

Abstract
Spinal Muscular Atrophy (SMA) is an autosomal recessive neurodegenerative disease that is a result of a deletion or mutation of the SMN1 (Survival Motor Neuron) gene. A duplicated and nearly identical copy, SMN2, serves as a disease modifier as increasing SMN2 copy number decreases the severity of the disease. Currently many therapeutic approaches for SMA are being developed. Therapeutic strategies aim to modulate splicing of SMN2-derived transcripts, increase SMN2 gene expression, increase neuroprotection of motor neurons, stabilize the SMN protein, replace the SMN1 gene and reconstitute the motor neuron population. It is our goal to develop a pig animal model of SMA for the development and testing of therapeutics and evaluation of toxicology. In the development of a SMA pig model, it was important to demonstrate that the human SMN2 gene would splice appropriately as the model would be based on the presence of the human SMN2 transgene. In this manuscript, we show splicing of the human SMN1 and SMN2 mini-genes in porcine cells is consistent with splicing in human cells, and we report the first genetic knockout of a gene responsible for a neurodegenerative disease in a large animal model using gene targeting with single-stranded DNA and somatic cell nuclear transfer.
Keywords: Transgenic pigs, Spinal Muscular Atrophy (SMA), Neurodegenerative disease, Animal model, For gene therapy
Introduction
Spinal Muscular Atrophy (SMA) is the second most common autosomal recessive disorder in humans and is the leading genetic cause of infantile death; however, no cure currently exists. The carrier frequency is approximately 1:35 with an incidence of 1 in 6,000–10,000 live births. SMA is characterized by the progressive degeneration of spinal motor neurons and muscle atrophy. Due to the broad range in motor function, SMA is defined by five clinical types (Type O: severe/birth, Type I: severe; Type II: intermediate; Type III: adolescent/mild; Type IV: adult onset/mild) that are based upon age of disease onset, severity of the symptoms and motor ability (Wirth 2000; Elsheikh et al. 2009; Prior 2010; Prior et al. 2009).
The gene responsible for SMA is called Survival Motor Neuron-1 (SMN1). A nearly identical gene, SMN2 is present only in humans; however, mutations in SMN2 have no clinical consequence if SMN1 is retained. SMN2 cannot prevent disease development in the absence of SMN1 but SMN2 does modify disease severity. SMN1 and SMN2 are distinguished in sequence by a few non-polymorphic nucleotide differences that do not alter the coding capacity of SMN2. However, a single C to T transition results in the disruption of an exonic splice enhancer (ESE) in exon 7 of SMN2 (Lorson et al. 1999; Monani et al. 1999; Cartegni and Krainer 2002). As a result, the majority of SMN2 transcripts are alternatively spliced, resulting in a truncated protein that lacks the 16 amino acids derived from SMN exon 7 (Δ7); only approximately ten percent of SMN2 transcripts are full-length. SMNΔ7 protein is unstable and rapidly degraded. SMN1 alleles almost exclusively produce full-length SMN transcripts. All SMA patients retain one or more copies of SMN2 and there is an inverse correlation between SMA severity and SMN2 copy number. The relationship between disease severity and SMN2 is associated with the increase in full-length SMN produced by each additional SMN2 gene.
Several mouse models have been developed to study SMN function and SMA disease pathology. Deletion of Smn in mice, as in all mammals but humans, results in embryonic lethality; however, the lethality can be rescued by the introduction of the human SMN2 transgene (Schrank et al. 1997; Hsieh-Li et al. 2000; Monani et al. 2000). Increasing SMN2 copy number decreases the disease severity such that mice with two SMN2 copies have a severe and very progressive phenotype with death within 6 days while mice with eight copies of SMN2 show no disease phenotype. The addition of SMN Δ7, the major splice product of SMN2, in a Smn−/−;SMN2 background increases survival to approximately 13 days and demonstrates that the Δ7 product is not detrimental to survival (Le et al. 2005). Mice with a mild SMA phenotype and a lifespan of over 15 months were generated using the SMN1 (A2G) mutation in a Smn−/−;SMN2 background with a single copy of SMN2 (Monani et al. 2003). Recently, a new mouse model Smn−/−;SMN2 with three copies of SMN2 in a C57BL/6 N background produces a slightly less progressive disease phenotype with neuromuscular and breathing defects and a median survival of 14 days (Michaud et al. 2010). While SMA mouse models have proven quite valuable in our understanding of SMN function and SMA, mouse models of SMA have limitations in testing the efficacy of therapeutics. The rapid and progressive post-natal phenotype observed in Smn−/−;SMN2 mice make them in general too severe for therapeutic delivery. As a result, most therapeutic studies to date utilize the SMNΔ7 SMA mice. While the lifespan is slightly longer in SMNΔ7 mice, the translation of therapeutics towards clinical application has been difficult.
We have chosen to generate a pig animal model for SMA specifically for the purpose of evaluating the efficacy of SMA therapeutics and for studying their toxicology. We believe a pig SMA model will provide an animal model that closely models humans in size, physiology, metabolism, cardiac output, lung capacity, immune system and nervous system to identify the most efficacious therapeutics. It is anticipated that since pigs have many biological and physiological similarities to the developing human that a pig SMA model will more closely mimic the human condition and expand the therapeutic window. Therefore, the delivery of therapeutics and the analysis of potency, expression and outcome of therapeutics over a prolonged period of time can be studied.
Previous studies have shown that expression of pig SMN in cells and tissues is consistent with that observed in human and mouse cells. Pig SMN co-localizes with human SMN in the cytoplasm and to nuclear gems, and transient transfection of pig SMN1 into 3813 SMA type I fibroblasts rescues the deficiencies of total SMN protein and gem formation (Lorson et al. 2008). Pig SMN can also rescue deficiencies in snRNP assembly in SMA patient fibroblast extracts (unpublished data). Here we report the production of SMN+/− pigs using gene targeting of single-stranded SMN DNA and somatic cell nuclear transfer.
Materials and methods
RT–PCR assays
Subconfluent human HeLa or pig PK15 (kidney) cells were transfected with 5 μg of the human SMN1, human SMN2, or the human SMN1-ΔSE2 mini genes using PEI (poylethylenimine). Cells were harvested 24 h post-transfection. Cells were then subjected to a TRIzol (Invitrogen) extraction and RNA was isolated. First-strand cDNA synthesis was performed by using Super Script III (Invitrogen) at 42°C for 50 min followed by 70°C for 10 min. Two microliters of the first-strand synthesis was used in the subsequent PCR amplification [94°C for 5 min, (94°C for 45 s, 60°C for 1 min, 68°C for 2 min) × 30] using Vent exo-(Invitrogen). pCI-SMN1 and pCI-SMN2 cDNAs served as controls for full-length SMN and SMNΔ7 respectively. The plasmid-specific primers used were pCI Reverse 5′-agctcgtctgtactattctatgtaa and pCI Forward2 5′-cactataggctagcctcgagaat.
Targeting vector construction
Genomic DNA was isolated from pig fetal fibroblasts. An 8,640 bp PCR product encompassing SMN exons 2a through exon 6 was amplified using LA Taq (Takara). The PCR product was cloned into pCR-Topo TA (Invitrogen) and verified by sequencing. This plasmid is pML. The pML plasmid served as the template to generate the SMN recombination arms. pKW2 is a synthesized promoter trap vector with multiple cloning sites 5′ and 3′ of the loxP (locus of X-ing over) sites. Within the loxP sites is an IRES (internal ribosome entry site) sequence, neoR cassette and SV40 poly A. To build pML8, a 2,538 bp Acc65I/BsrGI restriction fragment was isolated and cloned into pKW4 to generate the 5′ recombination arm upstream of the neoR cassette. A 3,468 bp SacI/NotI restriction fragment was isolated and cloned into pKW4 to generate the 3′ recombination arm downstream of the neoR cassette. pKW4 is a synthesized vector containing a neoR cassette (based on mammalian codon usage) driven by the pgk (phosophoglycerate kinase) promoter. LoxP sites flank the pgk promoter and neoR cassette. The sequence junctions of pML8 were verified by sequencing. pML8 was linearized with PmeI for electroporation. To generate single-stranded DNA the linearized template was boiled for 5 min and placed on ice.
Electroporation and selection
Pig fetal fibroblasts were resuspended at a concentration of 1 × 106 cells/ml in electroporation media (25% optimem +75% cytosalts (120 mM KCl; 15 mM CaCl2; 10 mM KPO4; 5 mM MgCl2) (Ross et al. 2010). Four hundred μl of the cell suspension and linearized pML8 DNA were placed in an electroporation cuvette (4 mM) and the cells were electroporated using 490 volts × 3 pulses for 1 ms/ pulse. The cells were immediately plated to forty 100 mm tissue culture dishes. Thirty-six hours after plating cells were selected using G418 (400 mg/liter) for 11 days. Colonies were harvested using cloning cylinders and screened by PCR.
PCR screening
Cells were placed in 96-well PCR plates, spun, resuspended in 5 μl of embryo lysis buffer (ELB) (40 mM Tris, pH 8.9; 0.9% Triton X-100; 0.9% Nonidet P-40; 0.4 mg/ml proteinase K) and incubated at 65°C for 15 min and then 95°C for 10 min. For 5′ PCR analysis, fragments were amplified using LA Taq (Takara) and 1 μl of cell lysate using the following parameters: 30 cycles of 30 s at 94°C, 30 s at 60°C and 4 min increasing 10 s/cycle at 68°C with a final extension of 8 min at 72°C. The 5′ PCR primers were: 360L 5′-gcaggtaggcttcttgtggttt 6632R 5′-aagacctgct cctggcatagg. For 3′ PCR analysis, fragments were amplified using LA Taq (Takara) and 1 μl of cell lysate using the following parameters: 30 cycles of 15 s at 94 °C, 30 s at 60°C and 8 min increasing 10 s/cycle at 68°C with a final extension of 10 min at 72°C. The 3′ PCR primers were: 1610L 5′-ggcccgaagatatttccacagt 460R 5′-tgcagaacccatggatacagag. For LR-PCR analysis: LA Taq (Takara) and 1.25 μl of cell lysate using the following parameters: 30 cycles of 15 s at 94°C, 30 s at 59°C and 10 min increasing 5 s/cycle at 68°C with a final extension of 10 min at 72°C. The LR-PCR primers were: 360L 5′-gcaggtaggcttcttgtggttt 460R 5 ′-tgcagaacccatggatacagag.
Southern blot analysis
Piglet tail snips were digested in approximately 1 ml of genomic isolation buffer (50 mM Tris pH8, 100 mM EDTA, 100 mM NaCl, 1% SDS; 0.4 mg/ ml proteinase K) overnight at 55°C. Genomic DNA was isolated following organic extractions of phenol and chloroform and isopropanol precipitations. Ten μg of genomic DNA was digested with EcoRI and separated on a 0.8% agarose gel. Following electrophoresis the DNA was transferred to a nylon membrane. A 1,139 bp fragment (5′NcoI and 3′ NotI) containing the neoR cDNA from the KW4 plasmid served as the template to generate a 32P-αdATP random prime labeled probe. The membrane was prehybridized in Church’s Buffer (0.5 M NaP04, 7% SDS, 10 mM EDTA) at 65°C for 1 h. The membrane was hybridized overnight at 63°C with boiled probe and Church’s buffer. The membrane was washed successively with 10× SSC/1% SDS, 2× SSC/1% SDS, 0.5× SSC/1% SDS each for 20 min at 63°C and then exposed to film.
Nuclear transfer and embryo transfer
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Missouri. Oocytes were purchased from ART Inc. (Madison, Wisconsin). Oocytes were resuspended and matured in maturation medium and cultured for a total of 42–44 h at 38.5°C in 5% CO2 in air. Cumulus cells were removed from oocytes by gentle vortexing in 0.5 mg/ml hyaluronidase. Oocytes were resuspended in micromanipulation medium at 38.5°C. SCNT was performed in micro-manipulation medium supplemented with 7.5 μg/ml cytochalasin B. Metaphase II chromosomes and the polar bodies were aspirated by inserting a micropipette through the zona pellucida. Donor cells were transferred by inserting the pipette into the previously made hole in the zona pellucida and the donor cell was deposited under the zona pellucida. Cells were fused in a low Ca2+ solution (0.3 M mannitol, 0.1 mM CaCl2.2H2O, 0.1 mM MgCl2.6H2O, and 0.5 mM HEPES), activated with 200 μM thimerosal for 10 min in the dark and incubated with 8 mM DTT for 30 min (Machaty et al. 1997). Oocytes were then washed 3× with PZM3 for 30 min. Fused oocytes were cultured overnight in 500 nM Scriptaid and then washed 3× in PZM3 before being transferred into the recipient (Zhao et al. 2009). The embryonic cleavage rate was determined before transferring the reconstructed embryos into recipients. Recipients on the first day of estrus or the first day after standing estrus were used. Embryo transfer was performed surgically as previously described (Lai and Prather 2003). Recipients were checked for pregnancy by ultrasound. Cesarean section was performed to deliver the piglets on 115–119 days of gestation.
Quantitative RT–PCR
Fibroblast cells cultured from tail snips of piglets 3-3 and 3-4 were subjected to a TRIzol (Invitrogen) extraction and RNA was isolated. RNA was quantified and one microgram of RNA was used for first-strand cDNA synthesis using Super Script III (Invitrogen) and random primers as described by the manufacturer. One microliter of the first-strand synthesis was used in the subsequent PCR amplification. Sequence specific primers to exons 4 and 5 of SMN (left primer 5′-cctatgccaggagcaggtct and right primer 5′-agccagcatgacaggaagtg) and GAPDH (left primer 5′-actcactcttctacctttgatgct and right primer 5′-tgttgctgtagccaaattca) were used. Three independent first-strand cDNA synthesis reactions were performed for each, JR1 wildtype, piglet 3-3 and piglet 3-4. Each qRT–PCR was performed in triplicate. The PCR reaction was carried out using SYBR green PCR mix (ABI) and the ABI 7500 system. To determine the fold difference between JR1 wildtype and SMN+/− piglets the following formulas were calculated. The average of each first-strand cDNA synthesis reaction using SMN or GAPDH was calculated. The difference between GAPDH and SMN was determined for each reaction and then the average for all three RT reactions was determined for the JR1 wildtype and SMN+/− piglets. Transgenic minus control was calculated with JR1 wildtype at zero and then the fold difference determined. When calculating the fold difference between JR1 (one fold), piglet 3-3 (0.704 fold) and piglet 3-4 (0.996) the variation was not significant when factoring the deviation between piglets of 0.146 fold.
Results
Conserved splicing of the human SMN1 and SMN2 mini-genes in pig cells
Multiple SMA therapeutic approaches are based on increasing the production of full-length SMN protein from human SMN2. Therefore, to produce a successful transgenic pig model of SMA, it was necessary to demonstrate that human SMN1 and, more importantly, human SMN2 alternative splicing events are conserved in this animal model.
While SMN1 homologs exist in all vertebrate species, SMN2 is unique to humans. Since the human SMN2 gene will be an important transgene in the development of a pig SMA model, pre-mRNA splicing patterns from previously characterized human mini-genes were examined in the pig kidney cell line PK15 (Fig. 1). SMN1 and SMN2 mini-genes correspond to the human genomic region spanning exon 6 through exon 8. The SMN1-ΔSE2 mini-gene carries mutations in the essential splice enhancer SE2 (the hTra2β1 binding element) region and therefore exclusively produces SMNΔ7 transcripts (Lorson et al. 1999). Pig PK15 and human HeLa cells were transfected with human SMN1, SMN2 or SMN1 exon7ΔSE2 mini-genes. Transfected HeLa cells served as positive control for splicing of the mini-genes. The full-length SMN and SMNΔ7 cDNAs were used as PCR controls for full-length and exon 7 skipped products, respectively (Fig. 1). The RT–PCR analysis of the mini-genes expressed in PK15 cells and HeLa cells demonstrated similar splicing patterns for each cellular context. Cells transfected with the human SMN1 mini-gene produced 100% full-length SMN product (Fig. 1, lanes 4 and 7). The transfected human SMN2 mini-gene produced reduced full-length and high levels of the exon7-skipped product in HeLa and PK15 cells (Fig. 1, lanes 5 and 8); while the human SMN1 exon7ΔSE2 mini-gene produced a single exon7-skipped product (Fig. 1, lanes 6 and 9). Amplification of previously cloned SMN full-length and SMNΔ7 cDNAs produced the appropriately sized products (Fig. 1, lanes 2 and 3). These results demonstrate that the splicing patterns for the human mini-genes are similar in human and pig, providing evidence that the regulation of splicing and splicing factors are conserved and the construction of a porcine SMA model based upon a human SMN2 transgene would be feasible.
Fig. 1.
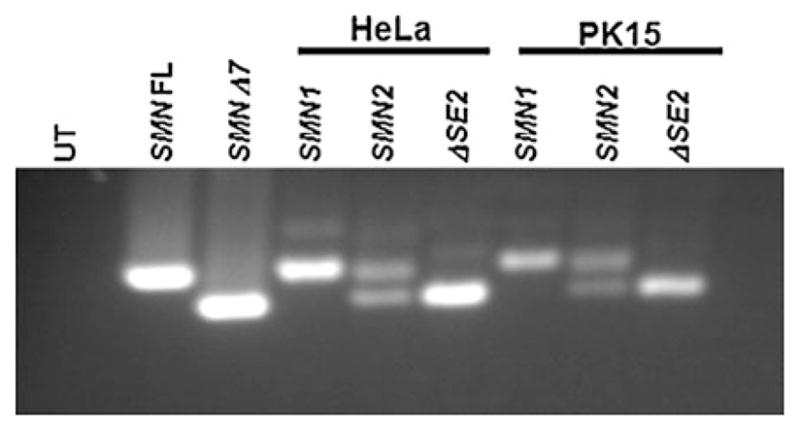
Splicing of the human SMN1 and SMN2 mini-genes. Pig kidney (PK15) and human HeLa cells were transfected with the SMN1, SMN2 or SMN1ΔSE2 mini-genes. The relative ratio of full-length to Δ7 SMN RNA was analyzed by RT-PCR. SMN1 and SMNΔ7 correspond to full-length and exon 7 skipped products, respectively (lanes 2 and 3). Cells transfected with the human SMN1 mini-gene produced exclusively full-length SMN product in HeLa and PK15 cells (lanes 4 and 7). The transfected human SMN2 mini-gene produced full-length and exon7-skipped products (lanes 5 and 8); while the human SMN1 exon7ΔSE2 mini-gene produced entirely exon7-skipped product (lanes 6 and 9) in human and pig cells
Development of vectors to target the SMN gene in pig fibroblasts
Due to the complex genetics surrounding SMA, the pig model of SMA will be accomplished in three steps. The first step, knockout of the SMN allele to produce SMN+/− pigs is reported here. The second step, addition of the human SMN2 transgene, is in progress. The third step is to produce SMN−/−;SMN2 animals through breeding or a second round of nuclear transfer.
We initially chose to target SMN using a promoter-trap vector via electroporation, as a promoter-trap strategy was used successfully in targeting the α-1,3-galactosyltransferase gene in pigs (Dai et al. 2002; Lai et al. 2002). A promoter-trap strategy is advantageous as it typically decreases the number of random integration events that give rise to G418-resistant colonies, thereby decreasing the number of false positive G418-resistant colonies. Previous studies demonstrated that SMN was robustly expressed in pig fetal fibroblast cells; therefore a SMN promoter-trap strategy was possible (Lorson et al. 2008).
We generated a SMN promoter trap vector, pML2, with a 5′ recombination arm encompassing exons 2a through 2b and a 3′ recombination arm including part of exon 4 through exon 6. Early passage KW6 and JR1 (Large White, male) fetal fibroblasts were chosen as primary cells based on colony-forming assays (unpublished data). Large White animals were chosen for primary cells due to the larger litter size, piglet weight at birth and that Large White is a maternal breed. Male primary cells were chosen as male pigs can more quickly establish a breeding colony as compared to females.
Fibroblasts were electroporated with linearized, double-stranded pML2 SMN DNA. Thirty-six hours following electroporation, cells were selected for 11 days using 0.4 mg/ml G418. G418-resistant colonies were harvested and cell lysates were screened by PCR using a 3-step process (screening the 5′ and then the 3′ recombination arms and then confirming with PCR of the entire targeted region) to reduce the number of false-positive PCR results. In two different fetal fibroblast primary cells (KW6 and JR1) we screened a total of 1,000 G418-resistant colonies and did not identify a SMN targeted clone. We postulated that the strength of the SMN promoter might not be sufficient to provide significant levels of G418 resistance; therefore, we developed a new SMN targeting vector, pML8, which contains the pgk promoter driving the neoR cDNA and flanked by 5′ and 3′ SMN targeting arms different than those in pML2 (Fig. 2a). In this vector, the neo selection cassette is flanked by loxP sites. Homologous recombination of the pML8 SMN targeting sequence with the endogenous SMN allele results in a 3,467 bp deletion of SMN exons 2b, 3 and part of exon 4 with the insertion of the neoR cDNA between exons 2b and 4. The loss of the Tudor domain, required for U snRNP assembly and Sm protein binding, and the significant deletion and disruption of the SMN reading frame is predicted to result in functional inactivation of the SMN gene.
Fig. 2.

Disruption of the pig SMN locus by gene targeting. a Diagram of the pig SMN locus, the corresponding pig SMN genomic sequence used to construct the 5′ and 3′ recombination arms for the pML8 targeting vector and the targeted locus following homologous recombination with the pML8 targeting sequence. The primers 360L and 6632R were used to screen for recombination of the 5′ arm. The primers 1610L and 460R were used to screen for recombination of the 3′ arm. The primers 360L and 460R were used for long range PCR. Primers 360L and 635neoR and 764 neoL and 460R were used to screen for the presence of neo. b PCR of pig fetal fibroblast cells from colony X8D2. Lanes 1, 8 and 15 are 1 kb DNA ladders. For each primer pair, the templates are no template control, JR1 wildtype and X8D2 pig cell lysates respectively. Lanes 2–4 are PCRs using primers 360L and 6632R. Lanes 5–7 are PCRs using primers 360L and 635neoR. Lanes 9–11 are PCRs using primers 1610L and 460R. Lanes 12–14 are PCRs using primers 764 neoL and 460R. Lanes 16–18 are PCRs using primer pairs 360L and 460R
Disruption of the pig SMN gene by gene targeting using single-stranded SMN DNA
Initially JR1 primary cells were electroporated with linearized, double-stranded pML8 DNA. However, after screening more than 500 colonies we did not successfully confirm targeting of the SMN locus. We hypothesized and tested whether electroporating single-stranded, linearized pML8 DNA might facilitate homologous recombination and therefore targeting of the SMN locus.
G418-resistant colonies were harvested and lysates were screened using the 3-step PCR process. We expected that colonies would be heterozygous for the SMN locus with one wild-type allele (endogenous) and one disrupted (targeted) SMN allele. All lysates were initially screened using primers (360L-6632R) located outside of the 5′ recombination arm (Fig. 2a). These primers amplify a 5.6 kb endogenous, wildtype SMN allele and a smaller 4.8 kb targeted SMN allele. All positive lysates were then PCR screened using primers (1610L-460R) located outside of the 3′ recombination arm (Fig. 2a). These primers amplify a 7.5 kb endogenous SMN allele and a 6.7 kb targeted SMN allele. Lysates positive for targeting both the 5′ and 3′ recombination arms were then confirmed using the long-range PCR (LR-PCR) (Fig. 2a). The LR-PCR amplifies the entire targeted region using primers (360L-460R) outside of the 5′ and 3′ recombination arms. Targeting of the SMN locus using LR-PCR is expected to result in a 9.4 kb endogenous product and an 8.6 kb targeted product. Using primers outside of the recombination arms for each PCR was advantageous, as amplification of the endogenous allele served as an internal control for every cell lysate. Additional PCRs using primers (360L-635R and 764L-460R) outside of the 5′ or 3′ recombination arms and within neo were used to confirm SMN targeting. These PCRs generate single amplimers from the targeted SMN allele (Fig. 2a).
Three clones (X8C5, X8D2, Z3A4) were identified and confirmed as positive for SMN targeting using primer pairs to the 5′ and 3′ SMN targeting arms as well as primers for the LR-PCR and primers to the 5′ and 3′ recombination arms in combination with neo primers (Fig. 2b and not shown). The resulting recombination targeting efficiency was 0.9% using linearized, single-stranded pML8 SMN DNA. The average targeting efficiency in pigs using electroporation is approximately one to two percent.
Cells from the three positive clones were expanded and frozen. Early passage SMN+/− fetal fibroblast cells from clones X8C5, X8D2 and Z3A4 were used for somatic cell nuclear transfer (SCNT). We transferred between 120 and 250 SCNT embryos to each of the 16 recipient females. Six of the recipients of embryo transfer became pregnant. Four of those pregnancies were from clone X8D2 and two were from Z3A4. We did not obtain a pregnancy from X8C5 cells following two embryo transfers. Of those pregnancies, two pregnancies developed to term (~116 days of gestation). Three pregnancies were used for fetal collection and one pregnancy was lost. The first SMN+/− piglets, from clone Z3A4, were born on July 19, 2010 (Fig. 3). Of the six piglets born, three piglets survived PND (post natal day) 1. One piglet was undersized and died from acute respiratory distress. Two other piglets were developmentally deficient and were euthanized. The three surviving SMN+/− piglets, referred to as 3-2, 3-3 and 3-4 ranged in weight from 0.9 kg to 1.33 kg, grew normally and were weaned. This type of survival rate is not unusual in the generation of clones and does not raise any concerns regarding the targeting event related to this specific clone.
Fig. 3.
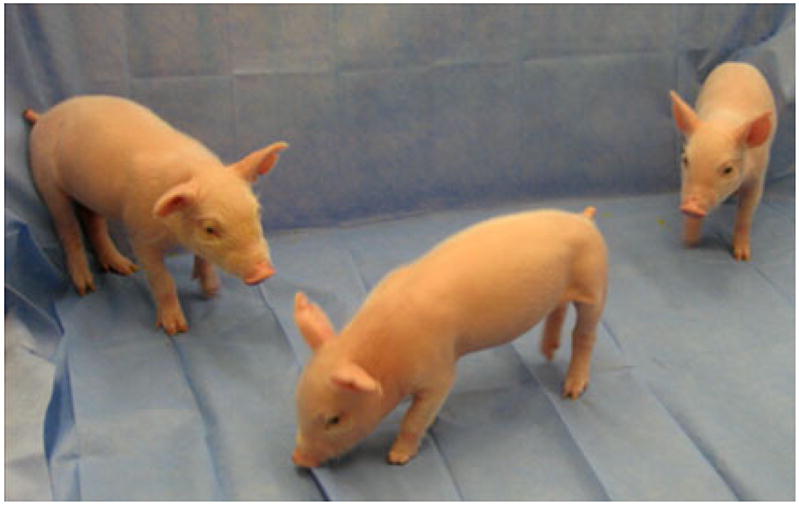
SMN gene knockout piglets at 10 days of age. The first SMN+/− piglets, from clone Z3A4, were born July, 2010. Of the six piglets born, three piglets survived PND (post natal day) 1. The three surviving SMN+/− piglets are 3-2, 3-3 and 3-4
SMN+/− offspring
To demonstrate that the Z3A4 piglets were heterozygous at the SMN locus we performed PCR using DNA isolated from tail snips using the same primers used to identify the original targeted clones. PCR analysis demonstrates that all six of the offspring delivered on July 19, 2010, were heterozygous for the SMN locus with one endogenous wild-type and one targeted allele (Fig. 4).
Fig. 4.
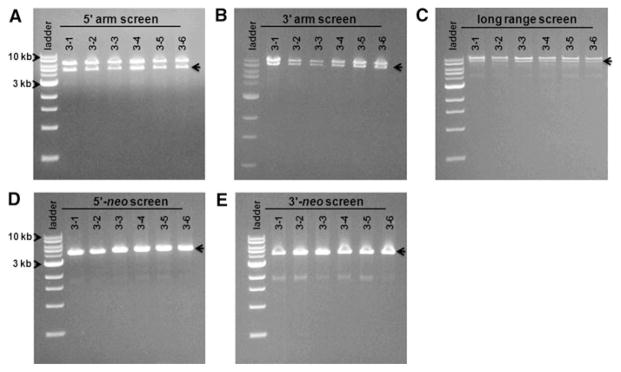
Piglets generated from colony Z3A4 are heterozygous at the SMN locus. DNA isolated from tail snips from piglets 3-1 to 3-6 was used for PCR with the same primers as in Fig. 2. Lane 1 for each set is a 1 kb DNA ladder. a PCR demonstrating recombination at the 5′ arm using primers 360L-6632R. A larger 5.6 kb wildtype allele and a smaller 4.8 kb targeted allele were amplified. b Recombination at the 3′ arm using primers 1610L-460R. A 7.5 kb wildtype allele and a 6.7 kb targeted allele were amplified. c LR-PCR amplification of the SMN targeted region. Wildtype allele is 9.4 kb while the SMN targeted allele is 8.6 kb. d, e Amplification of SMN targeted alleles using primers to 5′ and 3′ recombination arms and neo, respectively. Negative controls for the primer pairs are in Fig. 2 and not shown. The arrow indicates the targeted amplimer
Genomic DNA was isolated from tail snips of the SMN+/− offspring and used for Southern blot analysis. In an attempt to optimize conditions for Southern blot analysis we generated multiple probes but due to the large regions of highly repetitive sequence in SMN we did not obtain a SMN probe that worked well for Southern blot analysis. As a result, we used sequences specific for neo as a probe. Genomic DNA was isolated from tail snips from each piglet and digested with EcoRI. A 1.9 kb EcoRI band is expected for piglets with a disrupted SMN locus due to targeted insertion of the pgk neo cassette (Fig. 5a). No bands should be produced from non-targeted, wild-type DNA (JR1). All piglets confirmed as SMN targeted by PCR analysis were also confirmed positive for the insertion of neo by Southern blot analysis (Fig. 5b). The slightly upshifted band for piglet 3-2 is a result of a slight electrophoresis irregularity on this gel.
Fig. 5.
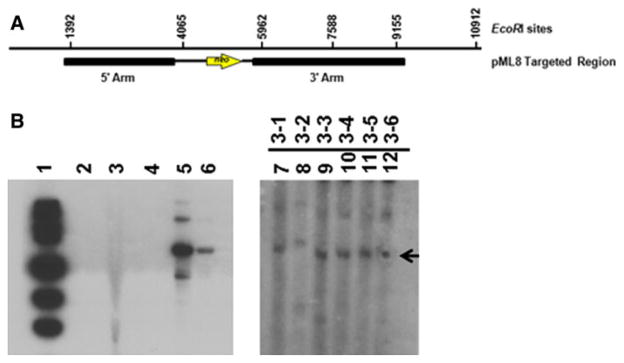
Southern blot analysis of SMN+/− piglets. a Diagram depicting the EcoRI sites within the SMN targeted sequence with the pML8 targeted genomic region as reference. b Genomic DNA was isolated from tail snips of piglets 3-1 to 3-6 and digested with EcoRI. Lane 1 is the 1 kb DNA ladder. Lane 2 is no DNA control. Lane 3 is wildtype (SMN+/+) JR1 genomic DNA digested with EcoRI. Lane 4 is a blank lane. Lanes 5 and 6 are a BlpI digested control plasmid containing neo with 10 or 1 molecules of DNA, respectively. Lanes 7–12 contain genomic DNA from piglets 3-1 to 3-6 digested with EcoRI. A 1,139 bp NcoI/NotIDNA fragment isolated from the KW4 plasmid that contains the neoR cDNA served as a probe. Exposure for lanes 1–6 was 1 day. Exposure for lanes 7–12 was 6 days
To determine whether there was a discernable difference in SMN mRNA production in the SMN+/−pigs as compared to wild-type pigs, we used quantitative RT–PCR. We did not detect a significant variation in SMN RNA abundance between wildtype JR1 and two SMN+/− animals (3-3 and 3-4) (Fig. 6). When comparing SMN RNA between wildtype (100%), piglet 3-3 (85%) and piglet 3-4 (100%) the percent difference was not substantial when factoring in the deviation between the SMN+/− piglets.
Fig. 6.
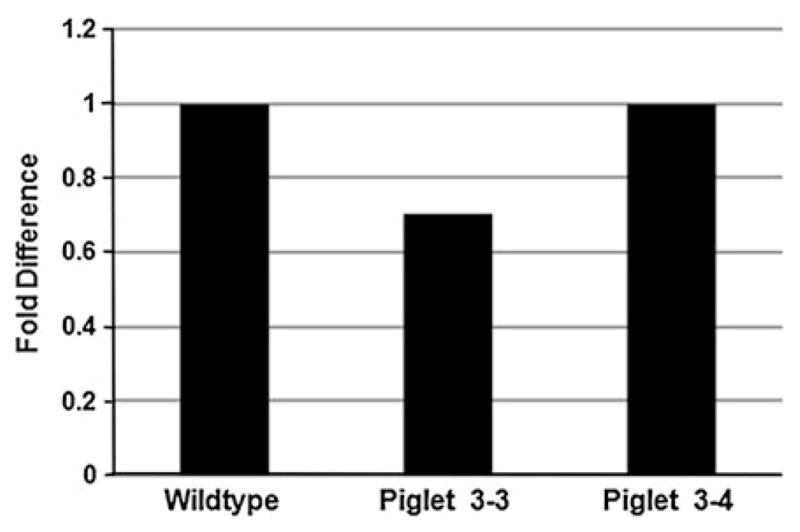
SMN RNA expression in SMN+/− piglets. Total RNA was isolated from tail fibroblast cells cultured from JR1 (SMN+/+ cells) and piglets 3-3 and 3-4. SMN RNA was quantified by qRT-PCR in wildtype and SMN+/− cells
Discussion
Our objective is to generate a large animal model of SMA purposely for evaluating the efficacy of therapeutics. We anticipate that a pig SMA model will more closely recapitulate SMA disease progression in humans, expand the window for therapeutic delivery and allow the analysis of delivery, potency and expression, in order to more efficiently translate bench to bedside.
While a cure for SMA does not currently exist, a number of factors strongly suggest that SMA is well positioned for translational success: (1) The gene deleted in SMA, SMN1, has been identified. Mutations and deletions in SMN1 account for nearly all cases of SMA. (2) The nearly identical copy gene, SMN2, is present and functions as a disease modifier. (3) Diagnostic screens are available and are being improved for newborn screening. (4) Advancements in drug development, gene therapy and SMN2 modulation are progressing toward clinical application (Lorson et al. 2010; MacKenzie 2010; Stavarachi et al. 2010). Reports indicate that compounds can increase either total transcription from SMN2 and/or exon 7 inclusion (Andreassi et al. 2001; Chang et al. 2001; Zhang et al. 2001; Brichta et al. 2003; Sumner et al. 2003; Andreassi et al. 2004; Wolstencroft et al. 2005; Mattis et al. 2006; Thurmond et al. 2008; Hastings et al. 2009; Mattis et al. 2009a, b; Butchbach et al. 2010; Riessland et al. 2010). Modulation of SMN2 splicing patterns using nucleic acid based therapeutics has shown to increase full-length SMN (Lim and Hertel 2001; Madocsai et al. 2005; Baughan et al. 2006; Coady et al. 2008; Baughan et al. 2009; Geib and Hertel 2009; Singh et al. 2009; Coady and Lorson 2010; Lorson et al. 2010; Hau et al. 2008, 2010). Recent gene replacement studies have demonstrated that delivery of AAV8SMN or AAV9SMN into SMA mice resulted in improvement in muscle function and lifespan (Foust et al. 2010; Passini et al. 2010; Valori et al. 2010; Dominguez et al. 2011). A large animal model of SMA to test these translational approaches would be a very powerful tool towards clinical application. To this end, we report the targeted disruption of the SMN gene in pigs.
We have demonstrated through SMN2 splicing assays that a SMA pig model based on the SMN2 transgene is feasible and that the therapeutic benefit of compounds and small molecules based on exon 7 inclusion and total full-length SMN should be readily assayed using traditional methods. Splicing of SMN1 and SMN2 appears to be highly conserved as similar ratios of full-length and exon 7 skipped products are produced in a variety of experimental contexts such as mice, rats, zebrafish and now swine.
Towards a porcine model of SMA we have generated three SMN+/− knockout fetal fibroblast clones, X8C5, X8D2 and Z3A4 by homologous recombination of a SMN targeting vector. We found that homologous recombination of the pML8 targeting construct was greatly enhanced by electroporation of single-stranded, linearized DNA as compared to double-stranded, linearized DNA from the same construct. One potential explanation is that homologous recombination is more efficient when providing a single-stranded DNA. We are investigating whether providing a single-stranded DNA fragment with homologous sequence on both ends will increase SMN gene targeting.
Nuclear transfer of SMN targeted clone Z3A4 produced three healthy SMN+/− piglets demonstrating that like their human counterparts SMN+/− pigs are phenotypically normal. PCR and Southern blot analysis have confirmed that we have targeted the SMN locus. While we did not detect a significant reduction in SMN RNA between wildtype and SMN+/− animals, these results are consistent with the levels of SMN RNA being tightly regulated through an autoregulatory feedback mechanism and in keeping with the housekeeping role of SMN in snRNP assembly. Loss of one SMN allele is not detrimental as upregulation in expression of the second SMN allele can maintain cellular activities. Importantly, these studies remain consistent with knockout of a single SMN allele.
An additional nuclear transfer from clone Z3A4 has resulted in a pregnancy and, on November 1, 2010, the birth of six healthy and phenotypically normal SMN+/− piglets. All six piglets are healthy to date. SMN+/− animals will be grown to sexual maturity and outcrossed to wild-type females to generate SMN+/− animals for breeding.
Our goal is to develop SMA pigs to specifically address the need for a large SMA animal model for therapeutic analysis. We have completed the first step towards this goal in generating SMN+/− pigs. We are currently using the SMN+/− fetal fibroblast cells to introduce the human SMN2 transgene, step two, towards generating SMA SMN−/−; SMN2 pigs.
Acknowledgments
Thank you to members of the C. Lorson, K. Wells and R. Prather laboratories for their assistance and generosity especially Martha Bennett, Chad O’Gorman, Erik Osman, Armedia Stump, Hans Rindt, August Rieke and Jason Dowell. This work was supported by FightSMA and National Institutes of Health [grant number NS059510 to MAL].
Footnotes
Conflict of interest. The authors declare that they have no competing financial interests.
Contributor Information
Monique A. Lorson, Department of Veterinary Pathobiology, Bond Life Sciences Center, University of Missouri, 1201 Rollins Road, Room 440C, Columbia, MO 65211-7310, USA
Lee D. Spate, Division of Animal Science, Animal Science Research Center, University of Missouri, 920 East Campus Drive, Columbia, MO 65211, USA
Melissa S. Samuel, Division of Animal Science, Animal Science Research Center, University of Missouri, 920 East Campus Drive, Columbia, MO 65211, USA
Clifton N. Murphy, Division of Animal Science, Animal Science Research Center, University of Missouri, 920 East Campus Drive, Columbia, MO 65211, USA
Christian L. Lorson, Email: lorsonm@missouri.edu, Department of Veterinary Pathobiology, Bond Life Sciences Center, University of Missouri, 1201 Rollins Road, Room 440C, Columbia, MO 65211-7310, USA. Department of Molecular Microbiology and Immunology, University of Missouri, Columbia, MO 65211, USA. Department of Veterinary Pathobiology, Bond Life Sciences Center, University of Missouri, 1201 Rollins Road, Columbia, MO 65211-7310, USA
Randall S. Prather, Division of Animal Science, Animal Science Research Center, University of Missouri, Columbia, MO 65211, USA
Kevin D. Wells, Division of Animal Science, Animal Science Research Center, University of Missouri, Columbia, MO 65211, USA
References
- Andreassi C, Jarecki J, Zhou J, Coovert DD, Monani UR, Chen X, Whitney M, Pollok B, Zhang M, Androphy E, et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10:2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12:59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- Baughan T, Shababi M, Coady TH, Dickson AM, Tullis GE, Lorson CL. Stimulating full-length SMN2 expression by delivering bifunctional RNAs via a viral vector. Mol Ther. 2006;14:54–62. doi: 10.1016/j.ymthe.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- Butchbach ME, Singh J, Thorsteinsdottir M, Saieva L, Slominski E, Thurmond J, Andresson T, Zhang J, Edwards JD, Simard LR, et al. Effects of 2, 4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA. 2001;98:9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coady TH, Lorson CL. Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J Neurosci. 2010;30:126–130. doi: 10.1523/JNEUROSCI.4489-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coady TH, Baughan TD, Shababi M, Passini MA, Lorson CL. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS One. 2008;3:e3468. doi: 10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Vaught TD, Boone J, Chen SH, Phelps CJ, Ball S, Monahan JA, Jobst PM, McCreath KJ, Lamborn AE, et al. Targeted disruption of the alpha 1, 3-galactosyl-transferase gene in cloned pigs. Nat Biotechnol. 2002;20:251–255. doi: 10.1038/nbt0302-251. [DOI] [PubMed] [Google Scholar]
- Dominguez E, Marais T, Chatauret N, Benkhelifa-Ziyyat S, Duque S, Ravassard P, Carcenac R, Astord S, de Moura AP, Voit T, Barkats M. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- Elsheikh B, Prior T, Zhang X, Miller R, Kolb SJ, Moore D, Bradley W, Barohn R, Bryan W, Gelinas D, et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve. 2009;40:652–656. doi: 10.1002/mus.21350. [DOI] [PubMed] [Google Scholar]
- Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Geib T, Hertel KJ. Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS One. 2009;4:e8204. doi: 10.1371/journal.pone.0008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings ML, Berniac J, Liu YH, Abato P, Jodelka FM, Barthel L, Kumar S, Dudley C, Nelson M, Larson K, et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci Transl Med. 2009;1:5ra12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Hua Y, Vickers T, Okunola H, Bennett C, Krainer A. Antisense masking of an hnRNP A1/A2 intronic splice silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahaski K, Hung G, Rigo F, Passini M, Bennett C, Krainer A. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Prather RS. Creating genetically modified pigs by using nuclear transfer. Reprod Biol Endocrinol. 2003;1:82. doi: 10.1186/1477-7827-1-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Kolber-Simonds D, Park KW, Cheong HT, Greenstein JL, Im GS, Samuel M, Bonk A, Rieke A, Day BN, et al. Production of alpha-1, 3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science. 2002;295:1089–1092. doi: 10.1126/science.1068228. [DOI] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J Biol Chem. 2001;276:45476–45483. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson MA, Spate LD, Prather RS, Lorson CL. Identification and characterization of the porcine (Sus scrofa) survival motor neuron (SMN1) gene: an animal model for therapeutic studies. Dev Dyn. 2008;237:2268–2278. doi: 10.1002/dvdy.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19:111–118. doi: 10.1093/hmg/ddq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaty Z, Wang WH, Day BN, Prather RS. Complete activation of porcine oocytes induced by the sulfhydryl reagent, thimerosal. Biol Reprod. 1997;57:1123–1127. doi: 10.1095/biolreprod57.5.1123. [DOI] [PubMed] [Google Scholar]
- MacKenzie A. Genetic therapy for spinal muscular atrophy. Nat Biotechnol. 2010;28:235–237. doi: 10.1038/nbt0310-235. [DOI] [PubMed] [Google Scholar]
- Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 Pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12:1013–1022. doi: 10.1016/j.ymthe.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Mattis VB, Rai R, Wang J, Chang CW, Coady T, Lorson CL. Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum Genet. 2006;120:589–601. doi: 10.1007/s00439-006-0245-7. [DOI] [PubMed] [Google Scholar]
- Mattis VB, Ebert AD, Fosso MY, Chang CW, Lorson CL. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum Mol Genet. 2009a;18:3906–3913. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis VB, Fosso MY, Chang CW, Lorson CL. Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA. BMC Neurosci. 2009b;10:142. doi: 10.1186/1471-2202-10-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud M, Arnoux T, Bielli S, Durand E, Rotrou Y, Jablonka S, Robert F, Giraudon-Paoli M, Riessland M, Mattei MG, et al. Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol Dis. 2010;38:125–135. doi: 10.1016/j.nbd.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Bu J, Roskelley EM, Richards AM, Sardi SP, O’Riordan CR, Klinger KW, Shihabuddin LS, Cheng SH. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior TW. Perspectives and diagnostic considerations in spinal muscular atrophy. Genet Med. 2010;12:145–152. doi: 10.1097/GIM.0b013e3181c5e713. [DOI] [PubMed] [Google Scholar]
- Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgman SJ, Burghes AH, Kissel JT. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85:408–413. doi: 10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riessland M, Ackermann B, Forster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- Ross JW, Whyte JJ, Zhao J, Samuel M, Wells KD, Prather RS. Optimization of square-wave electroporation for transfection of porcine fetal fibroblasts. Transgenic Res. 2010;19:611–620. doi: 10.1007/s11248-009-9345-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrank B, Gotz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NN, Shishimorova M, Cao LC, Gangwani L, Singh RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6:341–350. doi: 10.4161/rna.6.3.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavarachi M, Apostol P, Toma M, Cimponeriu D, Gavrila L. Spinal muscular atrophy disease: a literature review for therapeutic strategies. J Med Life. 2010;3:3–9. [PMC free article] [PubMed] [Google Scholar]
- Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AH, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- Thurmond J, Butchbach ME, Palomo M, Pease B, Rao M, Bedell L, Keyvan M, Pai G, Mishra R, Haraldsson M, et al. Synthesis and biological evaluation of novel 2, 4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J Med Chem. 2008;51:449–469. doi: 10.1021/jm061475p. [DOI] [PubMed] [Google Scholar]
- Valori CF, Ning K, Wyles M, Mead RJ, Grierson AJ, Shaw PJ, Azzouz M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2:35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL. A non-sequence-specific requirement for SMN protein activity: the role of aminoglycosides in inducing elevated SMN protein levels. Hum Mol Genet. 2005;14:1199–1210. doi: 10.1093/hmg/ddi131. [DOI] [PubMed] [Google Scholar]
- Zhang ML, Lorson CL, Androphy EJ, Zhou J. An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: potential therapy of SMA. Gene Ther. 2001;8:1532–1538. doi: 10.1038/sj.gt.3301550. [DOI] [PubMed] [Google Scholar]
- Zhao J, Ross JW, Hao Y, Spate LD, Walters EM, Samuel MS, Rieke A, Murphy CN, Prather RS. Significant improvement in cloning efficiency of an inbred miniature pig by histone deacetylase inhibitor treatment after somatic cell nuclear transfer. Biol Reprod. 2009;81:525–530. doi: 10.1095/biolreprod.109.077016. [DOI] [PMC free article] [PubMed] [Google Scholar]