHA-408 at the maximum dose soluble in HEPES buffered isotonic saline (pH 7.3) was therefore performed to determine the relative efficacy of parenteral administration of the drug. PHA-408 was administered daily at a dose of 0.8 mg/kg (30 days) beginning at 1 month of age by performing two ip injections spaced at approximately 12 hours. This procedure would maintain a relatively constant, low dose parenteral exposure to the drug. The results indicated that the drug-treated mdx mice did exhibit a significant (p < 0.05) reduction in nuclear p65 expression in comparison to the vehicle injected mice (Figure 7). These results indicate that administration of PHA-408 did effectively reduce the basal activation of the NF-κB pathway in adult mdx muscle, both in vitro (Figures 2, 3) and following prolonged ip administration (Figure 7).
Figure 7.
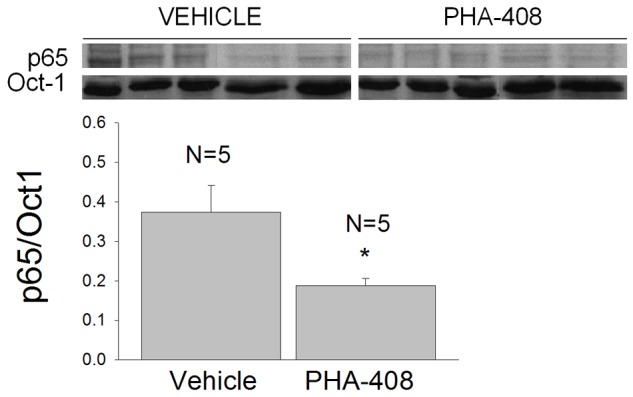
Long term intraperitoneal administration of PHA-408 reduces the nuclear expression of p65 in mdx costal diaphragm. PHA-408 (0.8 mg/kg) or vehicle (HEPES buffered isotonic saline) was administered daily to 1 month old mdx mice for a period of 30 days. Inset shows Western blots obtained from costal hemi-diaphragms of vehicle and drug-treated mice following the treatment period. N refers to the number of mice and costal hemi-diaphragms. Densitometric analyses (bar graph; p65/0ct-1) revealed a significant (*-p < 0.05) 50% reduction in nuclear p65 expression in the mice treated daily with PHA-408.
Discussion
The purpose of these studies was to assess the efficacy of two highly specific and potent IKKβ inhibitors in inhibiting NF-κB signaling in the mdx mouse, a model for Duchenne muscular dystrophy [1]. An important difference between the present study and previous investigations establishing the efficacy of these IKKβ inhibitors [25,26] is that, in the present study, the effect of the inhibitors was examined in a disease characterized by chronic over-activation [2,14] and up-regulation of many of the signaling mediators of the NF-κB pathway [27]. Previous studies were conducted using models of acute inflammation induced by treatment with LPS or streptococcal cell wall [25,26].
The results show that BMS-345541 at concentrations of 1 to 50 μM was ineffective at either increasing cytosolic IκB-α or reducing nuclear p65 expression (Table 1). Acute in vivo exposure to BMS-345541 at 100 mg/kg by either oral gavage or intraperitoneal injection was also ineffective at significantly reducing p65 expression (Figures 4, 5), although each of these routes of administration produced a 17 to 22% reduction in nuclear p65 levels that failed to reach significance. It should be emphasized that an oral dose of 100 mg/kg BMS-345541 is over 3 times the dose needed to inhibit 90% of the positive TNF-α response to LPS administration [25].
The more potent IKKβ inhibitor, PHA-408, was effective at reducing nuclear p65 and increasing cytosolic IκB-α (Figures 2, 3) in isolated mdx costal diaphragms at a concentration that was approximately 500 times larger than the IC50 for purified IKKβ activity, and approximately 20 times larger than the concentration required for inhibiting greater than 95% of IKKβ activity in other cellular systems [26]. A single oral administration of PHA-408 was ineffective at reducing nuclear p65 expression at a dose which was 3 times larger than a chronically administered dose which reduced 80% of the IKKβ activity following injection of streptococcal cell wall (30 mg/kg; [26]). However, long term oral administration of PHA-408 at that approximate dose (50 mg/kg) [26] did slightly increase cytosolic IκB-α, but failed to influence the nuclear expression of p65 (Figure 6). Long term parenteral exposure to a low dose administered twice daily was effective at producing a 50% reduction in nuclear p65 expression (Figure 7). These results suggest that a period of prolonged treatment with highly potent IKKβ inhibitors in vivo may be required before achieving a reduction in nuclear p65 expression in the dystrophic signaling environment.
In comparison to the use of these specific IKKβ inhibitors in models of acute inflammation [25,26], the present results clearly show a reduced efficacy in decreasing basal NF-κB signaling in dystrophic muscle. This result is consistent with our earlier demonstration that several critical mediators of the NF-κB signaling pathway are upregulated in freshly isolated adult mdx muscle [27]. In particular, there was a significant 6 fold elevation in IKKβ phosphorylation and a significant doubling of IKKβ expression at 5 months of age [27]. Furthermore, steady state cytosolic and whole cell expression of IκB-α was increased and associated with rate limited proteasomal degradation of phosphorylated IκB-α [27]. This signaling environment was maintained by steady state increases in whole cell and cytosolic p65 that would limit the therapeutic efficacy of agents designed to reduce the proportion of whole cell p65 residing within the nucleus [27]. These considerations and the present results suggest that agents that block NF-κB signaling by reducing p65 expression or directly inhibiting nuclear transactivation may be more immediately effective than upstream NF-κB inhibitors which increase cytosolic IκB-α. Combined use of such agents with specific and potent IKKβ inhibitors, however, may ultimately provide an optimal combination therapy for improving the phenotype of patients with Duchenne or Becker muscular dystrophy [8,9,14].
Acknowledgements
This work was supported by grants to CGC from the Association Francaise contre les Myopathies (AFM #13980) and the NIH (1R15AR055360-01A2). The authors gratefully acknowledge the support and assistance of the AT Still Research Institute and the KCOM Physiology and technical staff, especially Mrs. Bonnie King, Mrs. Janet Dabney, Mrs. RaElla Wiggins, and Mr. Alan Coonfield. This research was conducted in accordance with Material Transfer Agreements between AT Still University and Bristol Myers Squibb and AT Still University and Pfizer Pharmaceuticals. The authors are grateful to Bristol Myers Squibb and Pfizer for the opportunity to investigate the compounds cited in this study.
Disclosure of conflict of interest
None.
References
- 1.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar A, Boriek AM. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 2003;17:386–396. doi: 10.1096/fj.02-0542com. [DOI] [PubMed] [Google Scholar]
- 3.Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G. Activation of nuclear factor-kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology. 2003;60:993–997. doi: 10.1212/01.wnl.0000049913.27181.51. [DOI] [PubMed] [Google Scholar]
- 4.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 6.Barnes PJ. Nuclear factor-kappa B. Int J Biochem Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- 7.Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlson CG, Samadi A, Siegel A. Chronic treatment with agents that stabilize cytosolic IkappaB-alpha enhances survival and improves resting membrane potential in MDX muscle fibers subjected to chronic passive stretch. Neurobiol Dis. 2005;20:719–730. doi: 10.1016/j.nbd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Messina S, Bitto A, Aguennouz M, Minutoli L, Monici MC, Altavilla D, Squadrito F, Vita G. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp Neurol. 2006;198:234–241. doi: 10.1016/j.expneurol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 10.Siegel AL, Bledsoe C, Lavin J, Gatti F, Berge J, Millman G, Turin E, Winders WT, Rutter J, Palmeiri B, Carlson CG. Treatment with inhibitors of the NF-kappaB pathway improves whole body tension development in the mdx mouse. Neuromuscul Disord. 2009;19:131–139. doi: 10.1016/j.nmd.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Siegel AS, Henley S, Zimmerman A, Miles M, Plummer R, Kurz J, Balch F, Rhodes JA, Shinn GL, Carlson CG. The influence of passive stretch and NF-kappaB inhibitors on the morphology of dystrophic muscle fibers. Anat Rec (Hoboken) 2011;294:132–144. doi: 10.1002/ar.21294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miura T, Ouchida R, Yoshikawa N, Okamoto K, Makino Y, Nakamura T, Morimoto C, Makino I, Tanaka H. Functional modulation of the glucocorticoid receptor and suppression of NF-kappaB-dependent transcription by ursodeoxycholic acid. J Biol Chem. 2001;276:47371–47378. doi: 10.1074/jbc.M107098200. [DOI] [PubMed] [Google Scholar]
- 13.Graham KM, Singh R, Millman G, Malnassy G, Gatti F, Bruemmer K, Stefanski C, Curtis H, Sesti J, Carlson CG. Excessive collagen accumulation in dystrophic (mdx) respiratory musculature is independent of enhanced activation of the NF-kappaB pathway. J Neurol Sci. 2010;294:43–50. doi: 10.1016/j.jns.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buetler TM, Renard M, Offord EA, Schneider H, Ruegg UT. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr. 2002;75:749–753. doi: 10.1093/ajcn/75.4.749. [DOI] [PubMed] [Google Scholar]
- 16.Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol. 2008;586:2003–2014. doi: 10.1113/jphysiol.2007.148338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Assereto S, Stringara S, Sotgia F, Bonuccelli G, Broccolini A, Pedemonte M, Traverso M, Biancheri R, Zara F, Bruno C, Lisanti MP, Minetti C. Pharmacological rescue of the dystrophin-glycoprotein complex in Duchenne and Becker skeletal muscle explants by proteasome inhibitor treatment. Am J Physiol Cell Physiol. 2006;290:C577–582. doi: 10.1152/ajpcell.00434.2005. [DOI] [PubMed] [Google Scholar]
- 18.Hnia K, Hugon G, Rivier F, Masmoudi A, Mercier J, Mornet D. Modulation of p38 mitogen-activated protein kinase cascade and metalloproteinase activity in diaphragm muscle in response to free radical scavenger administration in dystrophin-deficient Mdx mice. Am J Pathol. 2007;170:633–643. doi: 10.2353/ajpath.2007.060344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visconti R, Cerutti J, Battista S, Fedele M, Trapasso F, Zeki K, Miano MP, de Nigris F, Casalino L, Curcio F, Santoro M, Fusco A. Expression of the neoplastic phenotype by human thyroid carcinoma cell lines requires NFkappaB p65 protein expression. Oncogene. 1997;15:1987–1994. doi: 10.1038/sj.onc.1201373. [DOI] [PubMed] [Google Scholar]
- 20.Balcerczak M, Balcerczak E, Pasz-Walczak G, Kordek R, Mirowski M. Expression of the p65 gene in patients with colorectal cancer: comparison with some histological typing, grading and clinical staging. Eur J Surg Oncol. 2004;30:266–270. doi: 10.1016/j.ejso.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Weichert W, Boehm M, Gekeler V, Bahra M, Langrehr J, Neuhaus P, Denkert C, Imre G, Weller C, Hofmann HP, Niesporek S, Jacob J, Dietel M, Scheidereit C, Kristiansen G. High expression of RelA/p65 is associated with activation of nuclear factor-kappaB-dependent signaling in pancreatic cancer and marks a patient population with poor prognosis. Br J Cancer. 2007;97:523–530. doi: 10.1038/sj.bjc.6603878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 23.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 24.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi FC. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 26.Mbalaviele G, Sommers CD, Bonar SL, Mathialagan S, Schindler JF, Guzova JA, Shaffer AF, Melton MA, Christine LJ, Tripp CS, Chiang PC, Thompson DC, Hu Y, Kishore N. A novel, highly selective, tight binding IkappaB kinase-2 (IKK-2) inhibitor: a tool to correlate IKK-2 activity to the fate and functions of the components of the nuclear factor-kappaB pathway in arthritis-relevant cells and animal models. J Pharmacol Exp Ther. 2009;329:14–25. doi: 10.1124/jpet.108.143800. [DOI] [PubMed] [Google Scholar]
- 27.Singh R, Millman G, Turin E, Polisiakeiwicz L, Lee B, Gatti F, Berge J, Smith E, Rutter J, Sumski C, Winders WT, Samadi A, Carlson CG. Increases in nuclear p65 activation in dystrophic skeletal muscle are secondary to increases in the cellular expression of p65 and are not solely produced by increases in IkappaB-alpha kinase activity. J Neurol Sci. 2009;285:159–171. doi: 10.1016/j.jns.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 28.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 29.Miles MT, Cottey E, Cottey A, Stefanski C, Carlson CG. Reduced resting potentials in dystrophic (mdx) muscle fibers are secondary to NF-kappaB-dependent negative modulation of ouabain sensitive Na+-K+ pump activity. J Neurol Sci. 2011;303:53–60. doi: 10.1016/j.jns.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlson CG, Feng Y. Asynaptic expression of the adult nicotinic acetylcholine receptor in long-term cultures of mammalian myotubes. Brain Res Dev Brain Res. 1993;72:245–252. doi: 10.1016/0165-3806(93)90189-h. [DOI] [PubMed] [Google Scholar]
- 31.Carlson CG, Adkins SD, Blake MJ, Hasan AK, Loyland S. Differential influence of electrical blocking agents on embryonic acetylcholine receptor mRNA levels in long-term cultures of aneural mammalian myotubes. Synapse. 1994;18:281–287. doi: 10.1002/syn.890180402. [DOI] [PubMed] [Google Scholar]