

Abstract
Autophagy is ubiquitous in all forms of heart failure and cardioprotective miR-133a is attenuated in human heart failure. Previous reports from heart failure patients undergoing left ventricular assist device (LVAD) implantation demonstrated that autophagy is upregulated in the LV of the failing human heart. Studies in the murine model show that diabetes downregulates miR-133a. However, the role of miR-133a in the regulation of autophagy in diabetic hearts is unclear. We tested the hypothesis that diabetes exacerbates cardiac autophagy by inhibiting miR-133a in heart failure patients undergoing LVAD implantation. The miRNA assay was performed on the LV of 15 diabetic (D) and 6 non-diabetic (ND) heart failure patients undergoing LVAD implantation. Four ND with highly upregulated and 5 D with highly downregulated miR-133a were analyzed for autophagy markers (Beclin1, LC3B, ATG3) and their upstream regulators (mTOR and AMPK), and hypertrophy marker (beta-myosin heavy chain) by RT-qPCR, Western blotting and immunofluorescence. Our results demonstrate that attenuation of miR-133a in diabetic hearts is associated with the induction of autophagy and hypertrophy, and suppression of mTOR without appreciable difference in AMPK activity. In conclusion, attenuation of miR-133a contributes to the exacerbation of diabetes mediated cardiac autophagy and hypertrophy in heart failure patients undergoing LVAD implantation.
Keywords: Hypertrophy, LC3B, Beclin1, mTOR, AMPK, LVAD, Milrinone
Introduction
The incidence of diabetes is rapidly increasing [1,2] and accounts for ~30% of heart failure patients [3]. It exacerbates the risk of heart failure if accompanied by other heart disease [3-5]. As per The National Diabetes Statistics Report released on June 10, 2014 (American Diabetes Association), 29.1 million people (9.3% of population) were reported to have diabetes in United States, which accounts for $245 million of health care costs per year [6]. This number was dramatically increased from 25.8 million (8.3% of population) in 2010 suggesting that there is a dire need to elucidate the mechanism(s) of diabetes mediated cardiac complications. Diabetic cardiomyopathy is independent of coronary artery disease or valve defect in the heart [7], and is associated with the activation of myocardial autophagy [8]. Autophagy is a lysosome mediated catabolic process, where aged and damaged cytoplasmic organelles, and defective proteins are sequestered, degraded, and recycled for ATP generation [9]. It is initiated with the induction of Beclin1 that forms a double membrane crescent shape structure called the autophagophore, which with the help of microtubule-associated protein 1 light chain 3 B (LC3B) and autophagy related 3 (ATG3) forms a double membrane vesicle called the autophagosome. The autophagosome fuses with the lysosome to form an autolysosome, where cytoplasmic components are degraded by lysosomal enzymes [9,10]. An elevated level of cardiac autophagy is common in heart failure both in murine models [11-13] and human patients [14-16]. It is documented that in human heart failure patients undergoing mechanical unloading with a left ventricular assist device (LVAD), autophagy markers are upregulated [15]. However, the contribution of diabetes on the level of cardiac autophagy in these patients is unclear.
Recently, it is demonstrated that differential regulation of microRNAs (miRNAs) in end-stage failing hearts is associated with LVAD unloading [17]. MiRNAs are a newly emerging class of endogenous, evolutionary conserved, tiny, non-coding regulatory RNAs that modulate gene expression by either mRNA degradation (if seed sequences of the miRNA perfectly match with the mRNA) or translational repression [18-20]. MiRNAs have emerged as a therapeutic target for cardiovascular diseases [19,21]. There are more than 800 miRNAs in the heart and several dozens of them are differentially expressed in the failing heart [22]. However, miR-133a is the most abundantly expressed miRNA in the human myocardium [21]. MiR-133a is a muscle specific miRNA, which is expressed both in cardiac and skeletal muscle. There are two alleles of miR-133a, namely miR-133a-1 and miR-133a-2. Both are exactly identical in their sequences but are located on chromosome 18 and chromosome 2, respectively. MiR-133a differs from miR-133b (an isotype) by two nucleotides and the expression of the latter in only skeletal muscle [23,24].
Diabetes has been shown to be a miRNA associated disease [23] with the majority of the miRNAs being attenuated in the diabetic heart [25], and miR-133a is one of them [25-27]. It is a cardioprotective miRNA that prevents pathological remodeling by inhibiting cardiac hypertrophy [28] and fibrosis [29,30]. It also regulates beta-adrenergic activity in the heart by regulating several genes in the beta-adrenergic signaling cascade [31]. Although several miRNAs are implicated in the regulation of autophagy [11,32-34], the role of miR-133a in the regulation of cardiac autophagy is unclear. Therefore, the goal of this study was to determine whether: 1) Diabetes exacerbates cardiac autophagy in heart failure patients undergoing LVAD implantation; 2) Cardiac levels of miR-133a are associated with the levels of autophagy in diabetic heart failure patients; and 3) Diabetes augments cardiac hypertrophy in failing human hearts.
Methods
Patient population and tissue procurement
Tissue collection protocols, procedures and archival in the Nebraska Cardiovascular BioBank and Registry were duly approved by the Institutional Review Board (IRB) of The University of Nebraska Medical Center under strict ethical guidelines. All studies conducted on patient samples followed the declaration of Helsinki. Thoroughly informed written consent for the collection and use of tissue samples was obtained from individual patients prior to donation. The LV apex tissue from patients who underwent implantation were collected and flash frozen in liquid nitrogen until use. Patient’s samples were then grouped into diabetic and non-diabetic. Detailed patient demographics are presented in Table 1. LV apex tissue samples from 23 heart failure patients (20 male, 3 female) in a cohort who presented with class IV New York Heart failure were also assessed.
Table 1.
The demographics of diabetic and non-diabetic patients used for association of attenuation of miR-133a with cardiac autophagy
| Patients | Diabetic | Non-diabetic |
|---|---|---|
| Number | 5 | 4 |
| Age | 55.0 ± 8.34 | 60.75 ± 3.88 |
| Male | 5 | 3 |
| Female | 0 | 1 |
| BMI | 29.0 ± 1.49 | 33.4 ± 5.22 |
| LDL | 72.25 ± 6.32 | 65.75 ± 2.59 |
| HDL | 27.75 ± 2.59 | 40.0 ± 7.61 |
| Triglycerides | 95.25 ± 29.71 | 75.25 ± 16.83 |
| Glucose | 130.6 ± 11.64 | 108.0 ± 6.82 |
| Hemoglobin A1C | 6.76 ± 0.09 | 6.33 ± 0.09 |
| LV Mass | 170.4 ± 12.73 | 185.0 ± 25.10 |
| CHF Peptide | 669.25 ± 170.45 | 914.25 ± 378.21 |
| Diastolic dysfunction | 80% | 50% |
| % EF Average | 12-17 | 13.75-18.75 |
| History of tobacco | 75% | 80% |
There are no significant relationships between any pairs of variables (p > 0.05). Diastolic dysfunction was present to a greater extent in diabetic patients. H11, H12, H23 and H25 were not prescribed statins, whereas H13 and H23 were non-smokers. BMI = body mass index, LDL = low density lipid, HDL = high density lipid, LV = left ventricle, CHF = chronic heart failure, EF = ejection fraction.
RNA isolation and RT-qPCR assay
The total RNA was isolated by standard Trizol (Life Technology, USA) method. High quality RNA (A260/280 > 1.8 and 260/230 > 1.8) was used for cDNA preparation using iScript™ Reverse Transcription Supermix (Bio-Rad Laboratories, USA). The cDNA was amplified by thermal cycler (Bio-Rad Laboratories, USA) with an initial denaturation at 95°C for 5 minutes, and then 35 cycles of denaturation at 95°C for 30 seconds, annealing at 55°C for 45 seconds, extension at 72°C for 45°C seconds, and final extension at 72°C for 5 minutes. The 18S oligonucleotide primer was used as endogenous control. The primer sequences from 5’-3’ are as follows: Beclin1, Forward- GCTCCATTACTTACCACAGC, and Reverse- CCCAGTGACCTTCAGTCTT; LCB, Forward- GATACAAGGGTGAGAAGCAG, and Reverse- GAGATTGGTGTGGAGACG; 18S, Forward- GTAGTTCCGACCATAAACGAT, and Reverse- GCTATCAATCTGTCAATCCTG.
MiRNA assay
For the miRNA-133a assay, RNA was isolated from LV apex tissue using the mirVana™ miRNA Isolation Kit (Life Technology, USA). The RNA quality and quantity was determined by NanoDrop 2000 c (Thermo Scientific Inc., USA) and RNAs with 260/280 ~2 and 260/230 ~2 were used for miR-133a assay. U6 SnRNA was used as endogenous control for miR-133a. The RT-qPCR was performed using Taqman primers specific for miR-133a (Assay ID-002246) and U6 SnRNA (assay ID: 001973) (Life Technology, USA). The thermal cycle was initial denaturation at 95°C for 15 minutes, and then 40 cycles of denaturation at 95°C for 15 seconds, annealing and extension at 60°C for 60 seconds. The Bio-Rad CFX qPCR instrument was used for RT-qPCR and the data was analyzed by BioRad CFX Manager 3.0 software (Bio-Rad Laboratories, USA).
Protein isolation and Western blot analysis
Protein was extracted from whole snap frozen LV tissues. Tissues were crushed in RIPA buffer (Boston BioProducts, USA) in liquid nitrogen using mortal and pastel, transferred into an Eppendorf tube, centrifuged at 3,000 xg, and supernatant collected. Proteins were quantified by Pierce™ BCA Protein Assay Kit (Pierce Biotechnology, USA). Western blotting was performed following standard protocol. Briefly, equal amounts (40 μg) of each whole protein lysate were loaded on a SDS-PAGE gel. After gel electrophoresis, they were transferred onto nitrocellulose membrane and incubated with 5% non-fat dried milk in TBS for 1 hr at room temperature. The membrane is incubated with primary antibodies (dilution; 1:1000) for LC3B (cat #2775), and Beclin-1 (cat #3783) from Cell Signaling Technology, USA, and β-actin (cat #Sc-47778, Santa Cruz Biotechnology, USA) for overnight at 4°C with constant rocking. HRP conjugated secondary antibodies were diluted 1:4000 and incubated at room temperature for 2 h. The membranes were developed using Clarity Western ECL substrate (cat #170-5061, Bio-Rad Laboratories, USA) using a Molecular Imager Chemi-DocTM XRS version 3.0 (Bio-Rad Laboratories, USA). The relative band intensity was quantified using the Bio-Rad Image lab software.
Histology and immunohistochemistry
Tissue samples were embedded in OCT compound and 5 μM cryosections were obtained using the CryoStar NX50 (Thermo Scientific, USA). Immunohistochemistry was performed for β-MHC (Cat #172967, dilution; 1:500, Abcam) using standard protocol. In brief, sections were fixed in ice cold methanol for 10 minutes at -20°C and rehydrated in PBS. Then they were blocked in 1% BSA in TBS followed by washing and incubation with the primary antibody. Anti-rabbit Alexa Fluor488 secondary (Cat #A21441, Life Tech, USA) was used at a 1:1000 dilution. Sections were mounted by Fluoromount-G mounting medium (Cat# 0100-01, Southern Biotech, USA) and covered under coverslip. Images were captured by EVOS Cell Imaging Systems (Life Technology, USA).
WGA staining
The cryosections were hydrated by dipping in 1xPBS. The sections were fixed in 4% paraformaldehyde for 40 minutes and washed with PBS. A concentration of 5 μg/mL WGA solution was used to stain the cell boundaries. Sections were counter stained with DAPI for nuclear staining and mounted by Fluoromount-G. Stained sections were observed under fluorescent microscope (EVOS, Life Technology, USA) and the number of the cardiomyocytes per unit area was counted using EVOS Imaging System.
Statistical analyses
Student t-test analyses are performed for mRNA, protein and cellular expression of miR-133a, Beclin1, LC3B, and β-MHC; and number of the cardiomyocytes per unit area, LV mass, and percentage ejection fraction. Pearson correlation test was used to evaluate the correlation between the different groups. The values are expressed as the mean ± S.E, and P < 0.05 is considered statistically significant.
Results
Cardiac miR-133a is differentially expressed in the LV of diabetic and non-diabetic heart failure patients
To determine the association of miR-133a with cardiac autophagy, we first measured the levels of miR-133a in all the heart failure samples. It is obvious that miR-133a did not show a clear pattern and is differentially regulated in both diabetic and non-diabetic failing hearts (Figure 1A, 1B). This differential expression may be attributed variations in the physiological conditions as reflected in the patient demographics shown in Table 1. It is germane to mention here that the up-, and down-regulation of miR-133a is represented as the fold change with respect to a diabetic heart failure patient denoted by the coded symbol H8. It is demonstrated that miR-133a is downregulated in failing human hearts as compared to healthy hearts [28]. Since miR-133a is attenuated in diabetic hearts [26,27], we selectively analyzed the failing hearts from four ND and five D patients, where miR-133a is comparatively up-, and down-regulated, respectively (Figure 1Ci). The significant downregulation of miR-133a is found in the selected diabetic patients (Figure 1Cii).
Figure 1.

Expression of miR-133a in the left ventricle of non-diabetic (ND) and diabetic (D) heart failure patients. A. Expression of miR-133a in the diabetic and non-diabetic coded heart failure patients represented by colors (red is downregulated and green is upregulated). B. RT-qPCR of miR-133a normalized to U6 showing the differential expression of miR-133a between and amongst ND and D hearts. n = 15 for D and 6 for D. Ci. Five D (H2, H5, H7, H23, H25) and four ND (H11, H12, H13 and H15) hheart failure patients were selected for investigating the specific role of miR-133a in the regulation of autophagy and hypertrophy in diabetic hearts. The values are represented as the mean ± S.E. and expressed as fold change over H11. Cii. The average expression of miR-133a in the D and ND samples from Ci. The values are represented as mean ± S.E. and expressed as fold change over ND. D. The average percentage ejection fraction of the four ND and five D patients from Ci. The values were mean ± S.E. E. The average left ventricle (LV) mass of the four ND and five D patients from Ci. The values are mean ± S.E.
Both D and ND patients are at end-stage heart failure as they underwent LVAD. Therefore, no significant difference was expected in their cardiac dysfunction. The percentage ejection fraction and LV mass were compared between four ND and five D heart failure patients and they were very similar in both groups (Figure 1D and 1E).
Cardiac autophagy markers are upregulated in diabetic heart failure patients
To determine the specific association of miR-133a with cardiac autophagy, we measured the levels of cardiac autophagy markers, Beclin1 and LC3B. The analyses of mRNA levels revealed variations within the D and ND groups. However, cumulative data showed a trend of upregulation of Beclin1 in D as compared to the ND (Figure 2Ai, 2Aii). Similarly, Western blot results showed a trend of upregulation of Beclin1 protein in diabetic failing hearts (Figure 2Bi, 2Bii). The cellular levels of Beclin1 correlated with the protein expression (Figure 2C). The augmented expression of Beclin1 in the D group, where miR-133a was attenuated, suggests that attenuation of miR-133a exacerbates Beclin1 in diabetic heart failure patients.
Figure 2.
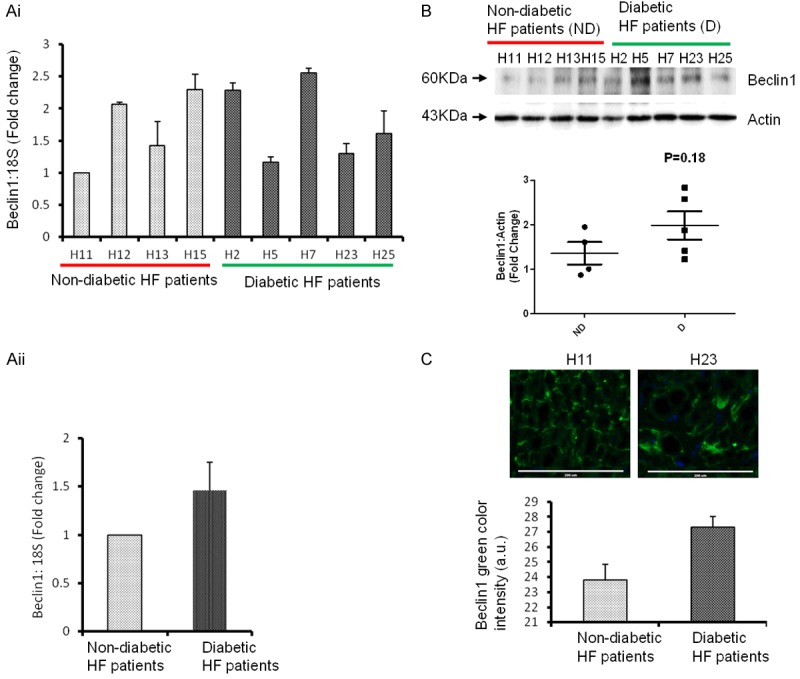
Cardiac level of Beclin1 is upregulated with attenuation of miR-133a in diabetic heart failure patients. Ai. RT-qPCR analysis of left ventricle (LV) of the non-diabetics, ND (H11, H12, H13 and H15) and diabetics, D (H2, H5, H7, H23, H25) heart failure patients. The values are presented as the mean ± SE and is represented as fold change over H11. Aii. The average level of Beclin1 in the four ND and five D patients from Ai. The values are mean ± SE and is represented as fold change over ND. B. Western blot analysis of Beclin1 from LV of above non-diabetics and diabetic heart failure patients. Top, representative bands of Beclin1 and actin (loading control). Bottom, the densitometric analyses of bands to show the relative Beclin1 expression in diabetic (D) and non-diabetic (ND) patients. The black horizontal bar represents the mean of all samples from ND and D groups. C. Top, representative immunohistochemistry of Beclin1 from D (H11) and ND (H23) patients. Bottom, bar graph of arbitrary Beclin1 pixel intensity from samples in Ai. Scale bar: 200 μm.
We also measured the level of LC3B as another marker of autophagy. Interestingly, the mRNA, protein and cellular expression of LC3B is similar to that of Beclin1 in the D group (Figure 3Ai-C). The level of ATG3, another marker of autophagy was significantly upregulated in the D group (Figure 3Di, 3Dii), suggesting that the inhibition of miR-133a contributes to upregulation of autophagy markers in the LV of diabetic heart failure patients. The correlationship of miR-133a with Beclin1 and LC3B mRNA and protein levels was also evaluated. There was a negative correlation of miR-133a with both Beclin1 and LC3B (statistically not significant), whereas Beclin1 was positively correlated with LC3B (statistically significant at the mRNA level) suggesting that inhibition of miR-133a contributes to the upregulation of autophagy markers in diabetic heart failure in humans (Tables 2 and 3). There was also a positive correlation between LC3B and AMPK proteins (statistically significant) suggesting that AMPK is involved in the induction of autophagy in the diabetic heart failure patients, when miR-133a is attenuated (Table 3). We also analyzed the correlation of the above molecules with the LV mass, diastolic dysfunction, CHF peptide, Milrinone, blood glucose level, hemoglobin A1C, mTOR, and AMPK. However, the majority of these parameters did not achieve statistical significance difference (Tables 2 and 3) suggesting that at end stage heart failure, both diabetic and non-diabetic patients behave very similarly at the molecular level, and there is no dramatic change between the groups. This is also attributed to the variability between the patient’s groups (Table 1).
Figure 3.
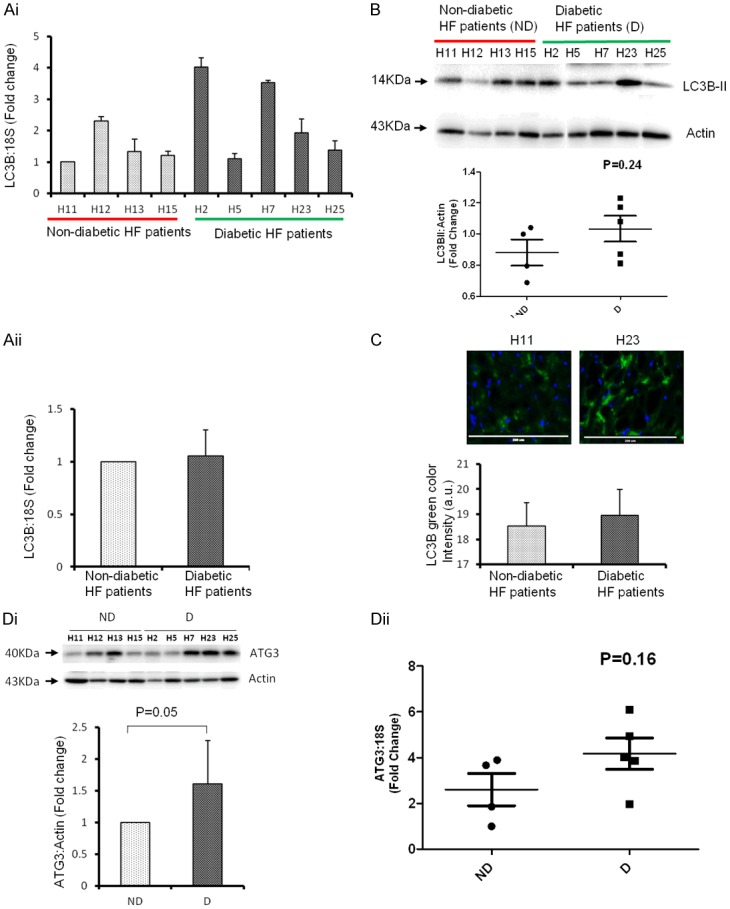
Cardiac level of LC3B is upregulated with attenuation of miR-133a in diabetic heart failure patients. Ai. RT-qPCR analysis of left ventricle (LV) of non-diabetic, ND (H11, H12, H13 and H15) and diabetic, D (H2, H5, H7, H23, H25) heart failure patients. The bar graph represents fold change over H11 sample. The values are mean ± SE. Aii. The average level of LC3B in the four ND and five D patients from Ai. The values are mean ± SE and is represented as fold change over ND. B. Western blot analysis of LC3B from LV of above ND and D heart failure patients. Top, representative bands of LC3B-II and actin (loading control). Bottom, the densitometric analyses of bands to show the relative LC3B expression in D and ND samples. The black horizontal bar represents the mean of all samples from ND and D groups. C. Top, representative immunohistochemistry of LC3B from D (H11) and ND (H23) patients. Bottom, bar graph of arbitrary LC3B pixel intensity from samples in Ai. Scale bar: 200 μm. Di. Western blot analysis of ATG3 from LV of above ND and D heart failure patients. Top, representative bands of ATG3 and actin (loading control). Bottom, the densitometric analyses of bands to show the relative ATG3 expression in D and ND samples. The values are mean ± SE and is represented as fold change over ND. Dii. RT-qPCR analyses of ATG3 in the same samples mentioned in Di and represented as relative value of 18s. The black horizontal bar represents the mean of all samples from ND and D groups.
Table 2.
Correlationship of miR-133a with mRNA of different genes in diabetic versus non-diabetic hearts failure patients
| LC3B | Beclin1 | LV mass | DF | CHF peptide | Milrinone | Glucose | HgbA1C | |
|---|---|---|---|---|---|---|---|---|
| miR-133a | ||||||||
| CC | -0.502 | -0.109 | 0.276 | -0.127 | 0.189 | 0.040 | -0.223 | -0.361 |
| P-value | 0.168 | 0.780 | 0.472 | 0.745 | 0.653 | 0.918 | 0.565 | 0.339 |
| LC3B | ||||||||
| CC | 0.715 | 0.063 | 0.111 | -0.079 | -0.545 | -0.046 | 0.287 | |
| P-value | 0.030 | 0.872 | 0.776 | 0.851 | 0.129 | 0.905 | 0.453 | |
| Beclin1 | ||||||||
| CC | 0.222 | -0.190 | 0.233 | -0.875 | -0.372 | 0.303 | ||
| P-value | 0.566 | 0.625 | 0.579 | 0.001 | 0.324 | 0.428 | ||
| LV mass | ||||||||
| CC | -0.149 | -0.209 | -0.307 | 0.119 | 0.417 | |||
| P-value | 0.703 | 0.619 | 0.422 | 0.761 | 0.265 | |||
| DF | ||||||||
| CC | 0.101 | 0.158 | 0.647 | 0.512 | ||||
| P-value | 0.811 | 0.685 | 0.059 | 0.159 | ||||
| CHF peptide | ||||||||
| CC | -0.110 | -0.015 | 0.070 | |||||
| P-value | 0.796 | 0.971 | 0.868 | |||||
| Milrinone | ||||||||
| CC | 0.200 | -0.344 | ||||||
| P-value | 0.606 | 0.364 | ||||||
| Glucose | ||||||||
| CC | 0.619 | |||||||
| P-value | 0.075 |
The number of samples are 9 for all except for CHF peptide, where the sample size is 8. CC = correlation co-efficient. LV mass = left ventricular mass, DF = diastolic function, CHF peptide = Congestive heart failure peptide (measurement of Brain Natriuretic Peptide), Milrinone = a drug for heart failure patients that improves heart rate and contractility, HgbA1C = Hemoglobin A1C, mTOR = mammalian target of rapamycin, AMPK = 5’Adenosine monophosphate (AMP) activated protein kinase.
Table 3.
Correlationship of different cardiac proteins in diabetic versus non-diabetic heart failure
| Beclin1 | LV Mass | DF | CHF Peptide | Milrinone | Glucose | HgbA1C | mTOR | AMPK | |
|---|---|---|---|---|---|---|---|---|---|
| LC3B | |||||||||
| CC | -0.253 | -0.465 | -0.006 | -0.378 | -0.107 | -0.357 | 0.014 | 0.070 | -0.688 |
| P-value | 0.512 | 0.207 | 0.987 | 0.356 | 0.784 | 0.346 | 0.971 | 0.858 | 0.040 |
| Beclin1 | |||||||||
| CC | 0.066 | 0.124 | 0.200 | -0.313 | 0.638 | 0.554 | 0.351 | 0.482 | |
| P value | 0.865 | 0.750 | 0.635 | 0.412 | 0.064 | 0.122 | 0.354 | 0.189 | |
| LV Mass | |||||||||
| CC | -0.149 | -0.209 | -0.307 | 0.119 | 0.417 | 0.068 | 0.192 | ||
| P-value | 0.703 | 0.619 | 0.422 | 0.761 | 0.265 | 0.861 | 0.620 | ||
| DF | |||||||||
| CC | 0.101 | 0.158 | 0.647 | 0.512 | -0.058 | -0.294 | |||
| P-value | 0.811 | 0.685 | 0.059 | 0.159 | 0.882 | 0.443 | |||
| CHF peptide | |||||||||
| CC | -0.110 | 0.015 | 0.071 | 0.143 | 0.222 | ||||
| P-value | 0.796 | 0.971 | 0.868 | 0.735 | 0.597 | ||||
| Milrinone | |||||||||
| CC | 0.200 | -0.344 | -0.208 | -0.318 | |||||
| P-value | 0.606 | 0.364 | 0.592 | 0.405 | |||||
| Glucose | |||||||||
| CC | 0.619 | .348 | 0.054 | ||||||
| P-value | 0.075 | 0.359 | 0.890 | ||||||
| HgbA1C | |||||||||
| CC | 0.348 | -0.186 | |||||||
| P-value | 0.359 | 0.632 | |||||||
| mTOR | |||||||||
| CC | 0.223 | ||||||||
| P-value | 0.564 |
The number of samples are 9 for all except for CHF peptide, where the sample size is 8. CC = correlation co-efficient. LV mass = left ventricular mass, DF = diastolic function, CHF peptide = Congestive heart failure peptide (measurement of Brain Natriuretic Peptide), Milrinone = a drug for heart failure patients that improves heart rate and contractility, HgbA1C = Hemoglobin A1C, mTOR = mammalian target of rapamycin, AMPK = 5’Adenosine monophosphate (AMP) activated protein kinase.
Diabetes inhibits mTOR activity is the failing hearts
It has been established that AMPK induces, while mTOR inhibits autophagy [9]. The activities of these upstream regulators of autophagy were determined in the ND and D groups. The mTOR activity was suppressed in diabetic hearts (Figure 4A). In contrast, AMPK activity showed very little differences between ND and D groups (Figure 4B). These data suggest that mTOR activity could have important role in regulation of autophagy in diabetic heart failure patients.
Figure 4.
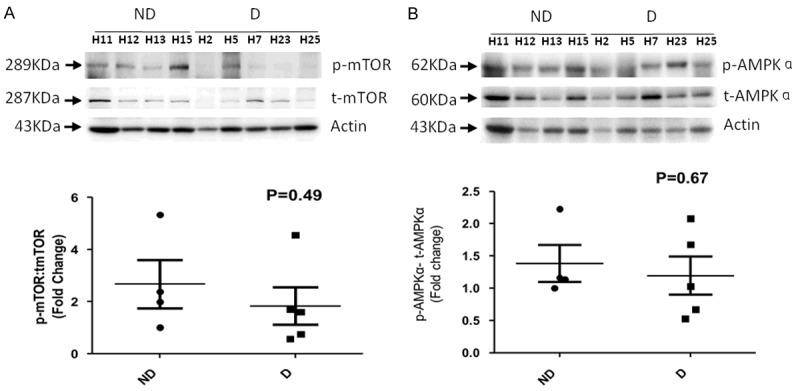
Activity of mTOR and AMPK in diabetic and non-diabetic heart failure patients. A. Western blotting to analyze the activity of mTOR. Top, representative blots for p-, and t-mTOR with actin as loading control. Bottom, the relative ratio of normalized p-mTOR to t-mTOR. B. Western blotting to analyze the activity of AMPK. Top, representative blots for p-, and t-AMPK with actin as loading control. Bottom, the relative ratio of normalized p-AMPK to t-AMPK. The values are mean ± SE.
MiR-133a and positive impact of Milrinone treatment on diabetic heart failure
Milrinone is a medication for heart failure patients. It is beneficial to the heart because it improves contractility and heart rate, acts as a vasodilator, and decreases blood pressure [35,36]. Our analyses showed positive but statistically insignificant correlation between miR-133a with Milrinone treated patients (Table 2) indicating that improvement in cardiac function by Milrinone treatment might be associated with upregulation of miR-133a. Conversely, the autophagy markers LC3B (insignificant) and Beclin1 (highly significant) are negatively correlated with the level of miR-133a in Milrinone treated patients (Table 2). Thus, the Milrinone mediated amelioration of cardiac function is negatively associated with the upregulation of autophagy markers.
Glycated hemoglobin (HbA1c) is formed by exposure of hemoglobin to plasma glucose and is a biomarker of glucose concentration in the blood for a prolonged period of time. The level of HbA1c increases in diabetic condition [37-39]. We found that miR-133a is negatively associated (insignificant), whereas autophagy markers are positively correlated (insignificant) with HbA1c (Table 2). These results further indicate that prolonged increase in blood glucose levels may be involved in induction of autophagy markers and thus the inhibition of miR-133a that ultimately leads to cardiac dysfunction.
Diabetes exacerbates cardiac hypertrophy in heart failure patients
To determine whether attenuation of miR-133a exacerbates cardiac hypertrophy in failing hearts, we measured the number of cardiomyocytes/unit area using Wheat Germ Agglutinin (WGA) staining. WGA stains cell membrane and is widely used for the measurement of the size of cardiomyocytes [40-43]. The analyses of number of cardiomyocytes per unit area between the ND and D groups revealed that attenuation of miR-133a in diabetic hearts increases the size of cardiomyocytes (Figure 5A). We also measured the level of β-MHC (hypertrophy marker) by immunohistochemistry, which showed a trend of upregulation in diabetic hearts (Figure 5B). These results reinforce the previous reports that attenuation of miR-133a is associated with the induction of cardiac hypertrophy [28].
Figure 5.
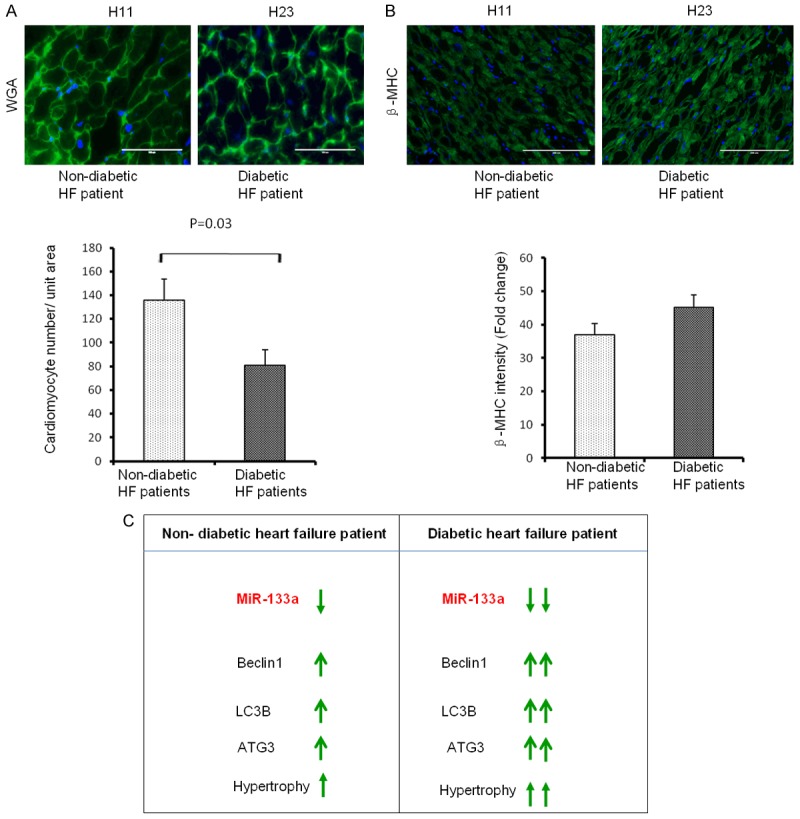
Cardiac hypertrophy in diabetic hearts with suppressed miR-133a. A. Top, Representative wheat germ agglutinin (WGA) staining of heart sections from ND (H11) and D (H23) patients. Scale bar : 100 μm. Bottom, average number of cardiomyocytes /unit area from the four ND and five D heart failure patients. The values are mean ± SE. B. Same as A, but for β-MHC. C. Diabetes exacerbates heart failure. In the failing heart, miR-133a is downregulated, autophagy markers Beclin1, LC3B, and ATG3 are upregulated, and cardiomyocytes show hypertrophy. On top of heart failure if the patient has diabetes, miR-133a will be further suppressed, and autophagy markers and hypertrophy will be robust.
Discussion
The cardioprotective role of miR-133a in diabetic hearts has been previously shown to be limited to the inhibition of cardiac hypertrophy and fibrosis [28,29,44]. Here, we report a novel role for miR-133a in cardiac autophagy in diabetic heart failure patients. Diabetes contributes to the inhibition of miR-133a that in turn upregulates the autophagy markers Beclin1 and LC3B, and exacerbates cardiac hypertrophy in heart failure patients undergoing LVAD implantation (Figure 5C). These autophagy markers are elevated in heart failure patients, but after mechanical unloading through LVAD and improvement of cardiac function, these markers are downregulated [15]. However, it is unclear how the up-, and down-regulation of autophagy occurs in these patients. Our findings provide insight into the regulation of autophagy markers in diabetic heart failure patients. We suggest that attenuation of miR-133a is associated with the upregulation of markers of autophagy in the heart failure patients undergoing LVAD implantation. Also, it is plausible that after mechanical unloading and improvement of cardiac function the level of miR-133a is normalized, which downregulates the markers of autophagy in LVAD implanted hearts. The rationale behind this is that miR-133a is downregulated in human heart failure [28] and miR-133a is upregulated in Milrinone treated hearts (Table 2). MiR-133a is cardioprotective and it suppresses pathological cardiac remodeling by inhibiting hypertrophy [28] and fibrosis [30], and regulating the β1-adrenergic receptor signaling cascade [31]. We and others have reported that miR-133a is downregulated in diabetic hearts [25-27]. Although diabetes causes cardiomyopathy independent of other heart diseases [7], miR-133a has a similar cardioprotective role in the diabetic heart and mitigates hypertrophy [27] and fibrosis [29]. Our results on attenuation of miR-133a (Figure 1Ci, 1Cii) and increased cardiac hypertrophy (Figure 5A, 5B) in diabetes heart failure patients are consistent with the results from other murine models. The elevated level of HbA1c [37-39], and attenuation of miR-133a [25-27] in diabetic hearts has been demonstrated. However, the relationship between the two is unclear. We found a negative correlation of miR-133a with HbA1c (Table 2). Conversely, autophagy markers have positive correlation with HbA1c, and miR-133a has a negative correlation with autophagy markers (Table 2). These results suggests that attenuation of miR-133a is associated with the upregulation of HbA1c, which is positively associated with the elevation of cardiac autophagy in diabetic hearts.
We believe that we did not observe a significant differences in the majority of cardiac parameters (Figures 2, 3, 4, 5, and Tables 1, 3) because both diabetic and non-diabetic patients are at end-stage heart failure. Their percentage ejection fraction (%FS) is below 20% (Figure 1D) and LV mass is less than 200 grams (Figure 1E). There were no significant difference between their % FS and LV mass (Figure 1D, 1E). The markers of autophagy such as Beclin1 and LC3B are downregulated in heart failure patients undergoing LVAD implantation [15]. To determine the contribution of diabetes on cardiac autophagy, we evaluated autophagy with the same markers in diabetic and non-diabetic heart failure patients undergoing LVAD implantation. To focus on the role of miR-133a, we categorically selected diabetic hearts with attenuated miR-133a and non-diabetic hearts with upregulated miR-133a. Our results show increased levels of Beclin1, LC3B (Figures 2A-C and 3A-C) and additional autophagy marker ATG3 in diabetic hearts (Figure 3D). These data suggest that attenuation of miR-133a contributes to exacerbation of autophagy in the diabetic hearts. To further analyze the contribution of upstream regulator of autophagy, we determined the activity of AMPK-an upstream inducer, and mTOR-an upstream inhibitor of autophagy. Our results demonstrate that mTOR activity is suppressed in diabetic hearts (Figure 4B), which contributes to the induction of autophagy. Nevertheless, there was no difference in AMPK activity between diabetic and non-diabetic hearts (Figure 4D). This suggests that mTOR inhibition may be the major mechanism underlying the induction of autophagy in diabetic hearts. Therefore, alleviation of miR-133a suppresses mTOR to up regulate autophagy in the diabetic heart failure patients. To validate the role of miR-133a as anti-hypertrophy, we measured the number of cardiomyocytes per unit area in diabetic and non-diabetic groups. Our results showed a decrease in the number of cardiomyocytes per unit area suggesting cardiac hypertrophy (Figure 5A). The data is consistent for beta-MHC, a molecular marker of cardiac hypertrophy (Figure 5B). The analyses of data using Pearson correlation show that miR-133a inhibits cardiac autophagy and hypertrophy in diabetic heart failure patients (Tables 2, 3). This study demonstrates a novel association of the attenuation of miR-133a and elevation of myocardial autophagy in failing human hearts with diabetes.
Despite advancement of medical science, a number of diabetic patients is rapidly increasing and heart failure is still the number one cause of mortality worldwide. Diabetes is detrimental at two levels, 1) it is an independent cause of cardiomyopathy, and 2) it increases the chances of heart failure as compared to non-diabetics. To improve the treatment regime, we need to have innovative strategies. MiRNAs are a novel class of non-coding RNAs that regulate biological processes by optimizing the level of different genes [18,19]. MiR-133a is anti-hypertrophy and anti-fibrotic miRNA [27-29]. It has been involved in the regulation of several other key biological processes such as the regulation of beta-adrenergic receptors [31,44]. Here, we evaluated the role of miR-133a in cardiac autophagy in diabetic patients and compared them with non-diabetes patients to elucidate that diabetes exacerbates heart failure through attenuation of miR-133a and induction of autophagy. Further mechanistic studies using loss-, and gain- of function experiments will provide insight on using miR-133a as therapeutic target for diabetic heart failure.
Acknowledgements
This work is supported in part by NIH grants HL-113281 and HL-116205 to Paras K. Mishra, and Institutional funds from UNMC to Daniel R. Anderson. SSN generated majority of the molecular data and contributed in writing of manuscript. MJD helps in collection of samples, and analyses of data. He also made all the tables. HRS contributed in IHC experiments and generation of data. MJD, GMT, and DRA contributed in drafting of manuscript. PKM conceived the idea and wrote the manuscript. PKM takes the responsibility for the work as a whole.
Disclosure of conflict of interest
None.
References
- 1.King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care. 1998;21:1414–31. doi: 10.2337/diacare.21.9.1414. [DOI] [PubMed] [Google Scholar]
- 2.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–53. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 3.Cohen-Solal A, Beauvais F, Logeart D. Heart failure and diabetes mellitus: epidemiology and management of an alarming association. J Card Fail. 2008;14:615–25. doi: 10.1016/j.cardfail.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Mathew V, Gersh BJ, Williams BA, Laskey WK, Willerson JT, Tilbury RT, Davis BR, Holmes DR Jr. Outcomes in patients with diabetes mellitus undergoing percutaneous coronary intervention in the current era: a report from the Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) trial. Circulation. 2004;109:476–80. doi: 10.1161/01.CIR.0000109693.64957.20. [DOI] [PubMed] [Google Scholar]
- 5.Pignone M, Alberts MJ, Colwell JA, Cushman M, Inzucchi SE, Mukherjee D, Rosenson RS, Williams CD, Wilson PW, Kirkman MS. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation. 2010;121:2694–701. doi: 10.1161/CIR.0b013e3181e3b133. [DOI] [PubMed] [Google Scholar]
- 6.American Diabetes Association. Economic costs of diabetes in the U. S. in 2012. Diabetes Care. 2013;36:1033–46. doi: 10.2337/dc12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavali V, Tyagi SC, Mishra PK. Predictors and prevention of diabetic cardiomyopathy. Diabetes Metab Syndr Obes. 2013;6:151–60. doi: 10.2147/DMSO.S30968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. 2011;50:1035–43. doi: 10.1016/j.yjmcc.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 11.Bo L, Su-Ling D, Fang L, Lu-Yu Z, Tao A, Stefan D, Kun W, Pei-Feng L. Autophagic program is regulated by miR-325. Cell Death Differ. 2014;21:967–77. doi: 10.1038/cdd.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, Bugger H, Frank D, Bevins J, Chen D, Perry CN, Dong XC, Valdez S, Rech M, Sheng X, Weimer BC, Gottlieb RA, White MF, Abel ED. In sulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest. 2013;123:5319–33. doi: 10.1172/JCI71171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–6. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 14.Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, Bauer EP, Klövekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–91. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 15.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120:S191–S197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kostin S, Pool L, Elsässer A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn WP, Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–24. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 17.Barsanti C, Trivella MG, D’Aurizio R, El Baroudi M, Baumgart M, Groth M, Caruso R, Verde A, Botta L, Cozzi L, Pitto L. Differential regulation of microRNAs in end-stage failing hearts is associated with left ventricular assist device unloading. Biomed Res Int. 2015;2015:592512. doi: 10.1155/2015/592512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 19.Mishra PK, Tyagi N, Kumar M, Tyagi SC. MicroRNAs as a therapeutic target for cardiovascular diseases. J Cell Mol Med. 2009;13:778–89. doi: 10.1111/j.1582-4934.2009.00744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tyagi AC, Sen U, Mishra PK. Synergy of microRNA and stem cell: a novel therapeutic approach for diabetes mellitus and cardiovascular diseases. Curr Diabetes Rev. 2011;7:367–76. doi: 10.2174/157339911797579179. [DOI] [PubMed] [Google Scholar]
- 21.Melman YF, Shah R, Das S. MicroRNAs in heart failure: is the picture becoming less miRky? Circ Heart Fail. 2014;7:203–14. doi: 10.1161/CIRCHEARTFAILURE.113.000266. [DOI] [PubMed] [Google Scholar]
- 22.Leptidis S, El Azzouzi H, Lok SI, de Weger R, Olieslagers S, Kisters N, Silva GJ, Heymans S, Cuppen E, Berezikov E, De Windt LJ, da Costa Martins P. A deep sequencing approach to uncover the miRNOME in the human heart. PLoS One. 2013;8:e57800. doi: 10.1371/journal.pone.0057800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guay C, Roggli E, Nesca V, Jacovetti C, Regazzi R. Diabetes mellitus, a microRNA-related disease? Transl Res. 2011;157:253–64. doi: 10.1016/j.trsl.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–54. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chavali V, Tyagi SC, Mishra PK. Differential expression of dicer, miRNAs, and inflammatory markers in diabetic Ins2+/- Akita hearts. Cell Biochem Biophys. 2014;68:25–35. doi: 10.1007/s12013-013-9679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chavali V, Tyagi SC, Mishra PK. MicroRNA-133a regulates DNA methylation in diabetic cardiomyocytes. Biochem Biophys Res Commun. 2012;425:668–72. doi: 10.1016/j.bbrc.2012.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab Res Rev. 2010;26:40–9. doi: 10.1002/dmrr.1054. [DOI] [PubMed] [Google Scholar]
- 28.Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 29.Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW 2nd, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med. 2014;18:415–21. doi: 10.1111/jcmm.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW 2nd. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106:166–75. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castaldi A, Zaglia T, Di Mauro V, Carullo P, Viggiani G, Borile G, Di Stefano B, Schiattarella GG, Gualazzi MG, Elia L, Stirparo GG, Colorito ML, Pironti G, Kunderfranco P, Esposito G, Bang ML, Mongillo M, Condorelli G, Catalucci D. MicroRNA-133 Modulates the beta1-Adrenergic Receptor Transduction Cascade. Circ Res. 2014;115:273–83. doi: 10.1161/CIRCRESAHA.115.303252. [DOI] [PubMed] [Google Scholar]
- 32.Huang J, Sun W, Huang H, Ye J, Pan W, Zhong Y, Cheng C, You X, Liu B, Xiong L, Liu S. miR-34a modulates angiotensin II-induced myocardial hypertrophy by direct inhibition of ATG9A expression and autophagic activity. PLoS One. 2014;9:e94382. doi: 10.1371/journal.pone.0094382. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Pan W, Zhong Y, Cheng C, Liu B, Wang L, Li A, Xiong L, Liu S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One. 2013;8:e53950. doi: 10.1371/journal.pone.0053950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao J, Zhu X, He B, Zhang Y, Kang B, Wang Z, Ni X. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J Biomed Sci. 2011;18:35. doi: 10.1186/1423-0127-18-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guglin M, Kaufman M. Inotropes do not increase mortality in advanced heart failure. Int J Gen Med. 2014;7:237–51. doi: 10.2147/IJGM.S62549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McFerson MC, McCanta AC, Pan Z, Collins KK, Jaggers J, da Cruz EM, Kaufman J. Tachyarrhythmias after the Norwood procedure: relationship and effect of vasoactive agents. Pediatr Cardiol. 2014;35:668–75. doi: 10.1007/s00246-013-0836-8. [DOI] [PubMed] [Google Scholar]
- 37.Fonarow GC. Diabetes Medications and Heart Failure: Recognizing the Risk. Circulation. 2014;130:1565–7. doi: 10.1161/CIRCULATIONAHA.114.012883. [DOI] [PubMed] [Google Scholar]
- 38.Grembowski D, Ralston JD, Anderson ML. Hemoglobin A, comorbid conditions and all-cause mortality in older patients with diabetes: A retrospective 9-year cohort study. Diabetes Res Clin Pract. 2014;106:373–82. doi: 10.1016/j.diabres.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 39.Kishimoto I, Makino H, Ohata Y, Tamanaha T, Tochiya M, Kada A, Ishihara M, Anzai T, Shimizu W, Yasuda S, Ogawa H. Hemoglobin A1c predicts heart failure hospitalization independent of baseline cardiac function or B-type natriuretic peptide level. Diabetes Res Clin Pract. 2014;104:257–65. doi: 10.1016/j.diabres.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 40.Chen S, Law CS, Grigsby CL, Olsen K, Hong TT, Zhang Y, Yeghiazarians Y, Gardner DG. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation. 2011;124:1838–47. doi: 10.1161/CIRCULATIONAHA.111.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goh JM, Bensley JG, Kenna K, Sozo F, Bocking AD, Brien J, Walker D, Harding R, Black MJ. Alcohol exposure during late gestation adversely affects myocardial development with implications for postnatal cardiac function. Am J Physiol Heart Circ Physiol. 2011;300:H645–H651. doi: 10.1152/ajpheart.00689.2010. [DOI] [PubMed] [Google Scholar]
- 42.Ma X, Takeda K, Singh A, Yu ZX, Zerfas P, Blount A, Liu C, Towbin JA, Schneider MD, Adelstein RS, Wei Q. Conditional ablation of nonmuscle myosin II-B delineates heart defects in adult mice. Circ Res. 2009;105:1102–9. doi: 10.1161/CIRCRESAHA.109.200303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, Pu WT. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–9. doi: 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mishra PK. Is miR-133a a promising therapeutic target for heart failure. J Diabetes & Metabolism. 2014;5:e118. [Google Scholar]