

Abstract
Phosphate homeostasis is coordinated and regulated by complex cross-organ talk through delicate hormonal networks. Parathyroid hormone (PTH), secreted in response to low serum calcium, has an important role in maintaining phosphate homeostasis by influencing renal synthesis of 1,25-dihydroxyvitamin D, thereby increasing intestinal phosphate absorption. Moreover, PTH can increase phosphate efflux from bone and contribute to renal phosphate homeostasis through phosphaturic effects. In addition, PTH can induce skeletal synthesis of another potent phosphaturic hormone, fibroblast growth factor 23 (FGF23), which is able to inhibit renal tubular phosphate reabsorption, thereby increasing urinary phosphate excretion. FGF23 can also fine-tune vitamin D homeostasis by suppressing renal expression of 1-alpha hydroxylase (1α(OH)ase). This review briefly discusses how FGF23, by forming a bone–kidney axis, regulates phosphate homeostasis, and how its dysregulation can lead to phosphate toxicity that induces widespread tissue injury. We also provide evidence to explain how phosphate toxicity related to dietary phosphorus overload may facilitate incidence of noncommunicable diseases including kidney disease, cardiovascular disease, cancers and skeletal disorders.
Introduction
All living organisms are dependent on exogenous sources of phosphorus. In humans and animals, dietary phosphorus must be acquired in sufficient amounts to avoid nutrition-related deficiencies such as rickets. In the body, exogenous phosphorus is metabolized to phosphate. Dietary phosphorus overload may contribute to dysregulation of phosphate metabolism leading to phosphate toxicity.1 In this review, we discuss the regulation of phosphate metabolism, and how its dysregulation can lead to phosphate toxicity. We define phosphate toxicity as excessive levels of extracellular and intracellular phosphate that are harmful to cellular function. In addition, several conditions associated with phosphate toxicity such as ectopic calcification, chronic kidney disease (CKD) and tumorigenesis are discussed.
The active roles of calciotropic hormones vitamin D and parathyroid hormone (PTH) to regulate phosphate homeostasis are well known.2,3 However, physiologic phosphate regulation cannot be fully explained by the actions of these hormones alone,4,5,6,7,8 and the identification of fibroblast growth factor 23 (FGF23), which can influence both vitamin D and PTH functions, helped us gain further insights into the physiologic regulation of phosphate homeostasis. Skeletal-derived FGF23 exerts phosphate-lowering effects in the kidney via a complex endocrine network; studies have identified osteoblasts and osteocytes as the main FGF23-producing cells.9,10 In addition to renal phosphate-lowering effects, FGF23 can lower 1,25-dihydroxyvitamin D production by suppressing the renal expression of 1-alpha hydroxylase (1α(OH)ase) and by increasing catabolism in the kidney. Studies have claimed that FGF23 may also suppress PTH synthesis.
FGF23 mediates its functions by interacting with an FGF receptor (FGFR) in a dependent manner using alpha-klotho as a cofactor. Although almost all tissues and organs express one or more isoforms of FGFRs, the restricted presence of alpha-klotho provides organ specificity to FGF23 functions.11,12,13,14 FGF23 was initially identified in the ventrolateral thalamic nucleus of the mouse brain,15 and its clinical importance was subsequently shown in patients with autosomal dominant hypophosphatemic rickets.16 The FGF23 gene has three exons separated by two introns, and the gene encodes a 32-kDa glycoprotein containing 251 amino acid residues: 24 amino acids constitute a hydrophobic signal sequence, an N terminus of 155 amino acids constitute the FGF core homology region and 72 amino acids form the C-terminal domain.17,18,19,20 After cleavage of the 24-amino-acid signal sequence, the mature protein (25–251)-FGF23 is secreted into the circulation where three distinct forms of the FGF23 protein can be detected: a full-length mature form (25–251)-FGF23, a shorter form (25–179)-FGF23 lacking the unique C-terminal tail of 72 amino acids and a C-terminal tail. The shorter form without a C-terminal tail arises from proteolytic cleavage at the 176RXXR179 site,17,18,19,20 and it can also be detected in the serum. Recent studies have shown that the C terminus is not only the potential alpha-klotho-interacting site, but it also determines the functionality of the FGF23 protein. The chimeric protein containing the N terminus of FGF2 and C terminus of FGF23 could act as a phosphatonin and could reduce the renal expression of 1α(OH)ase.19,20 It is worth mentioning that the presence of the C terminus of FGF23 in the chimeric protein paved the way for the alpha-klotho interaction. Recently, a family with sequence similarity 20, member C (Fam20C), was shown to phosphorylate FGF23. Such phosphorylation blocks O-glycosylation by polypeptide N-acetylgalactosaminyltransferase 3. This observation suggests that the interplay between phosphorylation and O-glycosylation of FGF23 may be a critical posttranslational modification by which the activity of secreted FGF23 protein is determined.21 Once secreted, FGF23 protein binds its receptors to exert diverse functions.
In vitro studies have shown that bioactive FGF23 protein can bind to FGFR1c, FGFR3c and FGFR4, but not to FGFR2c.22 In vivo studies have suggested that FGFR1c is the main target for FGF23 in the kidney23,24 as loss of FGFR3 or FGFR4 activity did not affect FGF23-mediated hypophosphatemia in Hyp mice.24 It is of relevance that Hyp mouse is the model for human X-linked hypophosphatemia related to mutations that inactivate the endopeptidases of the X chromosome (PHEX); the inactivation of PHEX leads to increased serum levels of FGF23, a phosphaturic hormone that induces excessive renal phosphate excretion and severe hypophosphatemia. Moreover, conditional deletion of FGFR1 in the kidney abolished phosphaturic effects of recombinant FGF23 administration.25 It is of relevance that no such abolition following administration of recombinant FGF23 was noted in FGFR3 or FGFR4 knockout mice.25 The in vivo functions of FGF23 are also elegantly demonstrated in genetically modified mouse models. For instance, FGF23 knockout mice develop severe hyperphosphatemia owing to increased renal reabsoption of phosphate,26,27 whereas FGF23-overproducing transgenic mice or Hyp mice are hypophosphatemic owing to excessive urinary phosphate excretion.28,29 Such FGF23-mediated hypophosphatemia is an alpha-klotho-dependent phenomenon, and miscommunication of FGF23 and alpha-klotho can lead to dysregulation of renal phosphate homeostasis.1,14,30,31
Renal Phosphate Regulation
Under normal physiologic conditions, the kidneys maintain serum phosphate balance by fine-tuning urinary phosphate excretion. Filtrated phosphate is reabsorbed in the renal proximal tubular epithelial cells using sodium-dependent phosphate (Na/Pi) cotransporters32,33,34 (Figure 1). Although these cells contain three types of Na/Pi cotransporters, the type 2 cotransporter family that includes Na/Pi2a (encoded by the SLC34a1 gene) and NaPi2c (encoded by the SLC34a3 gene) is believed to be most active in renal phosphate uptake. In contrast to the type 2 cotransporter system, the type 1 cotransporter (NPT1) is mostly an anion carrier, whereas the type 3 cotransporter system comprises two transporters, PiT1 (encoded by the SLC20a1 gene) and PiT2 (encoded by the SLC20a2 gene), and their role in phosphate transport is an active area of research. Studies have shown that FGF23 could suppress the expression of Na/Pi2a and Na/Pi2c, leading to less renal phosphate uptake with increased urinary phosphate excretion.25 In fact, transgenic mice overexpressing FGF23 showed severe urinary phosphate wasting owing to suppressed expression and reduced activity of renal Na/Pi2a,28,29 whereas Fgf23 knockout mice develop severe hyperphosphatemia owing to increased renal expression and activity of Na/Pi2a.26,27 It is of particular interest that genetically restoring FGF23 systemic actions in Fgf23 knockout mice reduced Na/Pi2a activity, resulting in reversal of hyperphosphatemia to hypophosphatemia.29 It is important to note that the segment of the nephron on which FGF23 exerts its most potent effects is not yet clear, as alpha-klotho, the cofactor for FGF23 bioactivities, is mostly present in the distal convoluted tubule, whereas Na/Pi cotransporter activity and vitamin D metabolizing enzymes are mainly present in the proximal tubular epithelial cells.35 Although further studies are needed to determine the exact molecular mechanisms of FGF23-mediated renal phosphate transport, miscommunication between FGF23 and alpha-klotho leads to altered phosphate balance.
Figure 1.
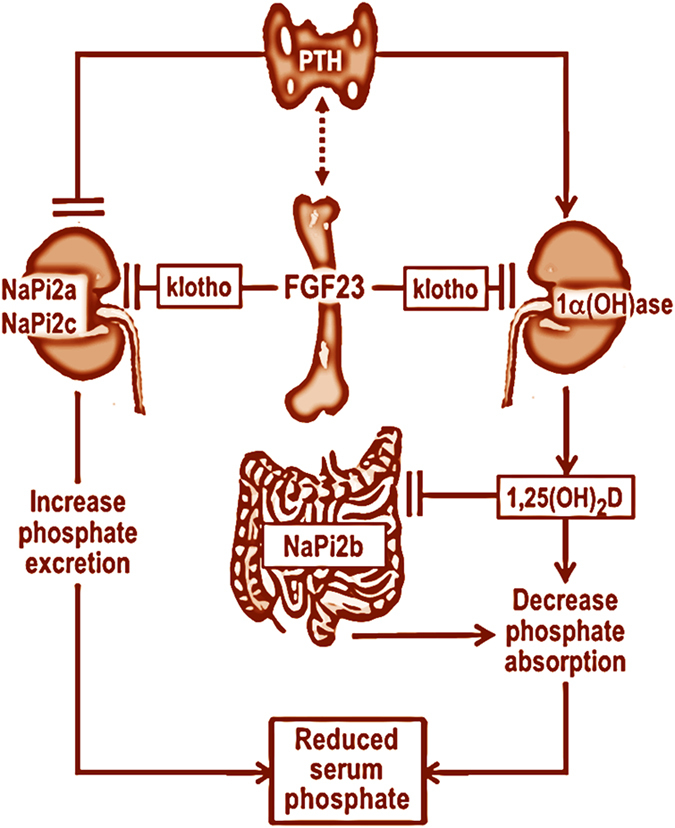
Simplified diagram showing multiorgan interactions in the regulation of phosphate homeostasis. Fibroblast growth factor (FGF) 23 produced in the bone cells can suppress renal Na/Pi2a and Na/Pi2c cotransporter activities to increase the urinary excretion of phosphate. Similarly, FGF23 can also suppress renal expression of 1-alpha hydroxylase (1α(OH)ase) to reduce the production of 1,25-dihydroxyvitamin D (1,25(OH)2D), which can suppress intestinal NaPi2b activities to reduce phosphate absorption, resulting in decreased serum phosphate levels. Of relevance, parathyroid hormone (PTH) can induce the expression of the 1α(OH)ase, and thereby it can increase the production of 1,25(OH)2D, which in turn can inhibit PTH and 1α(OH)ase expression. Such transcriptional repression feedback maintains vitamin D homeostasis. The figure is adopted with modification from our earlier publication.1,45 (Dashed lines indicate that the interactions either might not be direct or not scientifically validated by multiple groups).
Pathological conditions in CKD are mostly owing to miscommunication between bone-derived FGF23 and kidney-derived alpha-klotho, which leads to the development of phosphate toxicity. In CKD patients, reduced levels of alpha-klotho leaves FGF23 nonfunctional, thereby limiting the kidneys' ability to excrete urinary phosphate. In vivo studies conducted on Fgf23 knockout mice have shown that bioactive FGF23 protein could significantly reduce serum phosphate level in these mutant mice. It is of particular significance that bioactive FGF23 protein failed to exert phosphate-lowering effects in Fgf23/alpha-klotho double knockout mice,36,37 suggesting that FGF23 loses its phosphate regulating abilities without alpha-klotho. Moreover, FGF23-induced hypophosphatemia in Hyp mice is reversed to hyperphosphatemia in the Hyp mice without alpha-klotho activity (Hyp/alpha-klotho double mutant mice), despite high serum FGF23 levels in double mutant mice.3,36 In accordance with these animal studies, an inactivating mutation in the human alpha-klotho gene resulted in severe hyperphosphatemia, despite high serum FGF23 levels in a patient with tumoral calcinosis.38 From the above-cited human and experimental studies, it is clear that alpha-klotho has an indispensable role in FGF23-mediated phosphate metabolism.14,17,18,36,39,40 As mentioned, features of phosphate toxicity appear in various organs when there is a dysregulation of the FGF23-alpha-klotho system.
Phosphate Toxicity
Phosphate toxicity arising from a disproportionate accumulation of phosphate in the body has been shown to accelerate mammalian aging, produce bone deformities and reduce overall survival.41 Genetically altered alpha-klotho-knockout mice develop phosphate toxicity as early as at 3 weeks of age, which affects weight gain and the bone maturation process, produces a generalized soft tissue atrophy and results in reduced life span.1,14,40,41,42,43,44,45,46 In vivo studies found that phosphate toxicity in alpha-klotho ablated mice is associated with increased renal activity of NaPi2a. However, phosphate burden was lowered in hyperphosphatemic alpha-klotho-knockout mice by generating NaPi2a/alpha-klotho double-knockout mice, which resulted in prolonged survival.41 More importantly, compared with a normal-phosphate diet (0.6%), phosphate toxicity reappeared when a high-phosphate diet (1.2%) was fed to NaPi2a/alpha-klotho mutant mice, with the appearance of premature aging-like features leading to early death.41 These in vivo experimental observations clearly suggest that phosphate toxicity related to diet can contribute to the progression of the mammalian aging process, affecting both bone health and overall survival.
Phosphorous has a very strong electronegative attraction with calcium—having electronegative values on the Pauling scale of 2.1 for phosphorus and 1.0 for calcium.47 The strength of this attraction may help explain why phosphate toxicity owing to dietary phosphorus overload can so easily impair calcium metabolism. The chemical bonding of phosphorus with calcium is most evident in the body's hydroxyapatite mineral matrix of bone. These two minerals normally unite to form bone tissue in a calcium:phosphorus ratio of approximately 2:1 by weight, averaging 2.07:1 in spongy trabecular bone tissue48 and 2.17:1 in harder cortical bone tissue.49 Of relevance, the mean calcium:phosphorus ratio in studies of human milk is approximately in the range of 2:1 by weight.50
Referring to the calcium:phosphorus ratio, the Food and Nutrition Board that set the recommended dietary allowances (RDAs) as part of the Dietary Reference Intakes51 noted that ‘there is little or no evidence for relating the two nutrients, one to the other, during most of human life (p.154).' Nevertheless, the RDA that the Board set for calcium relative to that of phosphorus is equivalent to an ample calcium:phosphorus ratio of 1.4:1 for adults and 1.7:1 for adults aged 50 years and over. However, data from NHANES III reported by Chang et al.52 showed average intakes of 758 mg of calcium and 1242, mg of phosphorus for adults, which is equivalent to an average calcium:phosphorus ratio of only 0.61:1. As noted by Uribarri and Calvo,,53 the data reported by Chang et al.52 show a consistent association between increasing mortality in adult subjects and a decreasing calcium:phosphorus ratio as dietary phosphorus intake increases more than calcium intake.
In an earlier study, calcium retention increased when college women consumed 1500, mg of calcium and 800 mg of phosphorus with a calcium:phosphorus ratio of 1.88:1; however, increasing subjects' phosphorus intake to 1400, mg with a calcium:phosphorus ratio of 1.07:1 significantly reduced calcium utilization.54 More recent research showed that decreasing the calcium:phosphorus ratio in a diet fed to young women by increasing phosphorus intake disrupted calcium metabolism, resulting in increased bone resorption as indicated by higher levels of serum PTH and urinary calcium.55 Researchers used a calcium-adequate diet in an experimental animal model to demonstrate how lowering the calcium:phosphorus ratio by increasing the diet's phosphorus content produced defects in tooth enamel and dentin.56 The findings of these studies imply that lowering excessive phosphorus intake rather than increasing calcium intake beyond adequate levels is required to properly balance the dietary calcium:phosphorus ratio. Unfortunately, the US adult population's average calcium intake of 758 mg reported by Chang et al.52 is below RDA levels of 1000, mg and 1200, mg calcium for adults 50 years and older. Generally, the USDA Dietary Guidelines for Americans is incongruous with the latest findings on dietary phosphorus overload, with little indication for change in the 2015 Dietary Guidelines.57 For example, a 2000-calorie eating pattern recommended by the MyPlate program of the USDA Center for Nutrition Policy & Promotion58 averages 1884, mg of phosphorus a day—a daily overload far above the intake level where mortality effects are seen. Of particular concern, unrestricted consumption of phosphate can induce systemic complications related to phosphate toxicity (Table 1).
Table 1. Partial list of pathological events or diseases that can induce altered phosphate balance14,40,43,102.
| Hypophosphatemia |
| X-linked hypophosphatemic rickets |
| Vitamin D resistance/deficiency |
| Severe dietary deficiency |
| Sepsis |
| Respiratory alkalosis |
| Renal tubular defects (Fanconi syndrome) |
| Renal transplantation |
| Metabolic acidosis |
| Hyperparathyroidism |
| Hormones (insulin, glucagon and cortisol) |
| Diuretics |
| Diabetic ketoacidosis |
| Hyperphosphatemia |
| Vitamin D intoxication |
| Tumor lysis syndrome |
| Tumor calcinosis |
| Rhabdomyolysis |
| Respiratory acidosis |
| Phosphate enema |
| Metabolic acidosis |
| Magnesium deficiency |
| Intravenous/oral phosphate therapy |
| Hypoparathyroidism |
| Hemolysis |
| Chronic kidney disease |
| Bowel infarction |
| Bisphosphonate therapy |
| Acromegaly |
Please note that metabolic acidosis can induce either hypophosphatemia or hyperphosphatemia depending on clinical situations. It is believed that nonlactic metabolic acidosis associated with hypophosphatemia can be connected to impaired tubular reabsorption of bicarbonate and that phosphorus administration can improve such acidosis.103 On the other hand, respiratory or metabolic acidosis may hydrolyze intracellular organic phosphate-containing compounds and release them into the extracellular compartment. Cell lysis disorders, such as tumor lysis syndrome, hemolytic anemia and rhabdomyolysis, may all give rise to hyperphosphatemia.104
Phosphate Toxicity Associated with Ectopic Calcification
Calcium–phosphorus (CaxPi) product in serum, calculated by multiplying serum calcium and phosphorus levels, should be less than 55 mg2 dl−2 in adults.59 CaxPi product and other anions containing calcium normally account for 5% of serum calcium, whereas free ionized calcium accounts for 45% of serum calcium and the remaining serum calcium is bonded to albumin.60 Elevated levels of serum phosphorus in hyperphosphatemia—associated with impaired kidney function, dietary phosphorus overload or both—unite with free calcium to form an additional CaxPi product. Accumlated excess CaxPi product is likely to be deposited into soft tissue causing ectopic calcification.61 In addition, elevated serum levels of osteocalcin, a noncollagenous bone matrix protein and a marker for osteoblast function, are associated with FGF23 levels.62 Osteocalcin is also found in calcified atherosclerotic plaque.63 These associations provide evidence that rising serum calcium–phosphate product associated with hyperphosphatemia upregulates osteocalcin, which stimulates mineral deposition in soft tissue as ectopic calcification. An opposite action is seen in the protein osteopontin, which inhibits mineral deposition and regresses calcification.64 Vascular calcification in rats reversed when they were switched to a low-phosphate diet, which the researchers suggested was associated with calcification inhibition by osteopontin.65
One type of soft tissue that is susceptible to ectopic calcification from the deposition of CaxPi product is the endothelium of the arterial system. Arterial calcification increases mortality risk by threefold to fourfold.66 Calcification causes a hard or stable plaque to form in arterial vessels, which is related to arteriolosclerosis, hypertension, left ventricular hypertrophy and aortic valve disease. In vitro and in vivo experiments showed that a high phosphorus load acutely increases endothelial dysfunction by impairing vasodilation, raising the risk for cardiovascular disease.67 High levels of serum phosphorus in healthy young adults have been associated with coronary atherosclerosis68 and with left ventricular hypertrophy,69 and high levels of serum phosphorus in a cohort study were associated with a 40% greater risk of heart failure.70 A meta-analysis showed that serum phosphorus, but not serum calcium or PTH, was independently associated with mortality and risk for cardiovascular disease in CKD patients.71 Dietary phosphorus overload was found to raise systolic blood pressure, except from dairy products, which researchers attributed to nutrients in dairy other than phosphorus.72
The exact molecular mechanisms of how extracellular phosphate might exert its cytotoxicity is not yet clearly defined. Studies, however, have shown that extracellular phosphate can form insoluble nanoparticles with calcium and fetuin-A, commonly referred to as calciprotein particles (CPPs); these CPPs are highly bioactive ligands that can induce cytotoxicity, ranging from cell death to including osteogenic transformation of vascular smooth muscle cells (VSMCs). Furthermore, CPPs are detected in the circulation, both in human and animal models, particularly in patients with CKD, implicating a role in phosphate-mediated tissue injuries.73 Calcium phosphate and other crystals may be deposited in joints causing acute inflammation and damaged cartilage.74 Mice fed high-phosphate diets developed ectopic tumoral calcifications around joints.75 Other types of ectopic calcifications from calcium phosphate occur in the formation of kidney stones, often in combination with calcium oxalate,76 in bone tissue irregularities such as bone spurs or osteophytes77 and within visceral organs and the epidermis.61 Vascular calcification is a complex, ectopic biomineralization process, and phosphate toxicity can not only facilitate essential hydroxyapatite deposition in the calcifying vessels but also initiate the early events of apoptotic cell death, which is inflicted on both endothelial cells and VSMCs. Studies have shown higher risk for cardiovascular mortality in CKD patients undergoing hemodialysis treatment.
Phosphate Toxicity Associated with CKD
The estimated prevalence of CKD within the global population is 8–16%.78 The role of phosphate toxicity in the pathology of CKD and beyond is published elsewhere.1,45,73,79 A high concentration of extracellular phosphate is toxic to cells, and it leads to premature cellular aging. The kidneys regulate urinary excretion of phosphate to maintain the serum phosphate level that is circulated throughout the body. Dietary phosphate overload can lead to an increased phosphate burden, and it damages the kidneys through tubular injury and interstitial fibrosis; studies have found that subjects with high serum phosphorus had a higher risk for developing kidney diseases.80 Glomerular filtration rate decreases as serum phosphorus increases in kidney disease, and mortality from associated cardiovascular disease in dialysis patients is 20% annually. Of relevance, renal aging can accelerate systemic aging.81
As mentioned, increased serum calcium and phosphate levels are usually considered main contributors to vascular calcification, and the estimation of CaxPi product has long been used to predict vascular pathology in CKD patients.82 However, recent studies have found that serum phosphate levels in patients undergoing hemodialysis treatment correlates with vascular calcification more strongly than does the CaxPi product.83 In fact, the clinical utility of the CaxPi product has been questioned in recent studies, noting that the cause of calcification is more complicated than just the precipitation of the CaxPi product.84 It is of clinical relevance that, in contrast to hyperphosphatemia, associations between serum calcium levels and cardiovascular events or disease outcomes of CKD patients are not yet clearly established. However, a few studies have shown that hypercalcemia may have predictive value in determining the negative outcome of CKD patients.85
Although phosphate toxicity is known to promote disease progression in patients with CKD, its association with tumorigenesis is not studied in similar depth and detail. We next present evidence of this association and suggest some innovative theories of tumorigenesis. We believe that these suggestions are not based on deductive speculation, but rather on inductive theory generation grounded in reliable evidence.
Phosphate Toxicity Associated with Tumorigenesis
Hippocrates first observed that vessels attached to tumors appeared like legs on a crab. Thus, the name cancer, derived from the Latin word for crab, came into use to describe tumors.86 Today, the elaborate vessel system of tumors is considered to be complicit with the aggressive tendency for neoplasms to grow uncontrollably and metastasize throughout the body. However, a novel theory of tumor vessel function may be induced from biological findings related to phosphate toxicity. In biology, the mycorrhiza fungal root system of plants absorbs and sequesters phosphorus from soil.87 Evidence suggests that tumor vessels may function similarly in humans to absorb and sequester excess phosphorus from extracellular fluid. Cancer patients have been observed to retain and store phosphorus in tumors,88 and cancer cells accumulate up to twice as much phosphorus as normal cells.89 Just as tumorigenesis in tomato plants has been positively associated with phosphorus,90 a cellular environment high in phosphorus in humans has been found to induce tumor neovascularization and angiogenesis, or new blood vessel formation in neoplasms.91
Dietary phosphorus overload has been found to stimulate tumor growth in lung tissue.92 Increasing phosphorus levels in cultured breast cancer cells were observed to change cellular behavior and metabolism, thus supporting the role of inorganic phosphate in modulating tumor metabolism and metastasis.93 An average daily phosphorus intake of 1395, mg in men followed up for 22 years in the Health Professionals Follow-Up Study was associated with an increased overall risk of prostate cancer and with lethal and high-grade prostate cancer.94 Farmers of Liuchong Village in the Hubei province of China claimed that cancer cases increased in their village from drinking water contaminated by a nearby phosphate mine.95
Many studies have examined the association of meat and dairy intake with cancer, but a review of meta-analyses showed inconsistent findings.96 Few studies have examined the phosphate intake from meat and dairy in association with cancer. In addition, it is difficult to isolate the effect of phosphorus in one or two foods without considering the total phosphate content of the diet. For example, subjects with high intake of dietary phosphate from meat and dairy but with overall normal phosphate intake may show less increase in cancer incidence than subjects with low intake of phosphate from meat and dairy but with a high overall phosphate intake. Comparisons of diets containing similar amounts of total phosphate at baseline should be investigated for cancer risk as the phosphate content rises from meat and dairy intake. From the above-mentioned evidence, it is likely that dietary phosphate consumption might influence tumorigenesis, possibly as a storage mechanism to help maintain phosphate homeostasis, and further research using meaningful experimental designs is warranted to validate such speculation.
Conclusion
The endocrine communication between bone-derived FGF23 and kidney-derived alpha-klotho is essential for physiologic regulation of phosphate balance. As we have briefly discussed, dysregulation of FGF23/alpha-klotho system provokes phosphate imbalance and induces a wide range of organs/tissue damage in blood vessels, bone and kidney. Of clinical importance, phosphate toxicity induced by excessive exogenous phosphate administration in humans can be fatal.97,98,99 In fact, it is becoming more evident from experimental and human observations that features of phosphate toxicity can appear after consumption of a high-phosphate diet, even when serum phosphate levels are within the normal range.3,43,45,79 Recent survey results highlight that even future medical professionals and CKD patients undergoing hemodialysis are not adequately aware of the hidden source of phosphate in their diet, and emphasize the need for educational initiatives to raise awareness of the risk posed by dietary items with hidden phosphate ingredients.100,101 Taking into account human and animal observations, maintaining phosphate balance through adequate dietary intake appears to be important for a healthy life and for longevity. In addition, phosphate imbalance owing to bone–kidney miscommunication can induce serious debilitating complications.
Acknowledgments
Part of the original research that formed the basis of this review article was performed by Razzaque's lab members (Drs Teruyo Nakatani, Junko Akiyoshi, Satoko Osuka, Shigeko Kato, Kazuyoshi Uchihashi and Mutsuko Ohnishi) at the Harvard School of Dental Medicine, Boston, MA, USA.
Footnotes
The authors declare no conflict of interest.
References
- Razzaque MS. Phosphate toxicity: new insights into an old problem. Clin Sci (Lond) 2011; 120: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Felsenfeld AJ. PTH, FGF-23 and early CKD. Nephrol Dial Transplant 2008; 23: 3391–3393. [DOI] [PubMed] [Google Scholar]
- Razzaque MS. Osteo-renal regulation of systemic phosphate metabolism. IUBMB Life 2011; 63: 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Barron JL, Mirzazedeh M, Gallagher H, Hyer S, Cantor T et al. Changes in bone mineral parameters, vitamin D metabolites, and PTH measurements with varying chronic kidney disease stages. J Bone Miner Metab 2011; 29: 71–79. [DOI] [PubMed] [Google Scholar]
- Evenepoel P, Viaene L, Meijers B. PTH, FGF23, and calcium: it takes three to tango? Kidney Int 2011; 80: 1377. [DOI] [PubMed] [Google Scholar]
- Lanske B, Razzaque MS. Molecular interactions of FGF23 and PTH in phosphate regulation. Kidney Int 2014; 86: 1072–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RB, Haq A, Stanford CF, Razzaque MS. Vitamin D, phosphate and vasculotoxicity. Can J Physiol Pharmacol 2015;; doi: 10.1139/cjpp-2015-0083. [DOI] [PubMed] [Google Scholar]
- Burnett-Bowie SM, Henao MP, Dere ME, Lee H, Leder BZ. Effects of hPTH(1-34) infusion on circulating serum phosphate, 1,25-dihydroxyvitamin D, and FGF23 levels in healthy men. J Bone Miner Res 2009; 24: 1681–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitara D, Razzaque MS, St-Arnaud R, Huang W, Taguchi T, Erben RG et al. Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol 2006; 169: 2161–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 2006; 38: 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92: 131–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 2013; 75: 503–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Razzaque MS. Osteo-renal cross-talk and phosphate metabolism by the FGF23-Klotho system. Contrib Nephrol 2013; 180: 1–13. [DOI] [PubMed] [Google Scholar]
- Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol 2009; 5: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun 2000; 277: 494–498. [DOI] [PubMed] [Google Scholar]
- ADHR_Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat Genet 2000; 26: 345–348. [DOI] [PubMed] [Google Scholar]
- Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol 2007; 27: 3417–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci USA 2010; 107: 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz R, Ohnishi M, Ding X, Kurosu H, Wang L, Akiyoshi J et al. Klotho coreceptors inhibit signaling by paracrine fibroblast growth factor 8 subfamily ligands. Mol Cell Biol 2012; 32: 1944–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz R, Ohnishi M, Kir S, Kurosu H, Wang L, Pastor J et al. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J Biol Chem 2012; 287: 29134–29146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci USA 2014; 111: 5520–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 2005; 16: 107–137. [DOI] [PubMed] [Google Scholar]
- Li H, Martin A, David V, Quarles LD. Compound deletion of Fgfr3 and Fgfr4 partially rescues the Hyp mouse phenotype. Am J Physiol Endocrinol Metab 2011; 300: E508–E517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Vierthaler L, Tang W, Zhou J, Quarles LD. FGFR3 and FGFR4 do not mediate renal effects of FGF23. J Am Soc Nephrol 2008; 19: 2342–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 2009; 297: F282–F291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 2004; 113: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 2004; 23: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 2004; 314: 409–414. [DOI] [PubMed] [Google Scholar]
- DeLuca S, Sitara D, Kang K, Marsell R, Jonsson K, Taguchi T et al. Amelioration of the premature ageing-like features of Fgf-23 knockout mice by genetically restoring the systemic actions of FGF-23. J Pathol 2008; 216: 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanske B, Razzaque MS. Mineral metabolism and aging: the fibroblast growth factor 23 enigma. Curr Opin Nephrol Hypertens 2007; 16: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol 2007; 194: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia na/phosphate cotransporter. Annu Rev Nutr 2005; 25: 197–214. [DOI] [PubMed] [Google Scholar]
- Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto KI. Differential effects of Npt2a gene ablation and the X-linked Hyp mutation on renal expression of type IIc Na/Pi cotransporter. Am J Physiol Renal Physiol 2003; 2: 2. [DOI] [PubMed] [Google Scholar]
- Miyamoto KI, Haito-Sugino S, Kuwahara S, Ohi A, Nomura K, Ito M et al. Sodium-dependent phosphate cotransporters: lessons from gene knockout and mutation studies. J Pharm Sci 2011; 100: 3719–3730. [DOI] [PubMed] [Google Scholar]
- Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 2006; 17: 1305–1315. [DOI] [PubMed] [Google Scholar]
- Nakatani T, Ohnishi M, Razzaque MS. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J 2009; 23: 3702–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J 2009; 23: 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest 2007; 117: 2684–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant 2009; 24: 4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: is Klotho an essential player? Am J Physiol Renal Physiol 2009; 296: F470–F476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J 2010; 24: 3562–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390: 45–51. [DOI] [PubMed] [Google Scholar]
- Razzaque MS. Bone-kidney axis in systemic phosphate turnover. Arch Biochem Biophys 2014; 561: 154–158. [DOI] [PubMed] [Google Scholar]
- Nabeshima Y, Washida M, Tamura M, Maeno A, Ohnishi M, Shiroishi T et al. Calpain 1 inhibitor BDA-410 ameliorates alpha-klotho-deficiency phenotypes resembling human aging-related syndromes. Sci Rep 2014; 4: 5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuka S, Razzaque MS. Can features of phosphate toxicity appear in normophosphatemia? J Bone Miner Metab 2012; 30: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanske B, Razzaque MS. Premature aging in klotho mutant mice: cause or consequence? Ageing Res Rev 2007; 6: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TL, LeMay HEJ, Bursten BE, Murphy CJ, Woodward PM. Bond polarity and electonegativity. InChemisty: The Central Science 12th edn Pearson Prentice Hall: Boston, MA, USA, 2012; pp288–329. [Google Scholar]
- Zaichick V, Tzaphlidou M. Calcium and phosphorus concentrations and the calcium/phosphorus ratio in trabecular bone from the femoral neck of healthy humans as determined by neutron activation analysis. Appl Radiat Isot 2003; 58: 623–627. [DOI] [PubMed] [Google Scholar]
- Zaichick V, Tzaphlidou M. Determination of calcium, phosphorus, and the calcium/phosphorus ratio in cortical bone from the human femoral neck by neutron activation analysis. Appl Radiat Isot 2002; 56: 781–786. [DOI] [PubMed] [Google Scholar]
- Jenness R. The composition of human milk. Semin Perinatol 1979; 3: 225–239. [PubMed] [Google Scholar]
- IOM. Dietary Reference Intakes For Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride The National Academies Press: Washington, DC, USA, 1997;. [PubMed] [Google Scholar]
- Chang AR, Lazo M, Appel LJ, Gutierrez OM, Grams ME. High dietary phosphorus intake is associated with all-cause mortality: results from NHANES III. Am J Clin Nutr 2014; 99: 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribarri J, Calvo MS. Dietary phosphorus intake and health. Am J Clin Nutr 2014; 99: 247–248. [DOI] [PubMed] [Google Scholar]
- Leichsenring JM, Norris LM, Lamison SA. The effect of level of intake on calcium and phosphorus metabolism in college women. J Nutr 1951; 45: 407–418. [DOI] [PubMed] [Google Scholar]
- Kemi VE, Karkkainen MU, Rita HJ, Laaksonen MM, Outila TA, Lamberg-Allardt CJ. Low calcium:phosphorus ratio in habitual diets affects serum parathyroid hormone concentration and calcium metabolism in healthy women with adequate calcium intake. Br J Nutr 2010; 103: 561–568. [DOI] [PubMed] [Google Scholar]
- Jekl V, Krejcirova L, Buchtova M, Knotek Z. Effect of high phosphorus diet on tooth microstructure of rodent incisors. Bone 2011; 49: 479–484. [DOI] [PubMed] [Google Scholar]
- Hennessy M. Until phosphorus gets on the USDA's radar labeling policy won't change: NKF2014; http://www.foodnavigator-usa.com/Regulation/Until-phosphorus-gets-on-the-USDA-s-radar-labeling-policy-won-t-change-NKF.
- CNPP. 2011; MyPlate: Sample menus for a 2000 calorie food pattern http://www.choosemyplate.gov/food-groups/downloads/Sample_Menus-2000Cals-DG2010.pdf.
- NKF. 2003; KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease: Guideline 6. Serum calcium and calcium-phosphorus product. [PubMed]
- Mihai R, Farndon JR. Parathyroid disease and calcium metabolism. Br J Anaesth 2000; 85: 29–43. [DOI] [PubMed] [Google Scholar]
- NKF. 2003b; KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease: background. [PubMed]
- Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 2005; 90: 1519–1524. [DOI] [PubMed] [Google Scholar]
- Foresta C, Strapazzon G, De Toni L, Fabris F, Grego F, Gerosa G et al. Platelets express and release osteocalcin and co-localize in human calcified atherosclerotic plaques. J Thromb Haemost 2013; 11: 357–365. [DOI] [PubMed] [Google Scholar]
- Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H et al. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol 2002; 161: 2035–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch JL, Lee DH, Liapis H, Ritter C, Zhang S, Suarez E et al. Phosphate restriction significantly reduces mortality in uremic rats with established vascular calcification. Kidney Int 2013; 84: 1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennenberg R. J. M. W., Kessels AGH, Schurgers LJ, Engelshoven JMA, Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta analysis. Vasc Health Risk Manag 2009; 5: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto E, Taketani Y, Tanaka R, Harada N, Isshiki M, Sato M et al. Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol 2009; 20: 1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphorus levels associate with coronary atherosclerosis in young adults. J Am Soc Nephrol 2009; 20: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphorus and left ventricular hypertrophy in young adults: the coronary artery risk development in young adults study. J Am Soc Nephrol 2009b; 20: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsey PL, Alonso A, Michos ED, Loehr LR, Astor BC, Coresh J et al. Serum magnesium, phosphorus, and calcium are associated with risk of incident heart failure: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr 2014; 100: 756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer SC, Hayen A, Macaskill P, Pellegrini F, Craig JC, Elder GJ et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA 2011; 305: 1119–1127. [DOI] [PubMed] [Google Scholar]
- Alonso A, Nettleton JA, Ix JH, Boer IH, Folsom AR, Bidulescu A et al. Dietary phosphorus, blood pressure and incidence of hypertension in the Atherosclerosis Risk in Communities (ARIC) Study and the Multi-Ethnic Study of Atherosclerosis (MESA). Hypertension 2010; 55: 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M. A phosphate-centric paradigm for pathophysiology and therapy of chronic kidney disease. Kidney Int Suppl 2013; 3: 420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliviero F, Scanu A, Punzi L. Metabolism of crystals within the joint. Reumatismo 2011; 63: 221–229. [DOI] [PubMed] [Google Scholar]
- Ichikawa S, Gray AK, Padgett LR, Reilly AM, Unsicker TR. High dietary phosphate intake induces development of ectopic calcifications in a murine model of familial tumoral calcinosis. J Bone Miner Res 2014; 29: 2017–2023. [DOI] [PubMed] [Google Scholar]
- Worcester EM, Coe FL. Calcium kidney stones. N Engl J Med 2010; 363: 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto M, Imai T, Seki K, Nomura R, Otahara Y. The effect on the bones of condensed phosphate when used as food additives: its importance in relation to preventive medicine. Environ Health Prev Med 1997; 2: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B et al. Chronic kidney disease: global dimension and perspectives. Lancet 2013; 382: 260–272. [DOI] [PubMed] [Google Scholar]
- Razzaque MS. Phosphate toxicity and vascular mineralization. Contrib Nephrol 2013; 180: 74–85. [DOI] [PubMed] [Google Scholar]
- Uribarri J. Dietary phosphorus and kidney disease. Ann NY Acad Sci 2013; 1301: 11–19. [DOI] [PubMed] [Google Scholar]
- Razzaque MS. Does renal ageing affect survival? Ageing Res Rev 2007; 6: 211–222. [DOI] [PubMed] [Google Scholar]
- Stompor TP, Pasowicz M, Sulowicz W, Dembinska-Kiec A, Janda K, Wojcik K et al. Trends and dynamics of changes in calcification score over the 1-year observation period in patients on peritoneal dialysis. Am J Kidney Dis 2004; 44: 517–528. [PubMed] [Google Scholar]
- Jung HH, Kim SW, Han H. Inflammation, mineral metabolism and progressive coronary artery calcification in patients on haemodialysis. Nephrol Dial Transplant 2006; 21: 1915–1920. [DOI] [PubMed] [Google Scholar]
- O'Neill WC. Vascular calcification: not so crystal clear. Kidney Int 2007; 71: 282–283. [DOI] [PubMed] [Google Scholar]
- Slinin Y, Foley RN, Collins AJ. Calcium, phosphorus, parathyroid hormone, and cardiovascular disease in hemodialysis patients: the USRDS waves 1, 3, and 4 study. J Am Soc Nephrol 2005; 16: 1788–1793. [DOI] [PubMed] [Google Scholar]
- cancer.org. (2014c) The history of cancer..
- Jansa J, Finaly R, Wallander H, Smith FA, Smith SE. Role of mycorrhizal symbioses in phosphorus cycling. In:Phosphorus in Action: Biological Processes in Soil Phosphorus Cycling eds Bünemann EK, Oberson A, Frossard E 137–168Springer: Heidelberg, Germany, 2011;. [Google Scholar]
- Fenninger LD, Waterhouse C, Keutmann EH. The interrelationship of nitrogen and phosphorus in patients with certain neoplastic diseases. Cancer 1953; 5: 930–941. [DOI] [PubMed] [Google Scholar]
- Elser JJ, Kyle MM, Smith MS, Nagy JD. Biological stoichiometry in human cancer. PLoS One 2007; 2: e1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain LW, Rier JPJ. Tumorigenesis on mineral deficient tomato plants. Cancer Res 1968; 28: 2496–2501. [PubMed] [Google Scholar]
- Lin Y, McKinnon KE, Ha SW, Beck GRJ. Inorganic phosphate induces cancer cell mediated angiogenesis dependent on Forkhead Box Protein C2 (FOXC2) regulated osteopontin expression. Mol Carcinog (e-pub ahead of print 2 April 2014; doi: 10.1002/mc.22153). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Xu CX, Lim HT, Park SJ, Shin JY, Chung YS et al. High dietary inorganic phosphate increases lung tumorigenesis and alters Akt signaling. Am J Respir Crit Care Med 2009; 179: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez C, Fiedler D. Investigating the role of inorganic phosphate in tumor metabolism and metastasis. Cancer Metab 2014; 2: 55. [Google Scholar]
- Wilson KM, Ma J, Giovannucci E. Abstract B99: Calcium and phosphorus intake and risk of prostate cancer: A 22-year follow-up study. Cancer Prev Res 2011; 4: B99–B99. [Google Scholar]
- Schmitz R. China's toxic harvest: a ‘cancer village' rises in protest. In marketplace.org2013;.
- Abid Z, Cross AJ, Sinha R. Meat, dairy, and cancer. Am J Clin Nutr 2014; 100: 386S–393S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail EA, Al-Mutairi G, Al-Anzy H. A fatal small dose of phosphate enema in a young child with no renal or gastrointestinal abnormality. J Pediatr Gastroenterol Nutr 2000; 30: 220–221. [DOI] [PubMed] [Google Scholar]
- Perlman JM. Fatal hyperphosphatemia after oral phosphate overdose in a premature infant. Am J Health Syst Pharm 1997; 54: 2488–2490. [DOI] [PubMed] [Google Scholar]
- Martin RR, Lisehora GR, Braxton M Jr., Barcia PJ. Fatal poisoning from sodium phosphate enema. Case report and experimental study. JAMA 1987; 257: 2190–2192. [PubMed] [Google Scholar]
- Shutto Y, Shimada M, Kitajima M, Yamabe H, Saitoh Y, Saitoh H et al. Inadequate awareness among chronic kidney disease patients regarding food and drinks containing artificially added phosphate. PLoS ONE 2013; 8: e78660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutto Y, Shimada M, Kitajima M, Yamabe H, Razzaque MS. Lack of awareness among future medical professionals about the risk of consuming hidden phosphate-containing processed food and drinks. PLoS ONE 2011; 6: e29105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Joshi SR. Disorders of calcium, phosphorus and magnesium metabolism. J Assoc Physicians India 2008; 56: 613–621. [PubMed] [Google Scholar]
- Palmese S, Pezza M, De Robertis E. Hypophosphatemia and metabolic acidosis. Minerva Anestesiol 2005; 71: 237–242. [PubMed] [Google Scholar]
- Loh TP, Saw S, Sethi SK. Hyperphosphatemia in a 56-year-old man with hypochondrial pain. Clin Chem 2010; 56: 892–895. [DOI] [PubMed] [Google Scholar]