

Abstract
Little is known about the interactions between nicotinic and muscarinic acetylcholine receptors (nAChRs and mAChRs). Here we report that methacholine (MCh), a selective agonist of mAChRs, inhibited up to 80% of nicotine-induced nAChR currents in sympathetic superior cervical ganglion neurons and adrenal chromaffin cells. The muscarine-induced inhibition (MiI) substantially reduced ACh-induced membrane currents through nAChRs and quantal neurotransmitter release. The MiI was time- and temperature-dependent. The slow recovery of nAChR current after washout of MCh, as well as the high value of Q10 (3.2), suggested, instead of a direct open-channel blockade, an intracellular metabotropic process. The effects of GTP-γ-S, GDP-β-S and pertussis toxin suggested that MiI was mediated by G-protein signalling. Inhibitors of protein kinase C (bisindolymaleimide–Bis), protein kinase A (H89) and PIP2 depletion attenuated the MiI, indicating that a second messenger pathway is involved in this process. Taken together, these data suggest that mAChRs negatively modulated nAChRs via a G-protein-mediated second messenger pathway. The time dependence suggests that MiI may provide a novel mechanism for post-synaptic adaptation in all cells/neurons and synapses expressing both types of AChRs.
Keywords: nAchRs, mAchRs, methacholine, nicotine, G-proteins
1. Introduction
Acetylcholine (ACh)-receptors are widely expressed in neurons, endocrine cells and muscle cells [1]. These receptors play essential roles in synaptic transmission, gland secretion and excitation–contraction coupling [2–7]. The nAChRs are a type of cation channel permeable to Na+, K+ and Ca2+ that are important to generate post-synaptic potential at synapses, endocrine cells and neuromuscular junctions [3,6,7]. The mAChRs are G-protein-coupled receptors with seven transmembrane domains, which modulate/gate other ion channels via G-proteins and second messengers [7–10]. Some cells (such as skeletal muscle) express only nAChRs, and others (such as lacrimal cells and cardiac muscle) express only mAChRs [11–13]. However, many central nervous system (CNS), autonomic neurons and endocrine cells co-express nAChRs and mAChRs [8,14], but little is known about the interaction of the two types of AChRs [2,10,15–17]. Here, we have studied the inhibition produced on nAChRs by activation of mAChRs in rat sympathetic superior cervical ganglion (SCG) neurons and rat adrenal chromaffin cells. SCG neurons and adrenal chromaffin cells develop from the same neural crest precursors. Thus, they express similar subtypes of nAChRs (mainly α3β4 [18–20]) as well as mAChRs [8,21]. Surprisingly, activation of mAChRs by ACh inhibited up to 80% of nAChRs' current evoked by their common natural ligand ACh. Muscarinic receptors may also have such effects in certain presynaptic endings because autoreceptor inhibition on further transmitter release was widely observed in presynaptic endings [22–24].
2. Material and methods
(a). Cell preparations
We dissociated SCG neurons from neonatal rats as described previously [25]. Briefly, ganglia from P3–7 sprague dawley (SD) rats were dissected and incubated in Ca2+-free solution containing collagenase (0.5 mg ml−1) and trypsin (1.5 mg ml−1) for 30 min at 31°C. Subsequently, the isolated cells were obtained by dispersing the ganglia on glass coverslips (50 × 50 mm2). For electrophysiological recordings, the cells were used between 20 min and 8 h after the plating. The incubation solution contained (in mM): 130 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose and 10 sucrose. pH was adjusted to 7.4. Rat adrenal chromaffin cells were prepared by digestion of adrenal medulla from adult SD rats (250 g) with collagenase D. The cells that were used for recordings had been cultured for 1–7 days [14,26].
(b). Electrophysiological recordings
The membrane currents were acquired with a patch-clamp amplifier (Axon 200B). Cell membrane capacitance and series resistance were electronically compensated. Data analysis was performed with the Igor software package. Graphs and current traces were also produced with the Igor software package [27].
The pipette solution contained (in mM): 135 CsCl, 8 NaCl, 1 MgCl2, 10 H-HEPES. The pH was adjusted to 7.2, and the osmolality was 299–301 mosm/kg H2O. The external solution consisted of (in mM): 141 NaCl, 2.8 KCl, 2 CaCl2, 1 MgCl2 and 10 HEPES. The pH was adjusted to 7.4, and the osmolality was 319–320.
Drugs were applied via RCP-2B multi-channels micro perfusion system (INBIO Inc., Wuhan, China), which had a fast exchange time (less than 100 ms) with electronic switching between seven solution channels [27,28]. The puffing pipette of 100 μm tip diameter was located approximately 120 μm from the cell. GTP-γ-S and GDP-ß-S were applied by dialysis from a whole-cell pipette. Perforated whole-cell recordings and standard whole-cell recordings were as described previously [29,30].
(c). Amperometric measurement of catecholamine release
Electrochemical amperometry using carbon fibre microelectrodes (CFEs) was done as described previously [26,29,31–34]. The sensor tips of 5 μm polypropylene-insulated CFE (ProCFE, Dagan, Minneapolis, MN, USA) were positioned to touch the cell gently. The CFEs were held at +780 mV by another patch-clamp amplifier, PC-2B (INBIO Inc.), to allow oxidation of released catecholamines [27]. The amperometric currents were acquired by the pClamp8 with a low-pass filter of either 10 Hz (for SCG neurons) or 500 Hz (for adrenal chromaffin cells). The acquired data were analysed with Igor software (AveMatrix, Lack Oswego, OR, USA).
3. Results
(a). Muscarinic inhibition of nAChRs currents
Methacholine (MCh) is a selective agonist of the mAChRs [35]. Application of MCh (1 mM) from a puffer pipette to a SCG neuron or a rat adrenal chromaffin cell failed to evoke any current (not shown), whereas application of 100 µM nicotine induced large inward whole-cell currents in both cell types. When MCh was applied for 10 s before Nic application, the Nic response was reduced by 80% (figure 1a). After wash of MCh for 2 min, the Nic-induced current recovered nearly completely. This muscarinic inhibition of nAChRs was observed in all SCG neurons (n = 57) and adrenal chromaffin cells (n = 30) tested. The concentration dependence of the MiI had a half maximal inhibitory concentration (IC50) at 4 µM, which is consistent with IC50 of selective muscarinic agonists in other studies [7,8]. The dose–response curve of nicotine showed a half maximal effective concentration (EC50) of 69 μM for nAChRs (figure 1b). This result implicated that stimulation with micromolar ACh induced both nAChRs current activation and mAChRs mediated inhibition on the nAChRs (see below).
Figure 1.

Inhibition of nAChRs currents by prepuff of muscarinic agonists. (a) Inhibition of nAChRs currents by a prepuff of MCh. Left panel shows nicotine (Nic)-induced currents in a SCG neuron via perforated whole-cell recording at –70 mV. The inward currents were evoked by Nic (100 µM) before applying MCh (left trace), during applying MCh (middle trace) and after wash of MCh (right trace). All traces were from the same neuron. Right panel shows the statistics of the experiments in both SCG neurons and adrenal chromaffin cells. The inhibition of nAChRs current by a 10 s prepuff of 1 mM MCh was 77 ± 4% in SCG neurons (n = 19), and 74 ± 3% in adrenal chromaffin cells (n = 11). (b) Dose-dependence of MCh inhibition of nAChRs currents. Left panel, the inhibition of 100 µM Nic-induced currents at –70 mV were 20% and 80% for 10 s-prepuff of 0.1 µM MCh (left traces, neuron 1) and 1 mM MCh (right traces, neuron 2), respectively. Right panel, the dose curves of MCh versus currents induced by Nic (100 μM) (n = 12), and of Nic versus nAChRs currents (n = 9). The EC50 and IC50 of maximum effects were 69 μM and 4 μM for nicotine and MCh, respectively.
(b). Kinetics of muscarine-induced inhibition
The MiI was time-dependent. Figure 2a shows the Nic-induced currents at various times after application of MCh (1 mM, the maximally effective concentration). Prepuff of MCh for 0.5 s and 6 s inhibited 25 ± 2% and 81 ± 1% of nAChRs currents in this cell, respectively. The Nic-induced currents were fully recovered after MCh wash for 150 s. The time course of MiI followed a single exponential curve. At 22°C, 0.85 s prepuff were required to produce half maximum MiI. The time constant of nAChR inhibition by 1 mM MCh was 1.3 s (figure 2b).
Figure 2.

Time course of the ‘MiI’ on inhibition of nAChRs currents at room temperature in SCG neurons. (a) The inhibition of 100 µM Nic-induced currents was strongly dependent on prepuff-time of MCh (1 mM) solution. In addition to the MCh-prepuff solution, MCh (1 mM) was also included in the Nic-puffing solutions, except the currents of ‘control’ and ‘recover’. Note that the ‘control’ current is nearly overlapped by the current of 0 s prepuff. MCh was washed out using standard external solution during and after the 150 s break. All traces in this panel were from one neuron. (b) Time course of the MCh effect in (a). The data points of the prepuff effect were well described by an exponential function with time constant of 1.3 s. Prepuff for 6 s was sufficient to get maximum MiI of 81 ± 1% (n = 8). (c) The recovery of nAChRs inhibition induced by MCh (1 mM) was strongly dependent on wash time. All traces in this panel were from another neuron. (d) Statistics of the wash experiments in (c). At 22°C, the data points were well fitted by a two exponential function with time constants of 10.0 s and 88.7 s, respectively (n = 7).
MiI was fully reversible after removing MCh from the bath. Half MiI of nAChRs was removed after wash of MCh for 18 s (figure 2c). The nAChR recovery followed a double-exponential curve with time constants of 13 ± 2 s and 88 ± 2 s, respectively (figure 2d). The relative amplitudes of the fast and slow recovery components were 76% and 24%, respectively. Wash for 3 min was sufficient to remove the MiI completely. Thus, the kinetics of both inhibition and recovery occurred on a time-scale of seconds to minutes. The inhibition was set approximately 10 times faster than recovery.
(c). Open-channel block cannot account for the muscarine-induced inhibition of nAChRs current
One possible mechanism of MiI could be that MCh block the opening nAChRs channels, i.e. a cholinergic ligand could occupy the channel pore and reduce the permeation of cations (Na+, K+ and Ca2+) through the channel, as observed in BC3H1 cells [36] and neuromuscular junction [37,38]. To test whether the MiI was owing to open-channel block, we first tested its temperature sensitivity. At room temperature, the pre-activation of mAChR is necessary for MiI of nAChRs current. Without prepuff of MCh or ACh, ACh or a mixture of MCh+Nic produced no inhibition of nAChR currents at 22°C. There was no difference in peak nAChRs currents induced by Nic (1 mM) and ACh (1 mM) at 22°C (figure 3a upper left panel). Similarly, peak nAChRs currents were smaller either when Nic (1 mM) alone or a mixture of MCh (1 mM) and Nic (1 mM) were applied (figure 3a, lower left). However, at a physiological temperature of 36°C, MiI of nAChRs was apparent even without pre-activation of mAChRs. nAChRs currents induced by ACh (1 mM) or the mixture of MCh (1 mM) + Nic (1 mM) were considerably smaller than those induced by Nic (1 mM) alone (figure 3a, middle panels). This suggested that the latency of MiI after activation of mAChRs is considerably reduced by the higher temperature and that the inhibition occurs during the short application time (1–2 s). The requirement of high temperature for visible MiI upon a puff of nicotine (or MCh, or ACh) might be the reason why the MiI was not discovered in previous studies [14,15,19,39]. Figure 3b shows, in contrast to the results obtained at room temperature, that the physiological temperature accelerated the muscarinic inhibition. With 0.5 s prepuff of MCh (1 mM), the MiIs were 25% at 22°C and 70% at 36°C, respectively. The time constants of MiI were 1.32 s, 0.58 s and 0.27 s at 22°C, 30°C and 36°C, respectively (figure 3b). By contrast, the known open-channel blockades of nAChRs by 1 μM atropine [40] as well as Ca2+ channels by 200 μM Cd2+ [41] had no detectable time or/and temperature dependence (n = 6, data not shown), suggesting that MiI of nAChRs here was not due to a direct open-channel blockade.
Figure 3.

Temperature sensitivity of the M-ihhibition. (a) In the absence of prepuff, MCh or ACh could inhibit nAChRs current only at 36°C but not 22°C. Left panels, Nic (1 mM), ACh (1 mM) and MCh (1 mM) + Nic (1 mM) induced currents at 22°C and 36°C, respectively. Right panel, the statistics of the experiments using protocol of left panel. Without prepuff at 36°C, the inhibition of nAChRs current was 33 ± 3% by 1 mM ACh (n = 5), and 34 ± 4% (n = 6) by 1 mM MCh. Note, there was no inhibition of nAChRs by MCh or ACh at 22°C. (b) The MiI of nAChRs was strongly dependent on temperature and prepuff time. Left two panels show currents induced by 100 µM-Nic at 22°C (neuron 1) and 36°C (neuron 2), respectively. MCh (1 mM) was prepuffed with MCh-prepuff-solution. MCh (1 mM) was also included in the Nic (100 µM)-puffing solution for the traces marked with the prepuff times (0 s, 0.5 s, etc.), except where only ‘Nic’ and ‘wash’ are marked. Right panel shows the statistics. The time constants of MiI versus prepuff time were 1.32 s, 0.58 s and 0.27 s, corresponding to 22°C (n = 11), 30°C (n = 8) and 36°C (n = 8), respectively.
(d). G-proteins and protein kinases are involved in muscarine-induced inhibition
To investigate whether the G-protein pathway is involved in MiI, we tested the effect of intracellular GTP-γ-S on nAChRs currents in SCG neurons and adrenal chromaffin cells. If MiI is mediated by G-protein activation, then including GTP-γ-S (a non-hydrolyzable GTP analogue) in the whole-cell pipette solution should mimic the MiI [42–45]. The experiments on GTP-γ-S in figure 4 required standard whole-cell recording, which caused a significant rundown of nAChRs currents induced by Nic (100 µM). This was evident in the absence of GTP-γ-S in both SCG neurons and adrenal chromaffin cells. In the absence of GTP-γ-S, the rundown of nAChRs currents after 8 min whole-cell dialysis (average series resistance Rs = 11 MΩ, membrane capacitance Cm = 14 pF) was approximately 42 ± 5% (figure 4a left panel). However, in the presence of GTP-γ-S (100 µM), the nAChRs current was reduced by 72 ± 2% (average Rs = 11 MΩ, Cm = 13 pF). Thus, like the MiI, in contrast to whole-cell control (figure 4a, left), GTP-γ-S inhibited nAChRs current significantly (p < 0.01, figure 4a, right). A similar result was observed in SCG neurons (data not shown).
Figure 4.

G-Proteins are involved in MiI of nAChRs currents in adrenal chromaffin cells. (a) Intracellular GTP-γ-S inhibited nAChRs currents. Left panel, rundown of whole-cell currents induced by Nic (100 μM). Middle panel, intracellular whole-cell dialysis (Rs = 11 MΩ, Cm = 14 pF) of GTP-γ-S (100 µM) for 8 min inhibited 72% of nAChRs (cell 2). Right panel, statistics of GTP-γ-S experiments. The GTP-γ-S inhibition of nAChRs is significant (p < 0.01). (b) GDP-ß-S removed MiI of nAChRs. Left panel shows that 5 µM MCh inhibited 35% of nAChRs under perforated whole-cell mode at –70 mV (cell 3). Middle panel, intracellular whole-cell dialysis (average Rs = 12, Cm = 12 pF) of GDP-ß-S (1 mM) for 8 min removed 5 µM MCh-inhibition to 10% (cell 4). Right panel, statistics of GDP-ß-S experiments. The effect of GDP-β-S on the MiI was significant (p < 0.01). (c) Effects of pertussis toxin (PTX) on MiI. PTX removed partial MiI of nAChRs under perforated whole-cell mode at –70 mV. Left two panels show that, after PTX treatment (150 ng ml−1 for overnight), MCh (5 µM) inhibition was reduced from 35% (left, cell 1) to 16% (middle, cell 2). Right panel, statistics of PTX experiments. On average, PTX removed MCh (5 µM) inhibition of nAChRs significantly from 34 ± 2% to 20 ± 3% (p < 0.01). (d) Effects of BAPTA on MiI. Left three panels show that, after intracellular dialysis of 20 mM BAPTA for 10 min, 5 µM MCh-inhibition was reduced from 34% (left first panel) to 21% (left second panel). The MiI was further reduced to 12% by combined PTX and intracellular dialysis of 20 mM BAPTA (left third pane). Right panel, statistics of BAPTA experiments. On average, BAPTA and BAPTA + PTX-treatment reduced 5 µM MCh-inhibition of nAChRs significantly from to 34 ± 2% to 21 ± 3% (p < 0.01) and 12 ± 2% (p < 0.01), respectively.
G-protein activation can be inhibited by GDP-ß-S (a non-hydrolyzable GDP analogue [42,44,45]). As shown in figure 4b, whole-cell dialysis of 1 mM GDP-ß-S (average Rs = 11 MΩ, Cm = 14 pF, dialysis for > 8 min) significantly reduced MCh-inhibition from 34 ± 2% to 16 ± 2% (p < 0.01).
Pertussis toxin (PTX) inhibits activation of PTX-sensitive G-proteins [9,44–46]. As shown in figure 4c, adrenal chromaffin cells were pre-treated with 150 ng ml−1 PTX overnight. In contrast to untreated cells, PTX reduced the MiI of nAChRs current by 5 µM MCh from 34 ± 2% to 20 ± 3% (p < 0.01). Control experiments ensured that the MiI of voltage-gated Ca channels was also reduced from 85 ± 6% (no PTX) to 35 ± 5% (PTX-treated, n = 5, data not shown).
In addition to the PTX-sensitive G-proteins, we tested PTX-insensitive but Ca2+-sensitive G-proteins. By including 20 mM BAPTA in the whole-cell internal solution, the MCh (5 μM) inhibition of nAChRs currents (34 ± 2%) was significantly reduced to 21 ± 3% (BAPTA) and to 12 ± 2% (PTX and BAPTA), respectively (figure 4d). This suggested that in addition to PTX-sensitive G-proteins, the PTX-insensitive but Ca2+-sensitive G-proteins were involved in the MiI of nAChRs. This finding was consistent with both M1 and M4 subtypes of mAChRs contributing to the modulation of Ca currents in SCG neurons [9]. Furthermore, the 0.5 μM muscarinic antagonist atropine removes the MCh-induced inhibition, indicating the involvement of mAChRs (data not shown). Taken together, these data suggested that G-proteins mediate the MiI.
Many G-protein-coupled receptors affect cell functions through activation of second messages and kinases [46]. To investigate whether kinases are involved in the MiI, we tested effects of agonists and antagonists of protein kinase A (PKA) and protein kinase C (PKC). Figure 5a,b demonstrates that the PKC antagonist BIS significantly reduces the MiI, whereas PKC agonist phorbol 12-myristate 13-acetate (PMA) mimicks only part of the MiI. Figure 5c,d shows that the PKA-specific antagonist H-89 reduces MiI significantly, whereas PKA agonist 8-Br-cAMP mimicks part of MiI, indicating that PKA signalling is also involved in MiI. In addition, the MiI pathway is via a cytoplasmic diffusible second messenger, because MiI of single channel recordings are persistent even when MCh is applied from extracellular patch area (data not shown). This is consistent with the finding that the M1-phospholipase C (PLC)-phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) pathway of the ‘M current’ (a K+ channel) [47] also mediates the MiI effect (50 nM Wortmanin blocked > 50% of MiI, data not shown; see also [48]). Taken together, these experiments provide strong evidence that PKA, PKC and M1-PLC-PIP2 signalling participate in MiI.
Figure 5.

Protein kinases A and C are involved in the MiI. (a) PKC-specific inhibitor, bisindolylmaleimide I (Bis) removes part of the MiI in SCG neurons and adrenal chromaffin cells under perforated whole-cell mode with holding potential –70 mV. Left two panels show 1 mM MCh-induced inhibition of nAChR currents were partially removed by Bis treatment (500 nM, in 37°C for 30 min). Bis reduced the MiI from 74% (left, chromaffin cell 1) to 46% (middle, chromaffin cell 2). Rightmost panel, Bis removed the MiI significantly from 77 ± 6% to 37 ± 9% and 74 ± 6% to 46 ± 5% in SCG neurons and adrenal chromaffin cells, respectively (p < 0.01). The digits in brackets in this and other histograms indicate numbers of cells tested in each condition. (b) Activation of PKC by phorbol 12-myristate 13-acetate (PMA) inhibits partial nAChRs currents. Left two panels, the protocol was similar to (a), except 10s-prepuff of MCh is replaced by 4 min-prepuff of PMA. Without Bis treatment, the nicotine-induced current was reduced 45% compared with that of control (left, SCG neuron 1). Bis removed the PMA-inhibition completely (middle, SCG neuron 2). Rightmost panel, Bis removed the PMA-inhibition significantly from 55 ± 10% to 2 ± 8% and 29 ± 6% to 4 ± 4% in SCG neurons and Adrenal chromaffin cells, respectively (p < 0.01). (c) PKA-specific inhibitor, H-89 removes part of the MiI in SCG neurons and Adrenal chromaffin cells under perforated whole-cell mode with holding potential –70 mV. Left two panels show 1 mM MCh-induced inhibition of nAChR currents were partially removed by H89 treatment (500 nM, in 37°C for 30 min). H89 reduced the MiI from 77% (left, chromaffin cell 3) to 50% (middle, chromaffin cell 4). Rightmost panel, H-89 removed the MiI significantly from 80 ± 5% to 40 ± 9% and 74 ± 6% to 50 ± 8% in SCG neurons and adrenal chromaffin cells, respectively (p < 0.01). (d) Activation of PKA by 8-Bromo-cyclic AMP (8Br-cAMP) inhibits partial nAChRs currents. Without H-89 treatment, the nicotine-induced current was reduced by 40% compared with that of control (left, chromaffin cell 5). H-89 removed partial 8Br-cAMP inhibition (middle, chromaffin cell 6). Rightmost panel, statistically H-89 removed the 8Br-cAMP-inhibition significantly from 47 ± 9% to 5 ± 18% and 40 ± 9% to 10 ± 8% in SCG neurons and adrenal chromaffin cells, respectively (p < 0.01).
(e). Physiological impacts
To further address the physiological relevance, we tested the effect of MiI on nAChRs-induced quantal catecholamine secretion. In SCG neurons [25,49], quantal secretion of catecholamines from somatic release sites can be detected by electrochemical amperometry using micro-CFEs. Figure 6a shows the MiI of quantal secretion evoked by Nic (100 μM) in a SCG neuron. The cell was pre-treated with 1 μM thapsigargin (Tg) for 10 min to remove intracellular Ca2+ stores sensitive to activation of mAChRs. Nic-induced quantal secretion was maximum when mAChRs were not activated (figure 6a(i)). Prepuff of MCh (1 mM) for 10 s inhibited all secretion (figure 6a(ii)). Nic-induced secretion recovered gradually after removing MCh (figure 6a(iii)). Nic-induced secretion recovered completely after wash of MCh for 3 min (figure 6a(iv)). Similar results were observed in eight cells.
Figure 6.
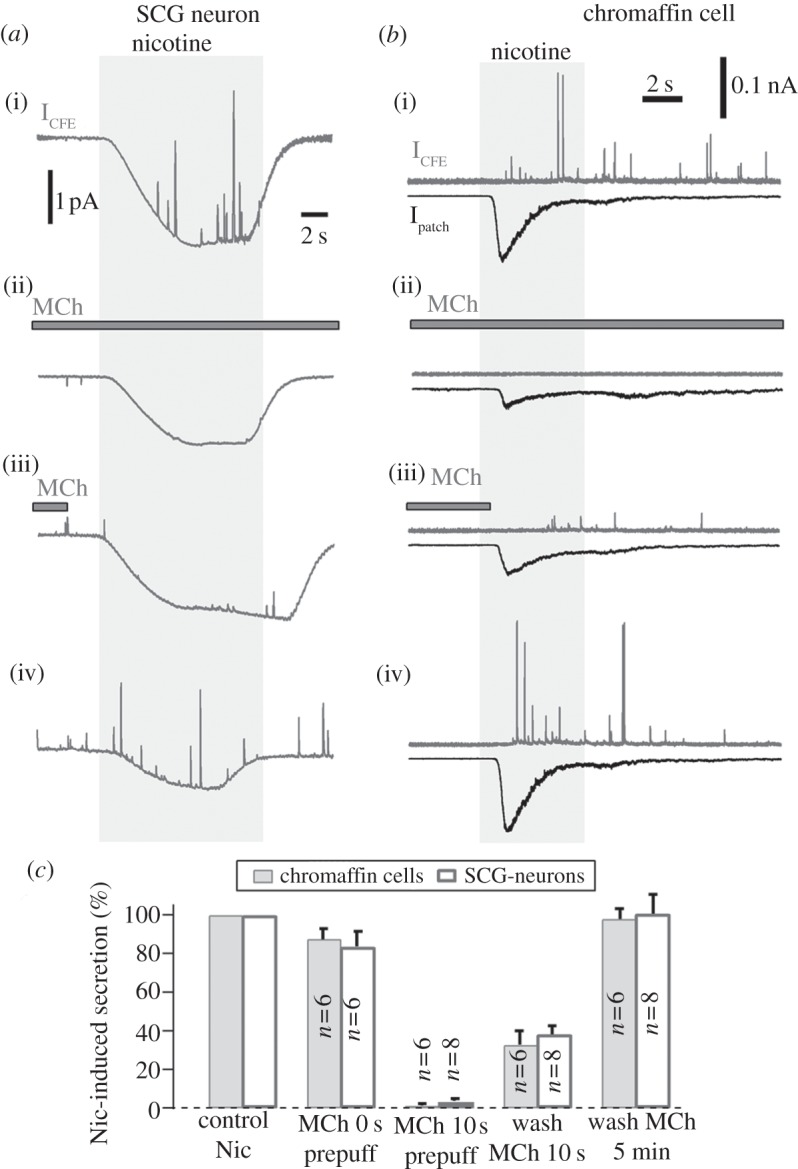
Methacholine inhibits the secretion in SCG neurons and adrenal chromaffin cells. (a) The activation of mAChRs inhibited nAChRs-induced quantal secretion of catecholamines in a SCG neuron. Representative amperometric spikes were induced by Nic (100 μM) in the presence and absence of MCh (1 mM). To reduce the baseline noise, the traces were low pass-filtered to 10 Hz. All traces are from the same neuron. (b) Similar experiments to (a), except the cell was a chromaffin cell under voltage-clamp at –70 mV. The nAChRs currents and quantal secretion were measured by combined patch-clamp and electrochemical amperometry in the presence of Tg (1 μM). Amperometric signals were low-pass filtered at 500 Hz. (c) Statistics of amperometry experiments shown in (a) and (b). On average, in contrast to Nic-induced secretion, MCh inhibited secretion by 16 ± 8% (Nic + MCh without MCh prepuff), 99 ± 1% (MCh prepuff for 10 s), 68 ± 7% (Nic with pre-wash for 10 s) and 2 ± 5% (washout MCh for 3 min) in SCG neurons, respectively (n = 8). In adrenal chromaffin cells, the corresponding inhibitions were 12 ± 6%, 97 ± 1%, 62 ± 5% and 0 ± 10%, respectively (n = 6).
The inhibition of secretion by MCh (figure 6a) could also be owing to the MiI of voltage-gated Ca2+ currents (data not shown), because the release site of the SCG neuron could not be voltage-clamped. To be sure that the MiI can directly block secretion induced by Ca2+ influx through nAChR channels [39,50], we did similar experiments in adrenal chromaffin cells, except that the cell was voltage clamped to exclude Ca2+ channels. Figure 6b shows the MiI of nAChRs currents and quantal secretion by combined patch-clamp and electrochemical amperometry in a chromaffin cell. Again, the cell was pre-treated with Tg (1 μM) for 10 min to remove intracellular Ca2+ stores sensitive to mAChRs. The cell was voltage clamped at –70 mV so that not all of the Ca2+ influx induced by nAChRs was through voltage gated Ca channels, but also through nAChRs channels, which contribute with a fractional Ca2+ current of 2.5% [37]. nAChRs currents and quantal secretion induced by 100 µM Nic were maximum when mAChRs were not activated (figure 6b(i)). Prepuff of 1 mM MCh for 10 s inhibited most Nic-induced current and eliminated secretion (figure 6b(ii)). After removing MCh, the Nic-induced nAChR current and secretion recovered gradually (figure 6b(iii)). nAchRs current and secretion recovered completely after wash of MCh for 3 min or longer (figure 6b(iv)). Similar results were observed in six cells tested.
For SCG neurons of figure 6a, in contrast to secretion induced by 100 µM Nic, the average MiI of the Nic-induced secretion was 16 ± 8% (Nic + MCh without MCh prepuff), 99 ± 1% (MCh prepuff for > 10 s), 68 ± 7% (removing MCh for 10 s) and 2 ± 5% (removing MCh for 3 min), respectively (n = 8, figure 6c). The statistics of the corresponding inhibitions in adrenal chromaffin cells shown in figure 6b were 12 ± 6%, 97 ± 1%, 62 ± 5% and 0 ± 10%, respectively (n = 6, figure 6c). This suggested that the MiI could abolish the nicotine-induced secretion nearly completely.
4. Discussion
We discovered that activation of mAChRs caused inhibition of nAChRs via a G-protein mediated pathway in SCG neurons and adrenal chromaffin cells. The present results suggest that, at room temperature, there was a delay of approximately 1 s for the MiI. However, if the cell was prepuffed with ACh or a muscarinic agonist, the MiI could reduce 80% of nAChRs current. At physiological temperature (36°C), the MiI was much faster so that 30% inhibition of nAChRs current occurred during a single brief puff of ACh.
(a). G-protein as the mechanism of muscarine-induced inhibition of nAChRs
ACh may cause some open-channel block in nAChRs channels in the neuromuscular junction [37,38]. However, because the MiI described here was sensitive to prepuff time (figure 2) and temperature (Q10 = 3.2, indicating a biochemical rather than a physical process, see figure 3), which was not observed for other known open-channel blocks, we concluded that the majority of MiI in this work is not owing to open-channel block. In addition, the MiI was present in all experiments testing the G-protein pathway. This is strongly supported by adding the agonist outside the pipette. These include GTP-γ-S, GDP-β-S and PTX (figure 4), suggesting that the mechanism of MiI is mediated by G-protein signal transduction. The G-protein pathway involves GTP-binding G-protein, hydrolyzation of GTP to GDP and breakdown of the G-protein into βγ-subunit and α-subunit [15,46,49]. There are two general signal pathways to affect ion channels. First, the G-protein βγ subunit produces a membrane-delimited short-cut action, in which the βγ-subunit diffuses to a nearby nAChR. Second, the second messenger pathway is activated by the α-subunit, which produces a diffusible second messenger in the cytoplasm and activates one or more kinases to phosphorylate the cytoplasmic site of nAChRs. For MiI on nAChR, we favour the diffusible second messenger pathway, because the MiI is preserved in cell-attached recordings when MCh was applied through the perfusion (data not shown).
PLC mediates depletion of PIP2 and modulates ion channels even in cell-attached recordings when muscarinic agonists are applied through the perfusion to other parts of the cell surface [47,48]. Similarly, it is likely that PIP2 depletion is involved in the MiI of nAChRs (data not shown).
(b). Subtypes of ACh-receptors
Superior cervical ganglion (SCG) neurons have at least two types of mAChRs: M1 and M4 [9,15]. M4 is PTX-sensitive and likely to be responsible for the PTX-sensitive MiI (figure 4). The M1 signal pathway is sensitive to Ca2+ and should be responsible for the BAPTA-sensitive fraction of MiI. According to figure 4c, the PTX-sensitive mAChRs including M4 should be responsible for about 50% of the MiI in rat SCG neurons, while the PTX-insensitive and Ca2+-sensitive mAChRs (including M1) took about 30% of MiI (figure 4).
(c). Crosstalk between ligand receptors
There are a few reports about crosstalk between different receptors in SCG neurons. For example, two ligand-gated receptor channels, nAChR and P2X, coexist in a sympathetic neuron. Activation of nAChR channels inhibits the nearby P2X channels [51,52]. Further crosstalk occurs was between prostaglandin E2 (PGE2) receptor and nAChR [53]. The metabolic inhibition of PGE2 is via G-protein [18]. Although more and more receptor crosstalks are being discovered in other neurons/cells, the crosstalk between nAChRs and mAChRs is the first example where both receptors are activated by a common ligand.
(d). Muscarine-induced inhibition of neurotransmitter release
Muscarine inhibits nicotine-induced secretion [54]. However, the MiI was interpreted exclusively as the inhibition of voltage-gated Ca2+ channels [15,46]. On the other hand, Ca2+ influx through nAChRs receptors is sufficient to trigger secretion [39,50]. In figure 6, we provide direct evidence that mAChRs blocked quantal release elicited by Ca2+ influx through nAChRs channels. Thus, in addition to MiI of voltage-gated Ca2+ channels, our data provide another mechanism for MiI of nAChR-induced secretion.
To produce large MiI, in some experiments (figures 2, 3, 5 and 6), we used a high concentration (1 mM) of ACh and MCh. However, as shown in the dose curves (figure 1 and figure 4), as much as 35% of MiI existed even at a physiological concentration of 5 μM MCh. This indicates that the MiI with 1 mM MCh and ACh should also apply to MiI with the physiological concentrations (micromoles), although the inhibition level under physiological concentrations was less profound. In conclusion, the muscarinic inhibition on nAChRs discovered in this work is sensitive to the history of mAChRs activation in the time window of seconds or sub-seconds. Release of presynaptic ACh stimulates both mAChRs and nAChRs in the post-synaptic membrane. The activation of mAChRs may then reduce nAChRs' response to subsequent ACh stimulations. Indeed, this phenomenon had been observed in adrenal chromaffin cells previously [14], although the mechanisms were not known until the present work. Since similar inhibition through autoreceptors exits in soma of cortical neurons (data not shown), as well as synapses [22–24], the MiI in this work might provide an adaptation upon repeated presynaptic transmitter release. Such adaptation should be gradually removed if presynaptic release stops for 1–3 min (figure 2). Future work should determine whether the MiI exists during synaptic neurotransmission.
Acknowledgement
We thank J. J. Zheng and M. Q. Hong for advice on cell preparations, X. K. Chen and W. Xiong for help with some experiments, Drs M. Lindau, H. P. Cheng and T. L. Xu for comments on the manuscript.
Author contributions
All authors contributed to the preparation of this manuscript and approved the final version. L.H., L.Z. and Z.Z., designed the work and wrote the paper. L.H., L.W., J.D., Q.Z., C.W. and L.Z. carried out the experiments. All authors provided interpretation of results presented.
Conflict of interests
The authors declare that no conflicts of interest exist.
Funding statement
This work was supported through funding from Chinese national grants supporting our laboratory since 1994, including recent grants from National Basic Research Program of China (2012CB518006), the National Natural Science Foundation of China (31228010, 31171026, 31100597, 31327901, 31400708, 31221002 and 31330024), the National Key Technology R&D Program (SQ2011SF11B01041) and a ‘985’ grant from the Department of Education of China.
References
- 1.Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. 1989. Distribution of alpha2, alpha3, alpha4, and beta2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J. Comp. Neurol. 284, 314–335. ( 10.1002/cne.902840212) [DOI] [PubMed] [Google Scholar]
- 2.Changeux JP, et al. 1998. Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res. Rev. 26, 198–216. ( 10.1016/S0165-0173(97)00040-4) [DOI] [PubMed] [Google Scholar]
- 3.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. 1996. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383, 713–716. ( 10.1038/383713a0) [DOI] [PubMed] [Google Scholar]
- 4.Lingle CJ, Maconochie D, Steinbach JH. 1992. Activation of skeletal muscle nicotinic acetylcholine receptors. J. Membr. Biol. 126, 195–217. ( 10.1007/BF00232318) [DOI] [PubMed] [Google Scholar]
- 5.MacDermott AB, Role LW, Siegelbaum SA. 1999. Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci. 22, 443–485. ( 10.1146/annurev.neuro.22.1.443) [DOI] [PubMed] [Google Scholar]
- 6.McGehee DS, Heath MJS, Gelber S, Devay P, Role LW. 1995. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269, 1692–1696. ( 10.1126/science.7569895) [DOI] [PubMed] [Google Scholar]
- 7.Taylor SC, Peers C. 2000. Three distinct Ca2+ influx pathways couple acetylcholine receptor activation to catecholamine secretion from PC12 cells. J. Neurochem. 75, 1583–1589. ( 10.1046/j.1471-4159.2000.0751583.x) [DOI] [PubMed] [Google Scholar]
- 8.Bernheim L, Mathie A, Hille B. 1992. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proc. Natl Acad. Sci. USA 89, 9544–9548. ( 10.1073/pnas.89.20.9544) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapiro MS, Loose MD, Hamilton SE, Nathanson NM, Gomeza J, Wess J, Hille B. 1999. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca2+ channels by using knockout mice. Proc. Natl Acad. Sci. USA 96, 10 899–10 904. ( 10.1073/pnas.96.19.10899) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slutsky I, Parnas H, Parnas I. 1999. Presynaptic effects of muscarine on ACh release at the frog neuromuscular junction. J. Physiol. Lond. 514, 769–782. ( 10.1111/j.1469-7793.1999.769ad.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Logothetis DE, Kim D, Northup JK, Neer EJ, Clapham DE. 1988. Specificity of action of guanine nucleotide-binding regulatory protein subunits on the cardiac muscarinic K+ channel. Proc. Natl Acad. Sci. USA 85, 5814–5818. ( 10.1073/pnas.85.16.5814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marty A, Tan YP. 1989. The initiation of calcium release following muscarinic stimulation in rat lacrimal glands. J. Physiol. Lond. 419, 665–687. ( 10.1113/jphysiol.1989.sp017892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soejima M, Noma A. 1984. Mode of regulation of the ACh-sensitive K-channel by the muscarinic receptor in rabbit atrial cells. Pflugers Arch. 400, 424–431. ( 10.1007/BF00587544) [DOI] [PubMed] [Google Scholar]
- 14.Zhou Z, Misler S. 1995. Action potential-induced quantal secretion of catecholamines from rat adrenal chromaffin cells. J. Biol. Chem. 270, 3498–3505. ( 10.1074/jbc.270.8.3498) [DOI] [PubMed] [Google Scholar]
- 15.Boehm S, Kubista H. 2002. Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol. Rev. 54, 43–99. ( 10.1124/pr.54.1.43) [DOI] [PubMed] [Google Scholar]
- 16.Noda M, et al. 1983. Structural homology of Torpedo californica acetylcholine receptor subunits. Nature 302, 528–532. ( 10.1038/302528a0) [DOI] [PubMed] [Google Scholar]
- 17.Sakmann B. 1992. Elementary steps in synaptic transmission revealed by currents through single ion channels. Science 256, 503–512. ( 10.1126/science.1373907) [DOI] [PubMed] [Google Scholar]
- 18.Du C, Role LW. 2001. Differential modulation of nicotinic acetylcholine receptor subtypes and synaptic transmission in chick sympathetic ganglia by PGE2. J. Neurophysiol. 85, 2498–2508. [DOI] [PubMed] [Google Scholar]
- 19.Kristufek D, Stocker E, Boehm S, Huck S. 1999. Somatic and prejunctional nicotinic receptors in cultured rat sympathetic neurones show different agonist profiles. J. Physiol. Lond. 516, 739–756. ( 10.1111/j.1469-7793.1999.0739u.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tachikawa E, Mizuma K, Kudo K, Kashimoto T, Yamato S, Ohta S. 2001. Characterization of the functional subunit combination of nicotinic acetylcholine receptors in bovine adrenal chromaffin cells. Neurosci. Lett. 312, 161–164. ( 10.1016/S0304-3940(01)02211-X) [DOI] [PubMed] [Google Scholar]
- 21.Malhotra RK, Wakade TD, Wakade AR. 1989. Cross-communication between acetylcholine and VIP in controlling catecholamine secretion by affecting cAMP, inositol triphosphate, protein kinase C, and calcium in rat adrenal medulla. J. Neurosci. 9, 4150–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cragg SJ, Greenfield SA. 1997. Differential autoreceptor control of somatodendritic and axon terminal dopamine release in substantia nigra, ventral tegmental area, and striatum. J. Neurosci. 17, 5738–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benoit-Marand M, Borrelli E, Gonon F. 2001. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J. Neurosci. 21, 9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Q, Reith ME, Walker QD, Kuhn CM, Carroll FI, Garris PA. 2002. Concurrent autoreceptor-mediated control of dopamine release and uptake during neurotransmission: an in vivo voltammetric study. J. Neurosci. 22, 6272–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Z, Misler S. 1995. Amperometric detection of stimulus-induced quantal release of catecholamines from cultured superior cervical ganglion neurons. Proc. Natl Acad. Sci. USA 92, 6938–6942. ( 10.1073/pnas.92.15.6938) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C, Zhou Z. 2002. Ca2+-independent but voltage-dependent secretion in mammalian dorsal root ganglion neurons. Nat. Neurosci. 5, 425–430. [DOI] [PubMed] [Google Scholar]
- 27.Wu JJ, He LL, Zhou Z, Chi CW. 2002. Gene expression, mutation, and structure−function relationship of scorpion toxin BmP05 active on SKCa channels. Biochemistry 41, 2844–2849. ( 10.1021/bi011367z) [DOI] [PubMed] [Google Scholar]
- 28.Wu B, Wang YM, Xiong W, Zheng LH, Fu CL, Bruce IC, Zhang C, Zhou Z. 2005. Optimization of a multi-channel puffer system for rapid delivery of solutions during patch-clamp experiments. Front. Biosci. 10, 761–767. ( 10.2741/1570) [DOI] [PubMed] [Google Scholar]
- 29.Chen XK, Wang LC, Zhou Y, Cai Q, Prakriya M, Duan KL, Sheng ZH, Lingle C, Zhou Z. 2005. Activation of GPCRs modulates quantal size in chromaffin cells through Gβγ and PKC. Nat. Neurosci. 8, 1160–1168. ( 10.1038/nn1529) [DOI] [PubMed] [Google Scholar]
- 30.Zhou Z, Neher E. 1993. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J. Physiol. 469, 245–273. ( 10.1113/jphysiol.1993.sp019813) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Z, Misler S. 1996. Amperometric detection of quantal secretion from patch-clamped rat pancreatic β-cells. J. Biol. Chem. 271, 270–277. ( 10.1074/jbc.271.1.270) [DOI] [PubMed] [Google Scholar]
- 32.Chen XW, et al. 2008. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J. Cell Biol. 181, 791–801. ( 10.1083/jcb.200711021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang HP, et al. 2007. Long latency of evoked quantal transmitter release from somata of locus coeruleus neurons in rat pontine slices. Proc. Natl Acad. Sci. USA 104, 1401–1406. ( 10.1073/pnas.0608897104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, et al. 2014. Modulation of dopamine release in the striatum by physiologically relevant levels of nicotine. Nat. Commun. 5, 3925 ( 10.1038/ncomms4925) [DOI] [PubMed] [Google Scholar]
- 35.Guerineau NC, Bossu JL, Gahwiler BH, Gerber U. 1995. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J. Neurosci. 15, 4395–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sine SM, Steinbach JH. 1987. Activation of acetylcholine receptors on clonal mammalian BC3H-1 cells by high concentrations of agonist. J. Physiol. Lond. 385, 325–359. ( 10.1113/jphysiol.1987.sp016496) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Legendre P, Ali DW, Drapeau P. 2000. Recovery from open channel block by acetylcholine during neuromuscular transmission in zebrafish. J. Neurosci. 20, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maconochie DJ, Steinbach JH. 1998. The channel opening rate of adult- and fetal-type mouse muscle nicotinic receptors activated by acetylcholine. J. Physiol Lond. 506, 53–72. ( 10.1111/j.1469-7793.1998.053bx.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Neher E. 1993. Calcium permeability of nicotinic acetylcholine receptor channels in bovine adrenal chromaffin cells. Pflugers Arch. 425, 511–517. ( 10.1007/BF00374879) [DOI] [PubMed] [Google Scholar]
- 40.Zwart R, Vijverberg HPM. 1997. Potentiation and inhibition of neuronal nicotinic receptors by atropine: competitive and noncompetitive effects. Mol. Pharmacol. 52, 886–895. ( 10.1124/mol.52.5.886) [DOI] [PubMed] [Google Scholar]
- 41.Chow RH. 1991. Cadmium block of squid calcium currents. Macroscopic data and a kinetic model. J. Gen. Physiol. 98, 751–770. ( 10.1085/jgp.98.4.751) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hampson AJ, Grimaldi M. 2002. 12-Hydroxyeicosatetrenoate (12-HETE) attenuates AMPA receptor-mediated neurotoxicity: evidence for a G-protein-coupled HETE receptor. J. Neurosci. 22, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neher E. 1988. The influence of intracellular calcium concentration on degranulation of dialysed mast cells from rat peritoneum. J. Physiol. Lond. 395, 193–214. ( 10.1113/jphysiol.1988.sp016914) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takano K, Yamashita N, Fujita T. 1996. Proadrenomedullin NH2-terminal 20 peptide inhibits the voltage-gated Ca2+ channel current through a pertussis toxin-sensitive G protein in rat pheochromocytoma-derived PC 12 cells. J. Clin. Invest. 98, 14–17. ( 10.1172/JCI118758) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teschemacher AG, Seward EP. 2000. Bidirectional modulation of exocytosis by angiotensin ii involves multiple G-protein-regulated transduction pathways in Chromaffin cells. J. Neurosci. 20, 4776–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hille B. 1994. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 17, 531–536. ( 10.1016/0166-2236(94)90157-0) [DOI] [PubMed] [Google Scholar]
- 47.Suh BC, Hille B. 2002. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35, 507–520. ( 10.1016/S0896-6273(02)00790-0) [DOI] [PubMed] [Google Scholar]
- 48.Selyanko AA, Stansfeld CE, Brown DA. 1992. Closure of potassium M-channels by muscarinic acetylcholine-receptor stimulants requires a diffusible messenger. Proc. R. Soc. Lond. B 250, 119–125. ( 10.1098/rspb.1992.0139) [DOI] [PubMed] [Google Scholar]
- 49.Koh DS, Hille B. 1997. Modulation by neurotransmitters of catecholamine secretion from sympathetic ganglion neurons detected by amperometry. Proc. Natl Acad. Sci. USA 94, 1506–1511. ( 10.1073/pnas.94.4.1506) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mollard P, Seward EP, Nowycky MC. 1995. Activation of nicotinic receptors triggers exocytosis from bovine chromaffin cells in the absence of membrane depolarization. Proc. Natl Acad. Sci. USA 92, 3065–3069. ( 10.1073/pnas.92.7.3065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khakh BS, Zhou XP, Sydes J, Galligan JJ, Lester HA. 2000. State-dependent cross-inhibition between transmitter-gated cation channels. Nature 406, 405–410. ( 10.1038/35019066) [DOI] [PubMed] [Google Scholar]
- 52.Nakazawa K. 1994. ATP-activated current and its interaction with acetylcholine-activated current in rat sympathetic neurons. J. Neurosci. 14, 740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan W, Du C, Siegelbaum SA, Role LW. 1998. Modulation of nicotinic AChR channels by prostaglandin E2 in chick sympathetic ganglion neurons. J. Neurophysiol. 79, 870–878. [DOI] [PubMed] [Google Scholar]
- 54.Wessler I. 1989. Control of transmitter release from the motor nerve by presynaptic nicotinic and muscarinic autoreceptors. Trends Pharmacol. Sci. 10, 110–114. ( 10.1016/0165-6147(89)90208-3) [DOI] [PubMed] [Google Scholar]