

Abstract
Since researches on the fate of highly excited triplet states demonstrated the existence of reverse intersystem crossing (RISC) from upper triplet levels to singlet manifold in naphthalene, quinoline, isoquinoline, etc. in the 1960s, this unique photophysical process was then found and identified in some other aromatic materials. However, the early investigations mainly focus on exploring the mechanism of this photophysical process; no incorporation of specific application was implemented. Until recently, our group innovatively used this ‘sleeping’ photophysical process to enhance the efficiency of fluorescent organic light-emitting diodes by simultaneously harvesting singlet and triplet excitons. Efforts are devoted to developing materials with high photoluminescence efficiency and effective RISC through appropriate molecular design in a series of donor–acceptor material systems. The experimental and theoretical results indicate that these materials exhibit hybridized local and charge-transfer excited state, which achieve a combination of the high radiation from local excited state and the high Tm→Sn (m≥2, n≥1) conversion along charge-transfer excited state. As expected, the devices exhibited favourable external quantum efficiency and low roll-off, and especially an exciton utilization efficiency exceeding the limit of 25%. Considering the significant progress made in organic light-emitting diodes with this photophysical process, we review the relevant mechanism and material systems, as well as our design principle in materials and device application.
Keywords: reverse intersystem crossing, organic light-emitting diodes, exciton statistics, hybridized local and charge-transfer state, hot exciton
1. Introduction
Reverse intersystem crossing (RISC) that is defined as a process of energy transfer from the excited triplet state back to the singlet is an important photophysical process [1]. Materials with efficient RISC could be used to increase the efficiency of fluorescent organic light-emitting diodes (FOLEDs) by harvesting singlet and triplet excitons simultaneously. Generally, this RISC occurs primarily from the lowest excited triplet state (T1) to the lowest excited singlet state (S1) of a molecule according to Kasha's rule [2]. Well-known examples of such RISC include the compounds with E- and P-type delayed fluorescence properties. E-type molecules usually possess a much smaller energy gap between S1 and T1, which allows the conversion of T1 excitons into the radiative S1 excitons through the thermally activated RISC (also called thermally activated delayed fluorescence, TADF) [3]. As for the P-type delayed fluorescence, the activation energy is provided by triplet–triplet annihilation (TTA), and therefore it does not require the small energy gap between S1 and T1 [4].
In some cases, RISC can also occur from upper triplet levels where intersystem crossing (ISC) to the singlet manifold competes with direct internal conversion (IC) to T1 [5]. Previous photophysical researches in some organic compounds have revealed and identified the existence of this photophysical process by the two-step two-colour laser technique [6]. This unique photophysical process should be also used in increasing the efficiencies of FOLEDs similar to the TADF and TTA materials. Here, we define the RISC process involving the low-lying excited states (T1→S1) as a ‘cold exciton’ process (for example, TADF and TTA mechanism), whereas the process involving high-lying excited states (Tm→Sn, m≥2, n≥1) is defined as a ‘hot exciton’ process. And ‘hot exciton’ process for FOLEDs may show some advantages over ‘cold exciton’. For example, TADF and TTA inevitably suffer from the problem of triplet–triplet or singlet–triplet quenching due to the accumulated T1 excitons (the low RISC rate); while the RISC from ‘hot exciton’ process can quickly transfer triplet in high levels to singlet states and avoid the accumulation of triplet exciton in T1 states. Unfortunately, the reported materials with RISC from ‘hot exciton’ process usually show low photoluminescence (PL) efficiencies and are difficult to be used as emitters for FOLEDs. Recently, our group found that some donor (D) and acceptor (A) constructed materials also showed RISC from hot exciton path [7,8]. These materials demonstrated a hybridized local and charge-transfer (HLCT) state, resulting in high PL efficiencies and effective RISC from upper triplet levels to singlet. As expected, the breakthrough of exciton statistics limit (25%) is confirmed by the high exciton utilization efficiency of the FOLEDs based on these materials. Due to the important application of RISC from upper triplet levels in FOLEDs, in this review article, we summarized the basic mechanism of RISC from upper triplet levels, the material systems with this photophysical process, as well as our new design principles of materials that combine high PL efficiencies together with effective RISC from upper triplet levels to singlet.
2. Early investigation and mechanism of reverse intersystem crossing from upper triplet levels
Since the electronic transitions from excited triplet states to higher excited triplet states in aromatic molecules were first observed and identified by Lewis and Lipkin in 1942 [9], interest in the fate of these highly excited triplet states has been widely expressed by researchers [10]. One possible path for the decay of these highly excited triplet states was predicted to be RISC into the singlet manifold and IC to the lowest excited singlet state. Until 1969, this presumable excited triplet–singlet ISC was experimentally confirmed by Keller based on the observation of weak fluorescence emission in several aromatic molecules, such as naphthalene, quinoline, isoquinoline, etc. after photo-excitation of metastable T1 [5]. Here, this weak fluorescence emission is identical to their continuous-wave fluorescence spectra, an indication of radiation decay from S1. However, because Keller's method is excited with a steady-state excitation source, it is difficult to obtain detailed information about the mechanisms of this photophysical process.
Due to the development of two-step two-colour laser technique, more and more organic materials are found showing such fluorescence when they are excited by a first laser with short wavelength and then probed by a second excitation of low-energy laser at a certain delayed time after the first laser pulse [11]. It should be emphasized that the excitation wavelength of the first laser in the experiment is in resonance with the S0→S1 transition, whereas the excitation wavelength of the second laser (probe pulse) is only resonant with the T1→Tm transition. This pronounced emission was only detected in two-colour, two-pulse experiments, namely two-step laser-induced fluorescence (TSLIF), and it did not occur when the second low-energy laser pulse was alone incident on the sample. These TSLIFs are further evidenced to originate from the RISC from upper triplet levels to singlet levels instead of TADF and TTA mechanisms by the following experimental results [6]. First, the energy gap between S1 and T1 was estimated to be very large in most of these compounds; thus, it is difficult to achieve the thermally activated RISC. Second, the intensity of this delayed fluorescence depends linearly on the population of the triplet state, instead of the square characteristic of this population in TTA process. Third, no emission was observed if only the first excitation pulse is applied (detection between pulses), which is contrasted with TADF and TTA. Fourth, the intensity of this delayed fluorescence increases with the probe pulse energy. Therefore, the previously speculated RISC from upper triplet levels to excited singlet states, Tm→Sn→S1 ISC (m≥2, n≥1), should be responsible for this kind of delayed fluorescence. A schematic description of this excited state process is shown in figure 1. After excitation by the first laser pulse, the ISC causes the conversion from the singlet states to the triplet states and the consecutive relaxation processes lead to a significant population of the lowest triplet state T1 [2]. By irradiation with the probe pulse for a delayed time less than the lifetime of the triplet state T1, a certain fraction of the triplet population is promoted to a higher triplet state, Tm. Due to the cooperation of large T1–T2 energy gap, energetic closeness between Tm and Sn, heavy atom effect, etc., RISC from the higher triplet states into the singlet manifold can successfully compete with the IC leading back to T1. Therefore, delayed fluorescence was consequently observed from the radiative decay of the lowest singlet state formed via RISC.
Figure 1.

Energy level diagram for RISC from upper triplet levels to singlet levels. (Online version in colour.)
3. Previous material systems with reverse intersystem crossing from upper triplet states
In the past four decades, several types of organic materials with RISC from upper triplet levels have been reported, such as anthracene derivatives [12], organic dyes [13], excited-state intramolecular proton transfer (ESIPT) materials [14–16], etc. The chemical structures of some representative materials are shown in figure 2.
Figure 2.

Chemical structures of organic compounds with RISC from upper triplet levels to singlet levels.
Anthracenes are representative examples of molecules showing TSLIFs [17]. Early researches on meso-substituted anthracenes found that their fluorescence yields increase with decreasing temperature and no triplet formation can be detected at 77 K [18,19]. These facts are explained by thermally activated ISC from S1 to T2. Out of interest in this ISC from S1 to T2, Kobayashi et al. [20] found that the RISC from T2 to S1 also occurred when the metastable T1 was further excited into higher triplet states. Through systematical study, they established the quantum yields of T2→S1 ISC (Φts) of anthracene, 9-methylanthracene, 9-phenylanthracene, 9,10-dichloroanthracene and 9,10-dibromoanthracene to be 2.6×10−5, 3.6×10−4, 4.7×10−4, 1.5×10−2 and 2.7×10−1, respectively. The significant increases of Φts in meso-derivatives are interpreted in terms of level inversion of T2 and S1 due to the substitution at the meso-position, assuming that  absorption cross section is negligible in comparison with
absorption cross section is negligible in comparison with  absorption cross section. As for the halogenated anthracenes, the increased Φts could be attributed to the internal heavy atom effect.
absorption cross section. As for the halogenated anthracenes, the increased Φts could be attributed to the internal heavy atom effect.
The RISC from upper triplet levels is also observed in some organic dyes [21], such as flavins [6], cyanine derivatives [13], erythrosin B, rose bengal and tetraphenylporphyrin [22,23]. Hub and co-workers reported that the flavin 10-phenylisoalloxazine can be excited with a sequence of two intense laser pulses (interpulse separation) of 337 and 580 nm, respectively, and the delayed fluorescence emission around 540 nm was detected. From the saturation effect of probe pulse induced emission intensity versus probe pulse energy, they derived an estimation of Φts to be 8×10−3. They further verified some other isoalloxazine derivatives also demonstrated TSLIFs, indicating of that RISC Tm→Sn is characteristic of isoalloxazines. The complicated photophysical processes of cyanine dyes also involved RISC from upper triplet levels. Redmond et al. observed TSLIFs in three cyanine dyes, merocyanine 540, BO-Se and QI-Se. The ratios of RISC to IC in promoted higher excited triplet state are estimated to approximately 0.7 for all cyanines, indicative of only 30% IC from Tm back to T1. The origin of RISC from upper triplet levels may be ascribed to both a large T2–T1 energy gap and the effect of heavy atom substitution (S, Se). Beside the flavins and cyanine dyes, Reindl and Penzkofer found that erythrosin B, rose bengal and tetraphenylporphyrin also show RISC from upper triplet levels [22,23].
ESIPT materials are another series of materials showing RISC from upper triplet levels. As shown in figure 3, the photophysical processes of ESIPT materials usually involve an intrinsic four-level cyclic proton-transfer reaction (E→E*→K*→K→E). Itoh and Chou groups have explored photophysical properties of a series of ESIPT materials, such as 2-(2′-hydroxyphenyl)-benzothiazole (HBT) [25], 3-hydroxyflavone [26,27], 7-hydroxy-1-indanone (7HIN) [24], etc., by the transient absorption and the two-step two-colour laser technique. Taking 7HIN as an example, a tentative mechanism of the complete photophysical process of TSLIF is schematically shown in figure 3. After excitation of the first pulse, the excited states of cis-enol form quickly evolve to excited states of cis-keto form accompanying proton transfer. It has been reported that in the tautomer form of 7HIN, the S′nπ* and T′nπ* states are possibly much higher in energy than the  (ππ*) states. As a result of the large spin–orbit coupling between T′nπ* and S′ππ* states, the stimulated triplet states from T′ππ* (T1) by the second pulse effectively transfer to S′ππ* states, resulting in delayed tautomer form emission. Because of the difficulty of obtaining the absolute yield of T2→S1 ISC without knowing the prepared concentration of the T1 state, they performed experiments to measure the relative tautomer emission intensities from TSLIF for 7HIN and 9,10-dibromoanthracene in which the relevant parameters had been determined. Accordingly, a value of 1.1×10−3 for Φts was calculated for 7HIN in methylcyclohexane.
(ππ*) states. As a result of the large spin–orbit coupling between T′nπ* and S′ππ* states, the stimulated triplet states from T′ππ* (T1) by the second pulse effectively transfer to S′ππ* states, resulting in delayed tautomer form emission. Because of the difficulty of obtaining the absolute yield of T2→S1 ISC without knowing the prepared concentration of the T1 state, they performed experiments to measure the relative tautomer emission intensities from TSLIF for 7HIN and 9,10-dibromoanthracene in which the relevant parameters had been determined. Accordingly, a value of 1.1×10−3 for Φts was calculated for 7HIN in methylcyclohexane.
Figure 3.

The proposed mechanism of the proton transfer cycle and dynamics of transient absorption and TSLIF for HBT at 77 K. Adapted from [24].
Although the above materials showed effective RISC from upper triplet levels, the low PL efficiencies restrict their application in FOLEDs. The low PL efficiencies of these materials may be attributed to the competitive non-radiative decay and/or ISC processes with the aid of heavy atom effects. Therefore, new materials should be developed for highly efficient emitters in FOLEDs with both effective RISC from upper triplet levels to singlet levels and high PL efficiencies.
4. The hot exciton process in electroluminescence
In electroluminescence (EL), the holes and electrons injected from the double electrodes recombine and generate excitons, and further the exciton radiation produces light [28–30]. The external quantum efficiency (EQE) of OLEDs is expressed as the following equation:
| 4.1 |
where ηrec is the charge balance factor (ηrec could reach 100% only under ideal conditions), ηPL is the intrinsic PL efficiency, ηout is light out-coupling efficiency (approx. 20%) and ηs is spin statistic factor. According to the spin statistics, the probability of forming singlet and triplet excitons is 25% and 75%, respectively, assuming the formation cross sections for singlet and triplet excitons are equal [31–34]. Phosphorescent OLEDs (POLEDs) could employ 100% excitons (including the singlet and triplet excitons) by means of enhanced ISC, and maximum EQE of over 20% has been achieved in POLEDs [35,36]. However, the high fabrication cost of POLEDs due to the utilization of rather expensive and unsustainable transition metals will critically restrict their extensive applications. Although the FOLEDs possess much lower fabrication cost, only singlet excitons (25% of the total excitons) can be used and triplet excitons decay to the ground state as non-radiative transition [37,38]. As a result, the maximum EQE of FOLEDs is limited to 5%. Recently, overcoming the limit of ηs in FOLEDs from triplet exciton RISC process has attracted increasing attention. Adachi's group at Kyushu University has made significant progress in this field by the design of the promising TADF materials [39–43]. T1 excitons of TADF emitters could be converted into S1 excitons under thermal activation because of the very small singlet–triplet energy splitting ΔEST (less than or equal to 100 meV), and ηs is further drastically increased. So far, employing nearly 100% excitons has been realized in some TADF FOLEDs, and their maximum EQE is comparable to that of POLEDs. Nevertheless, the efficiencies of most TADF devices suffer from a serious roll-off at high current density, which is probably due to the long lifetime of triplet exciton or bimolecular quenching. TTA is another approach to harvest the triplet excitons in FOLEDs [44–46]. As two triplet excitons fuse and form one singlet exciton in the TTA process, ηs can be increased only up to 62.5%, which will restrict the prospective applications of the TTA principle. Therefore, innovative approaches are still urgently required for the rapid development of the organic EL field.
In principle, it is feasible to increase ηs through RISC from high-lying triplet exciton (hot exciton process). Recently, we developed a series of highly efficient fluorescent materials (figure 4) according to the hot exciton principle, and ηs of the FOLEDs based on these emitters greatly broke through the theoretical limit of singlet excitons [7,8,33,34]. Particularly, employing approximately 100% excitons was obtained in a device of TPA-NZP [7,8]. Several distinct features in the excited state energy levels of these materials render the potential hot exciton RISC pathway, as shown in figure 5 [47]. In the case of TPA-NZP, firstly, a very large energy gap (ΔEST=1.22 eV) emerged between S1 and T1, which indicated the TADF process (RISC from T1 to S1) was impossible to occur. Alternatively, it can be found that both S2 and T2 belonged to a charge-transfer (CT) state, and a very small singlet–triplet energy splitting (ΔEST=0.29 eV) existed between them. Due to the fact that CT exciton possesses weak binding energy, correspondingly the spin flip between singlet state and triplet state is prone to occur for the CT excitons. The combination of the CT character of S2 and T2 and their identical energy level could provide an effective RISC channel from T2 to S2. More importantly, as a guarantee, the T1 and T2 states exhibited sufficient separation in energy (1.63 eV), which results in a relatively low IC rate (kIC) from T2 to T1 according to the energy gap law. Thus, in the case of TPA-NZP, it is possible that kRISC (T2→S2) is more competitive than kIC (T2→T1) due to its unique excited state properties. In the EL process of TPA-NZP, triplet excitons could be effectively converted into singlet excitons along the hot exciton channel as shown in figure 6, and as a result, using nearly all the excitons is realized in a TPA-NZP device.
Figure 4.

Examples of the hot exciton materials which have broken through the limitation of the spin statistics.
Figure 5.

The TPA-NZP energy diagram of the first 10 singlet and triplet excited states from TD-M06–2X calculations. (Online version in colour.)
Figure 6.

The hot exciton process in TPA-NZP. S: singlet state; T: triplet state; F: fluorescence; P: phosphorescence; kIC: internal conversion rate; kRISC: reverse intersystem crossing rate; CT: charge-transfer state; ΔEST: singlet–triplet energy splitting. (Online version in colour.)
Moreover, we can deduce that the unique excited state structures of hot exciton materials are crucial to their high ηs on the basis of the comparison between TPA-QAP and TPA-AC [7,8]. Both TPA-QAP and TPA-AC exhibit green emission, and they have quite similar highest occupied molecular orbital and lowest unoccupied molecular orbital levels determined from electrochemical measurements, which implies that the carrier injection and transport properties are nearly identical for these two molecules. Nevertheless, significantly different ηs was obtained for TPA-QAP and TPA-AC, and the corresponding values were 19% and 46%, respectively. As shown in figure 7, for TPA-QAP, closely spaced arrangement was presented in the singlet and triplet energy levels. According to Kasha's rule [2], the hot excitons will rapidly decay to the lowest excited states through IC relaxation. However, TADF cannot proceed in TPA-QAP due to the large energy gap (0.75 eV) between S1 and T1. As a consequence, only singlet excitons could be employed in a TPA-QAP device, and ηs obeys the spin statistics (ηs<25%). Differently, a large energy gap (1.12 eV) seriously suppressed kIC from T2 to T1 in the case of TPA-AC. The T2→S1 hot exciton channel could provide a more competitive kRISC than kIC due to the close energy levels and the CT-type excited states. Thus, the abnormally high ηs of 46% in a TPA-AC device could be attributed to the unique excited state structures similar to that in TPA-NZP. The different T2/T1 energy gaps for TPA-QAP and TPA-AC could be estimated from the different phosphorescence properties. At 77 K, TPA-QAP in methyltetrahydrofuran solutions exhibited strong phosphorescence with the emission peak at 560 nm. Nevertheless, the phosphorescence of TPA-AC cannot be observed even after much effort. The reason could be ascribed to that the high-lying triplet excitons can hardly populate the T1 state due to the blocking from the large T2–T1 energy gap. For TPA-QAP with a small T2/T1 energy gap, when it was excited to the S1 or a higher singlet state, the excitons in the S1 state can relax to the T1 state through a multistep process, including an ISC process from S1 to a close-lying higher triplet state (Tm), followed with an IC process to the lowest T1 state, finally the phosphorescence being emitted. Whereas in TPA-AC with large T2/T1 energy gaps, the excitons at the S1 state can transfer to a close-lying higher triplet state but cannot efficiently relax down to the T1 state due to the large T2/T1 energy gaps, which results in more competitive RISC (T2→S1) than IC (T2→T1). Thus no phosphorescence was observed in TPA-AC.
Figure 7.

(a) The energy levels of the first five singlet and triplet excited states in TPA-QAP and TPA-AC; (b) the exciton decay process after hole–electron recombination in OLEDs based on TPA-QAP and TPA-AC. (Online version in colour.)
We have been trying various measurements to explore the RISC process in the EL of hot exciton materials. First, magneto-electroluminescence (MEL) response experiments were carried out [48]. It is well known that when the interactions between electrons and holes (the spin–spin interaction and exchange interaction) are weak (small ΔEST), the singlet |↑↓−↓↑〉 and three triplet sub-states |↑↑〉, |↑↓+↓↑〉 and |↓↓〉 are degenerate, i.e. they are spin mixed, and their mixing rate depends on the hyperfine field that is intrinsic to organic materials, of several millitesla. In this case, all the three triplet sub-states are involved in the RISC [49]. When an external magnetic field (higher than the hyperfine field) is introduced, the degeneracy of the sub-states of the triplets is removed. As a result, spin mixing only occurs between the |↑↓−↓↑〉 and |↑↓+↓↑〉 states. Therefore, the conversion rate will be decreased, which causes a negative MEL. We have measured the MEL of TPA-NZP-based doping and non-doping devices: they both showed a rapid decrease with an increase of magnetic field in the range of the hyperfine field. These negative magnetic field effects implied that RISC indeed occurred in the TPA-NZP-based OLED. In addition, the transient PL spectrum of TPA-NZP neat film was measured with that of 5 wt% 4CzTPN-Ph:mCP (a red TADF emitter) co-deposited film as comparison shown in figure 8. The transient PL spectrum of 5 wt% 4CzTPN-Ph:mCP co-deposited film exhibited two-component decays, including the prompt decay (9.2 ns) and TADF decay (1.05 μs). As a contrast, only a short lifetime (12.2 ns) was observed for the TPA-NZP film, and there was no delayed fluorescence phenomenon. On the other hand, transient EL decay (figure 9) displayed a consistent trend with the PL decay. When electric voltage pulse was off, in the short-time range (0–0.5 μs), the EL delay was mainly dependent on the radiative decay of excitons formed before the voltage off. In this range, the long-time delayed decay was observed in 4CzTPN-Ph-based device, while only a sharp decay curve in TPA-NZP device. So we supposed that the different PL and EL decay behaviour between TPA-NZP and 4CzTPN-Ph may originate from the different mechanisms of triplet harvesting. For the TADF emitter 4CzTPN-Ph, the delayed fluorescence could be attributed to the relatively low kRISC and long lifetime of T1 excitons. In the case of TPA-NZP, due to the triplet excitons being harvested along the high-lying energy levels, kRISC could be possibly close to or even larger than the radiative rate according to Kasha's rule, and it is reasonable that no delayed fluorescence was observed in both PL and EL processes. In fact, the large kRISC of the hot exciton materials facilitates the enhancement of device stability, which is a critical evaluation for the OLED performance. So it is convincing that in TPA-NZP device, the high EQE of 1.7% and 1.6% was still maintained at the brightness of 100 and 1000 cd m−2, respectively, which were among the best results in non-doped deep-red OLEDs.
Figure 8.

PL lifetime measurement of (a) TPA-NZP and (b) 4CzTPN-Ph by using time-correlated single photon counting method under the excitation of a laser (471.0 nm) with 96.8 ps pulse width. (Online version in colour.)
Figure 9.

EL delay of TPA-NZP non-doped and 4CzTPN-Ph doped OLED under a transient electric voltage. (Online version in colour.)
5. The design principle for the S1 state of hot exciton materials
As mentioned above, high-performance OLEDs should take account of several factors, such as ηs, ηPL and ηrec. Traditional hot exciton materials can be hardly applied in the organic EL field, due to their low PL efficiencies and poor carrier injection and transport properties. To solve these problems, donor–acceptor structures with favourable bipolar transport properties are involved in our molecules. More importantly, we made significant efforts to adjust the nature of excited S1 state for the donor–acceptor molecules. In general, the excited states in organic molecular systems can be classified into two categories considering the binding energy: the CT state and local excited (LE) state [50,51]. The relatively weak binding energy of CT state benefits the spin mixing between singlet state and triplet state, and further affords the possibility to realize RISC [52]. Nevertheless, the molecules based on CT state always show low radiative transition rate and poor PL efficiency, which is mainly caused by the spatially separated transition orbital of CT state. On the contrary, the LE state with strong binding energy possesses a high radiative transition rate because of the large overlap between the transition orbitals. However, the LE type molecules usually only use the singlet excitons and cannot exceed the spin statistics. Thus, we proposed the HLCT state to combine the weak binding energy of CT state and large radiative transition rate of LE state [7,8]. According to the state-mixing principle in quantum chemistry (equations (5.1) and (5.2)), state mixing can be described as a linear combination of two states of ψ(CT) and ψ(LE), and the degree of mixing of the two states is related to the mixing coefficient (λ) [53]. Two important factors determine λ: (i) the energy gap (ECT−ELE) between the two states at the initial configurations, which is inversely proportional to λ; that is to say, the smaller the energy gap, the larger the mixing coefficient and (ii) the magnitude of the coupling matrix element between these two states, which is directly proportional to λ, and depends on the spatial wave function overlap of two states, symmetry properties and the nature of H (the electronic molecular Hamiltonian).
| 5.1 |
and
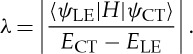 |
5.2 |
Therefore, in principle, the HLCT state could form on the condition that there is a small energy gap between LE state and CT state and a certain interstate coupling, as shown in figure 10. Recently, we achieved a series of donor–acceptor molecules with HLCT state after the careful manipulation of the distance, torsion angle and strength between the donor and acceptor moieties [7,8,33,34,47,54,55]. The quantified natural transition orbital (NTO) analysis indicated that the holes and particles of such molecules always exhibited obvious orbital separation and certain orbital overlap simultaneously. In experiment, the PL spectra usually displayed a large red-shift with increasing solvent polarity, while the HLCT state could ensure a relatively high PL efficiency.
Figure 10.

Illustration of the three possible cases of the LE and CT state energy levels in donor–acceptor molecules: (a,b) independent energy levels for their large energy gaps, and (c) possible state mixing for the small energy gap between the LE and CT states. (Online version in colour.)
For example, PTZ-BZP in figure 11 is a donor–acceptor molecule with HLCT state in S1 state [33,34]. The non-doped PTZ-BZP film exhibited a near-infrared emission with a peak at 700 nm, and a remarkable quantum efficiency of 16% was observed. The hole and particle NTOs of the S1 state showed an excellent balance between spatial separation and orbital overlap, indicating the coexistence of CT and LE components, which are consistent with the description of HLCT state. The fluorescence spectra become broadened and with a remarkable red-shift as the polarity of the solvent increased: from 590 nm in non-polar hexane to 790 nm in polar dichloromethane. With an increase of the solvent polarity, the Stokes shift versus the orientation polarizability plot shows two sets of linearity, which represent two different excited states in low- and high-polarity solvents, respectively. The dipole moment is calculated as 13.2 D and 31.1 D for low- and high-polarity solvents (figure 12), respectively. In high-polarity solvents, it is clear that CT state is dominant due to the large dipole moment (31.1 D) and the PL efficiency decreased rapidly as the solvent polarity increased (from 0.31 in isopropyl to 0.02 in dichloromethane). By contrast, the fluorescence behaviour could combine the relatively high PL efficiency (between 0.47 in hexane and 0.31 in isopropyl) and certain dipole moment (13.2 D) in low-polarity solvents. The HLCT character of PTZ-BZP is responsible for the unique PL properties.
Figure 11.

Natural transition orbitals for the S1 state of PTZ-BZP. (Online version in colour.)
Figure 12.

Linear correlation of orientation polarization (Δf) of solvent media with the Stokes shift (υa–υf) for PTZ-BZP. In low- and high-polarity solvents, the S1 state is HLCT state and CT state, respectively.
Some high-performance non-doped OLEDs were fabricated using hot exciton materials as emitter based on HLCT state [7,8,33,34]. The EL emission of the devices ranged from blue to near-infrared. The detailed data are summarized in table 1. The TPA-NZP-based device emitted a deep-red light of 664 nm with the maximum EQE of 2.8% and maximum brightness of 4574 cd m−2. The PTZ-BZP-based device exhibited the maximum EQE of 1.5% and maximum brightness of 780 cd m−2 with a near-infrared EL emission peaked at 692 nm. A maximum EQE of 3.0% and brightness of 10 079 cd m−2 were observed for the non-doped blue OLED based on TPA-AN. Green and orange devices showed better performance compared with red and blue devices. The devices of TPA-AC and TPA-BZP displayed EQE of 3.2% and 3.8%, and corresponding EL emission peak located at 504 and 588 nm, respectively. The performances of the devices were among the best results of each non-doped OLEDs, and the ηs of all the molecules have broken through the spin statistics limit. The favourable device performance suggested hot exciton RISC and HLCT state provided a new way to design the next-generation OLEDs.
Table 1.
The EL performance summary of the OLEDs based on hot exciton materials.
| device | LEmax (cd A−1) | ηmaxext (%) | Lmax (cd m−2) | λmax (nm) | ηs (%) |
|---|---|---|---|---|---|
| TPA-AN | 5.06 | 3.0 | 10 079 | 460 | 30 |
| TPA-AC | 8.97 | 3.2 | 13 760 | 504 | 46 |
| TPA-BZP | 8.84 | 3.8 | 37 911 | 588 | 42 |
| TPA-NZP | 1.00 | 2.8 | 4574 | 664 | 93 |
| PTZ-BZP | 0.30 | 1.5 | 780 | 692 | 48 |
6. Conclusion
In summary, we reviewed the basic mechanism of the hot exciton RISC process, the early materials with such unique photophysical phenomenon, and the further design of hot exciton materials for promising application in FOLEDs. Different from E- and P-type delayed fluorescence, hot exciton RISC process occurs at the high-lying energy levels, and consequently its kRISC could be close to or even larger than the radiative rate of S1 state. On the one hand, this character facilitates avoiding the accumulation of T1 excitons in EL, and further enhancing device stabilities. On the other hand, the hot exciton process is proposed to greatly increase the ηs for highly efficient FOLEDs. Due to the low PL efficiency, the early materials with RISC mainly were focused on the photophysical process and barely on the FOLEDs. Here, HLCT state molecules, which achieve an incorporation of effective hot exciton RISC and high PL efficiency, are designed and used to maximize the EQE of the FOLEDs. A series of fluorescence materials based on this mechanism were developed with the EL spectra ranging from blue to near-infrared, and their excellent EQEs were harvested in the non-doped OLEDs using these materials, and their ηs all broke through the upper limit of the spin statistics. HLCT state molecules with hot exciton RISC provide a new strategy to design the next-generation organic electroluminescent materials with low cost and high performance.
Funding statement
We thank the financial support from the Natural Science Foundation of China (91233113, 51473063, 51273078), National Basic Research Program of China (973 Program) (2013CB834705, 2015CB655000), Introduced Innovative R&D Team of Guangdong (201101C0105067115) and the Ministry of Science and Technology of China (2013CB834801, 2013CB834805).
Author contributions
D.H. and L.Y. participated in the design of the study and drafted the manuscript; Y.M. and B.Y. directed the study and helped draft the manuscript. All authors gave final approval for publication.
Conflict of interests
We have no competing interests.
References
- 1.Rohatgi-Mukherjee KK. 1978. Fundamentals of photochemistry. New Delhi, India: Wiley Eastern. [Google Scholar]
- 2.Kasha M. 1950. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 9, 14–19. ( 10.1039/DF9500900014) [DOI] [Google Scholar]
- 3.Parker CA, Hatchard CG. 1961. Triplet–singlet emission in fluid solutions. Phosphorescence of eosin. Trans. Faraday Soc. 57, 1894–1904. ( 10.1039/TF9615701894) [DOI] [Google Scholar]
- 4.Birks J. 1970. Photophysics of aromatics molecules. New York, NY: Wiley. [Google Scholar]
- 5.Keller RA. 1969. Excited triplet–singlet intersystem crossing. Chem. Phys. Lett. 3, 27–29. ( 10.1016/0009-2614(69)80010-2) [DOI] [Google Scholar]
- 6.Richterw C, Hub A, Traber R, Schneider S. 1987. Excited triplet–singlet intersystem crossing in isoalloxazines. Photochem. Photobiol. 45, 671–673. ( 10.1111/j.1751-1097.1987.tb07397.x) [DOI] [Google Scholar]
- 7.Li W, et al. 2014. Employing ∼100% excitons in OLEDs by utilizing a fluorescent molecule with hybridized local and charge-transfer excited state. Adv. Funct. Mater. 24, 1609–1614. ( 10.1002/adfm.201301750) [DOI] [Google Scholar]
- 8.Li W, et al. 2014. A hybridized local and charge-transfer excited state for highly efficient fluorescent OLEDs: molecular design, spectral character, and full exciton utilization. Adv. Opt. Mater. 2, 892–901. ( 10.1002/adom.201400154) [DOI] [Google Scholar]
- 9.Lewis G, Lipkin D. 1942. Reversible photochemical processes in rigid media: the dissociation of organic molecules into radicals and ions. J. Am. Chem. Soc. 64, 2801–2808. ( 10.1021/ja01264a025) [DOI] [Google Scholar]
- 10.Keller RA. 1965. Extinction coefficients of triplet–triplet transitions in aromatic compounds. J. Chem. Phys. 42, 2382–2387. ( 10.1063/1.1696304) [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi S, Kikuchi K, Kokubun H. 1976. Fluorescence of the antharacenes following Tn←T1 excitation studied by a double excitation method. Chem. Phys. Lett. 42, 494–497. ( 10.1016/0009-2614(76)80660-4) [DOI] [Google Scholar]
- 12.Kikuchi K, Fukumra H, Kokubun H. 1986. The Sm←T1 absorption spectrum of 9,10-dibromoanthracene. Chem. Phys. Lett. 123, 226–228. ( 10.1016/0009-2614(86)80018-5) [DOI] [Google Scholar]
- 13.Redmond RW, Kochevar IE, Krieg M, Smith G, McGimpsey WG. 1997. Excited state relaxation in cyanine dyes: a remarkably efficient reverse intersystem crossing from upper triplet levels. J. Phys. Chem. A 101, 2773–2777. ( 10.1021/jp963001f) [DOI] [Google Scholar]
- 14.Brewer WE, Studer SL, Standiford M, Chou P-T. 1989. Dynamics of the triplet states and the reverse proton transfer of 3-hydroxyflavone. J. Phys. Chem. 93, 6088–6094. ( 10.1021/j100353a029) [DOI] [Google Scholar]
- 15.Tokumura K, Yagata N, Fujiwara Y, Itoh M. 1993. Two-step laser-induced-fluorescence study of 2′,3′,4′,5′,6′-pentamethyl-3-hydroxyflavone in solution at room temperature: contribution of triplet states and confirmation of ground-state tautomer. J. Phys. Chem. 97, 6656–6663. ( 10.1021/j100127a015) [DOI] [Google Scholar]
- 16.Tokumura K, Kurauchi M, Oyama O. 1994. Reverse intersystem crossing from higher triplet to excited singlet in 2,2′-bipyridine-3,3′-diol phototautomer. J. Photochem. Photobiol. A 81, 151–158. ( 10.1016/1010-6030(94)03794-9) [DOI] [Google Scholar]
- 17.McGimpsey WG, Scaiano JC. 1989. Photochemistry and photophysics from upper triplet levels of 9,10-dibromoanthracene. J. Am. Chem. Soc. 111, 335–340. ( 10.1021/ja00183a050) [DOI] [Google Scholar]
- 18.Bennett RG, Mccartin PJ. 1966. Radiationless deactivation of the fluorescent state of substituted anthracenes. J. Chem. Phys. 44, 1969–1972. ( 10.1063/1.1726971) [DOI] [Google Scholar]
- 19.Lim EC, Laposa J, Yu JMH. 1966. Temperature dependence of intersystem crossing in substituted anthracenes. J. Mol. Spectrosc. 19, 412–420. ( 10.1016/0022-2852(66)90263-3) [DOI] [Google Scholar]
- 20.Kobayashi S, Kikuchi K, Kokubun H. 1978. Nonradiative relaxation processes of the higher excited triplet states of anthracenes studied by double excitation method. Chem. Phys. 27, 399–407. ( 10.1016/0301-0104(78)87145-6) [DOI] [Google Scholar]
- 21.Ringemann C, Schönle A, Giske A, Middendorff CV, Hell SW, Eggeling C. 2008. Enhancing fluorescence brightness: effect of reverse intersystem crossing studied by fluorescence fluctuation spectroscopy. ChemPhysChem 9, 612–624. ( 10.1002/cphc.200700596) [DOI] [PubMed] [Google Scholar]
- 22.Reindl S, Penzkofer A. 1996. Higher excited-state triplet–singlet intersystem crossing of some organic dyes. Chem. Phys. 211, 431–439. ( 10.1016/0301-0104(96)00191-7) [DOI] [Google Scholar]
- 23.Larkin JM, Donaldson WR, Foster TH, Knox RS. 1999. Reverse intersystem crossing from a triplet state of rose bengal populated by sequential 532- +1064-nm laser excitation. Chem. Phys. 244, 319–330. ( 10.1016/S0301-0104(99)00130-5) [DOI] [Google Scholar]
- 24.Chou P-T, Martinez ML, Studer SL. 1991. Role of triplet states in the reverse proton transfer mechanism of 7-hydroxy-1-indanone. J. Phys. Chem. 95, 10306–10310. ( 10.1021/j100178a014) [DOI] [Google Scholar]
- 25.Chou P-T, Martinez ML, Studer SL. 1992. The role of the cis-keto triplet state in the proton transfer cycle of 2-(2′-hydroxyphenyl)benzothiazole. Chem. Phys. Lett. 195, 586–590. ( 10.1016/0009-2614(92)85567-T) [DOI] [Google Scholar]
- 26.Sepioł J, Kołos R. 1990. Two-step laser excitation of 3-hydroxyflavone with stimulated-emission pumping. Chem. Phys. Lett. 167, 445–449. ( 10.1016/0009-2614(90)85028-B) [DOI] [Google Scholar]
- 27.Martinez ML, Studer SL, Chou P-T. 1990. Direct evidence of the triplet-state origin of the slow reverse proton transfer reaction of 3-hydroxyflavone. J. Am. Chem. Soc. 112, 2421–2429. ( 10.1021/ja00162a058) [DOI] [Google Scholar]
- 28.Tang CW, VanSlyke SA. 1987. Organic electroluminescent diodes. Appl. Phys. Lett. 51, 913–915. ( 10.1063/1.98799) [DOI] [Google Scholar]
- 29.Grimsdale AC, Chan KL, Martin RE, Jokisz PG, Holmes AB. 2009. Synthesis of light-emitting conjugated polymers for applications in electroluminescent devices. Chem. Rev. 109, 897–1091. ( 10.1021/cr000013v) [DOI] [PubMed] [Google Scholar]
- 30.Saragi TPI, Spehr T, Siebert A, Fuhrmann-Lieker T, Salbeck J. 2007. Spiro compounds for organic optoelectronics. Chem. Rev. 107, 1011–1065. ( 10.1021/cr0501341) [DOI] [PubMed] [Google Scholar]
- 31.Friend RH, et al. 1999. Electroluminescence in conjugated polymers. Nature 397, 121–128. ( 10.1038/16393) [DOI] [Google Scholar]
- 32.Segal M, Baldo MA, Holmes RJ, Forrest SR, Soos ZG. 2003. Excitonic singlet-triplet ratios in molecular and polymeric organic materials. Phys. Rev. B 68, 075211 ( 10.1103/PhysRevB.68.075211) [DOI] [Google Scholar]
- 33.Yao L, Yang B, Ma YG. 2014. Progress in next-generation organic electroluminescent materials: material design beyond exciton statistics. Sci. China Chem. 57, 335–345. ( 10.1007/s11426-013-5046-y) [DOI] [Google Scholar]
- 34.Yao L, Zhang S, Wang R, Li W, Shen F, Yang B, Ma YG. 2014. Highly efficient near-infrared organic light-emitting diode based on a butterfly-shaped donor–acceptor chromophore with strong solid-state fluorescence and a large proportion of radiative excitons. Angew. Chem. Int. Ed. 53, 2119–2123. ( 10.1002/anie.201308486) [DOI] [PubMed] [Google Scholar]
- 35.Baldo MA, O'Brien DF, You Y, Shoustikov A, Sibley S, Thompson ME, Forrest SR. 1998. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 395, 151–154. ( 10.1038/25954) [DOI] [Google Scholar]
- 36.Ma YG, Zhang H, Shen JC, Che C. 1998. Electroluminescence from triplet metal-ligand charge-transfer excited state of transition metal complexes. Synth. Met. 94, 245–248. ( 10.1016/S0379-6779(97)04166-0) [DOI] [Google Scholar]
- 37.Baldo MA, O'Brien DF, Thompson ME, Forrest SR. 1999. Excitonic singlet–triplet ratio in a semiconducting organic thin film. Phys. Rev. B 60, 14422–14428. ( 10.1103/PhysRevB.60.14422) [DOI] [Google Scholar]
- 38.Zhen CG, Dai YF, Zeng WJ, Ma Z, Chen ZK, Kieffer J. 2011. Achieving highly efficient fluorescent blue organic light-emitting diodes through optimizing molecular structures and device configuration. Adv. Funct. Mater. 21, 699–707. ( 10.1002/adfm.201002165) [DOI] [Google Scholar]
- 39.Uoyama H, Goushi K, Shizu K, Nomura H, Adachi C. 2012. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238. ( 10.1038/nature11687) [DOI] [PubMed] [Google Scholar]
- 40.Méhes G, Nomura H, Zhang Q, Nakagawa T, Adachi C. 2012. Enhanced electroluminescence efficiency in a spiro-acridine derivative through thermally activated delayed fluorescence. Angew. Chem. Int. Ed. 51, 11311–11315. ( 10.1002/anie.201206289) [DOI] [PubMed] [Google Scholar]
- 41.Zhang Q, Li J, Shizu K, Huang S, Hirata S, Miyazaki H, Adachi C. 2012. Design of efficient thermally activated delayed fluorescence materials for pure blue organic light emitting diodes. J. Am. Chem. Soc. 134, 14706–14709. ( 10.1021/ja306538w) [DOI] [PubMed] [Google Scholar]
- 42.Lee SY, Yasuda T, Yang YS, Zhang Q, Adachi C. 2014. Luminous butterflies: efficient exciton harvesting by benzophenone derivatives for full-color delayed fluorescence OLEDs. Angew. Chem. Int. Ed. 53, 6520–6524. ( 10.1002/anie.201402992) [DOI] [PubMed] [Google Scholar]
- 43.Li J, Nakagawa T, Zhang Q, Nomura H, Miyazaki H, Adachi C. 2013. Highly efficient organic light-emitting diode based on a hidden thermally activated delayed fluorescence channel in a heptazine derivative. Adv. Mater. 25, 3319–3323. ( 10.1002/adma.201300575) [DOI] [PubMed] [Google Scholar]
- 44.Chiang C, Kimyonok A, Etherington MK, Griffiths GC, Jankus V, Turksoy F, Monkman AP. 2013. Ultrahigh efficiency fluorescent single and bi-layer organic light emitting diodes: the key role of triplet fusion. Adv. Funct. Mater. 23, 739–746. ( 10.1002/adfm.201201750) [DOI] [Google Scholar]
- 45.Sinha S, Rothe C, Güntner R, Scherf U, Monkman AP. 2003. Electrophosphorescence and delayed electroluminescence from pristine polyfluorene thin-film devices at low temperature. Phys. Rev. Lett. 90, 127402 ( 10.1103/PhysRevLett.90.127402) [DOI] [PubMed] [Google Scholar]
- 46.Wallikewitz BH, Kabra D, Gélinas S, Friend RH. 2012. Triplet dynamics in fluorescent polymer light-emitting diodes. Phys. Rev. B 85, 045209 ( 10.1103/PhysRevB.85.045209) [DOI] [Google Scholar]
- 47.Pan Y, Li W, Zhang S, Yao L, Gu C, Xu H, Yang B, Ma YG. 2014. High yields of singlet excitons in organic electroluminescence through two paths of cold and hot excitons. Adv. Opt. Mater. 2, 510–515. ( 10.1002/adom.201300467) [DOI] [Google Scholar]
- 48.Peng Q, Li W, Zhang S, Chen P, Li F, Ma YG. 2013. Evidence of the reverse intersystem crossing in intra-molecular charge-transfer fluorescence-based organic \hboxlight-emitting devices through magneto-electroluminescence measurements. Adv. Opt. Mater. 1, 362–366. ( 10.1002/adom.201300028) [DOI] [Google Scholar]
- 49.Hu B, Yan L, Shao M. 2009. Magnetic-field effects in organic semiconducting materials and devices. Adv. Mater. 21, 1500–1516. ( 10.1002/adma.200802386) [DOI] [Google Scholar]
- 50.Grabowski ZR, Rotkiewicz K, Rettig W. 2003. Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 103, 3899–4032. ( 10.1021/cr940745l) [DOI] [PubMed] [Google Scholar]
- 51.Pope M, Swenberg CE. 1999. Electronic processes in organic crystals and polymers. Oxford, UK: Oxford University Press. [Google Scholar]
- 52.Gorse AD, Pesquer M. 1995. Intramolecular charge transfer excited state relaxation processes in para-substituted N,N-dimethylaniline: a theoretical study including solvent effects. J. Phys. Chem. 99, 4039–4049. ( 10.1021/j100012a026) [DOI] [Google Scholar]
- 53.Turro NJ. 1991. Modern molecular photochemistry. Sausalito, CA: University Science Books. [Google Scholar]
- 54.Li W, Liu D, Shen F, Ma D, Wang Z, Feng T, Xu Y, Yang B, Ma YG. 2012. A twisting donor-acceptor molecule with an intercrossed excited state for highly efficient, deep-blue electroluminescence. Adv. Funct. Mater. 22, 2797–2803. ( 10.1002/adfm.201200116) [DOI] [Google Scholar]
- 55.Zhang S, Li W, Yao L, Pan Y, Shen F, Xiao R, Yang B, Ma YG. 2013. Enhanced proportion of radiative excitons in non-doped electro-fluorescence generated from an imidazole derivative with an orthogonal donor–acceptor structure. Chem. Commun. 49, 11302–11304. ( 10.1039/C3CC47130F) [DOI] [PubMed] [Google Scholar]