

Abstract
Extracellular adenosine triphosphate (ATP) serves as a signal for diverse physiological functions, including spread of calcium waves between astrocytes, control of vascular oxygen supply and control of ciliary beat in the airways. ATP can be released from cells by various mechanisms. This review focuses on channel-mediated ATP release and its main enabler, Pannexin1 (Panx1). Six subunits of Panx1 form a plasma membrane channel termed ‘pannexon’. Depending on the mode of stimulation, the pannexon has large conductance (500 pS) and unselective permeability to molecules less than 1.5 kD or is a small (50 pS), chloride-selective channel. Most physiological and pathological stimuli induce the large channel conformation, whereas the small conformation so far has only been observed with exclusive voltage activation of the channel. The interaction between pannexons and ATP is intimate. The pannexon is not only the conduit for ATP, permitting ATP efflux from cells down its concentration gradient, but the pannexon is also modulated by ATP. The channel can be activated by ATP through both ionotropic P2X as well as metabotropic P2Y purinergic receptors. In the absence of a control mechanism, this positive feedback loop would lead to cell death owing to the linkage of purinergic receptors with apoptotic processes. A control mechanism preventing excessive activation of the purinergic receptors is provided by ATP binding (with low affinity) to the Panx1 protein and gating the channel shut.
Keywords: ATP, Pannexin, conductance, permeability, potassium, allosteric
1. Introduction
Adenosine triphosphate (ATP) and its metabolites adenosine diphosphate (ADP), adenosine monophosphate (AMP) and adenosine serve as transmitter molecules in many physiological and pathological body functions [1–3]. While the function of ATP as cellular energy store was rapidly accepted after its discovery, the transmitter function of ATP took decades to become accepted in the scientific community. The signalling function of ATP was first postulated in 1929 [4] and reformulated in 1953 and 1954 [5,6]. Starting in 1970, the tenacity of Burnstock et al. [7–9] finally led to the establishment of the field of purinergic transmission. The field has now grown to such an extent that the ‘Purines 2014’ meeting was attended by approximately 550 researchers.
To act as a transmitter, ATP has to negotiate the plasma membrane of the ATP releasing cell before it can act—sometimes after breakdown as ADP, AMP or adenosine—on purinergic receptors (P1, P2X or P2Y) on a targeted cell. Four pathways for ATP release need to be considered.
(A) Like many other transmitters, ATP can be released by exocytosis. In fact, often ATP is co-packaged and co-released with classical transmitters, such as acetylcholine or norepinephrine [10–13]. Further support for exocytotic/vesicular ATP release is the identification of vesicular ATP transport proteins [14,15]. Exocytosis allows for fast and targeted release of ATP onto receptors and is the prominent if not exclusive ATP release mechanism at nerve terminals [16,17].
(B) Several observations indicate the existence of alternative ATP release pathways. ATP is released from cells, such as erythrocytes [18], that do not contain vesicles under physiological conditions. In many cell types, there is uptake of extracellular tracer molecules correlated with ATP release [19,20]. Several membrane channel blockers, including carbenoxolone, can attenuate or even completely inhibit ATP release from cells and block tracer uptake [21]. These observations are all suggestive of a channel-mediated ATP release mechanism allowing the passive efflux of ATP from the cell following the concentration gradient. Actually, channel-mediated ATP release is the most ancient form [22].
(C) Because certain inhibitors of proteins mediating active membrane transport—a prime example is the urate transport inhibitor probenecid—were observed to interfere with ATP release from cells, it has been suggested that transport proteins can be involved in the release process [23]. However, that probenecid also is a strong inhibitor of the pannexin1 (Panx1) channel [24] undermines the evidence for transport-mediated ATP release at the plasma membrane.
(D) A fourth mechanism is only seen under pathological conditions. Lytic release of ATP through a compromised plasma membrane can be a consequence of direct trauma or part of programmed cell death.
In many if not most cell types, including astrocytes, both vesicular and channel-mediated ATP release mechanisms coexist, complicating detailed analysis of either mechanism. Therefore, to illuminate channel-mediated ATP release as it occurs in astrocytes or neurons, it is imperative to elucidate basic aspects of this process in cells where vesicular release is absent. The mature human erythrocyte has no organelles and no vesicles. Erythrocytes, when stimulated by a low oxygen environment or shear stress, release ATP [18]. Thus, the erythrocyte is ideal for studying basic aspects of channel-mediated ATP release.
The focus of this review is exclusively on channel-mediated ATP release. While several membrane channel proteins have been proposed to mediate ATP release, special emphasis is on the pannexin1 (Panx1) channel. A broad set of experimental evidence indicates that Panx1 mediates ATP release under physiological and pathological conditions in several cell types. In the plasma membrane, Panx1 appears as a hexameric assembly of Panx1 subunits [25] to form a transmembrane channel called pannexon [26]. Throughout the text, this term is used to indicate the fully oligomerized state, whereas the term Panx1 is used when reference is made to the monomeric protein.
2. Requirements for a membrane protein to serve as an ATP release channel
Several membrane proteins have been presented as putative ATP release channels, including the cystic fibrosis transmembrane regulator (CFTR) [27], the maxi anion channel [28], connexin 43 hemichannels [29], calcium homeostasis modulator 1 (CALHM 1) [30] and pannexon channels [31]. To serve as an ATP release channel, specific requirements have to be fulfilled. As mundane as several of these requirements are, they are not exhibited by some of the ATP release channel candidates. (i) ATP must demonstrably permeate the channel. As basic as this property gets, CFTR has been shown not to be permeable to ATP [32]. This does not preclude a regulatory role for CFTR on a genuine ATP release channel. However, CFTR by itself does not qualify as a permeation pathway for ATP. (ii) The ATP-permeable channel must be expressed in the cells from which ATP is released. Connexin 43 hemichannels have been suggested to mediate the release of ATP in many cell types [20]. Yet, erythrocytes (and other cells) release ATP, but do not express connexin 43 [21]. (iii) Expression of the channel must be localized at the surface where ATP is secreted. In several polarized cells, including airway epithelia, ATP release occurs exclusively at the apical surface. The two connexins expressed in these cells (Cx 30 and Cx 31), consistent with their gap junction function, are exclusively expressed at the basolateral membrane and are not detectable at the apical membrane [33]. (iv) The ATP channel must be capable of being active under physiological conditions. ATP release is involved in multiple physiological functions that occur in a normal ionic environment and often at the resting membrane potential. Both connexin hemichannels and CALHM 1 require removal of extracellular calcium ions to levels that may even not occur under pathological conditions. Furthermore, these channels open at positive potentials, which occur in excitable cells only for brief moments. (v) For efficient ATP release, a candidate ATP release channel should be activatable at or close to the resting membrane potential. At this potential, an optimal electrochemical gradient for ATP flux exists, because both concentration gradient and electrical gradient are in the outward direction for the negatively charged ATP molecule. (vi) The candidate ATP release channel should exhibit poor or no charge selectivity, because dyes in the size range of ATP of either charge can be used as a surrogate measure for ATP release. This requirement excludes the anion-selective maxi-anion channel as a ubiquitous ATP release channel. (vii) Two arguments suggest that a candidate ATP release channel should be activated by ATP through purinergic receptors: 1. In several tissues including astrocytes, ATP and purinergic receptors are involved in the propagation of calcium waves. 2. In several cells, including erythrocytes, the phenomenon of ATP-induced ATP release is observed. With the exception of the Panx1 channel, no experimental evidence for an activation of a candidate ATP release channel by ATP through purinergic receptors has been provided. (viii) Because the activation of an ATP release channel by ATP through purinergic receptors represents a potentially dangerous positive feedback loop, there must be a control mechanism preventing excessive activation of cells. In particular, a combination of the ‘death receptor’ P2X7 with an ATP release channel would be dangerous in the absence of a control mechanism. The simplest form of such a control would be the inhibition of the ATP release channel by its own permeant, ATP, involving a low affinity binding site. Inhibition by ATP has been demonstrated for the Panx1 channel, but for no other candidate ATP release channel. (ix) The pharmacology of the candidate ATP release channel should match that of non-vesicular ATP release. Because of overlapping pharmacologies between the various candidate ATP release channels, this issue has created some confusion in the literature. However, a few drugs are of some limited value for identification of the channel involved in ATP release. For example, probenecid inhibits ATP release and inhibits pannexons, but does not affect channels formed by connexins. (x) Mutations or chemical modification of a candidate ATP release channel protein within the channel pore or in regions controlling the channel's patency should affect ATP release.
3. The pannexon channel
Before the discovery of pannexins (Panx1, Panx2 and Panx3), the pharmacology of connexin hemichannels, based on a limited number of drugs, best matched ATP release in a number of cell types. However, a series of arguments (some mentioned above) raised suspicion about connexins being responsible for ATP release under physiological conditions. Because pannexins were discovered in vertebrates on the basis of limited sequence similarity with the invertebrate innexins, it was to be expected that the pharmacology of pannexins would be similar to that of innexins and connexins. This expectation led to the initial motivation to analyse the ability of Panx1 to be an ATP release channel [31]. Indeed, most connexin and innexin gap junction inhibitors subsequently were found to also inhibit the Panx1 channel [34,35].
Although pannexins were discovered 15 years ago as a ‘second family of gap junction proteins’ in vertebrates [36], there still is no firm evidence for their ability to indeed form gap junctions. Cell–cell coupling reported as a consequence of Panx1 expression could have been mediated by connexin-based gap junction channels.
There is ample evidence to exclude a gap junction function of pannexins, including (i) their expression in cells that do not form gap junctions, such as erythrocytes [21]; (ii) the expression in polarized cells only at the apical membrane [37,38], which is not in contact with any other cell; and (iii) the glycosylation of the pannexin proteins, which prevents the docking of pannexons in apposed cell membranes [25,39].
Unquestionably, Panx1 forms a patent non-junctional membrane channel that allows the exchange of molecules smaller than 1.5 kDa between intra- and extracellular space [21,31,40,41]. (Of the three pannexins, only the properties of the Panx1 channel are sufficiently characterized for inclusion in this discussion.) Despite the lack of sequence homology to connexins and the very limited homology to innexins, pannexins fold in a similar way: both amino- and carboxy-termini are located intracellularly, four segments traverse the plasma membrane and two loops face the extracellular space [21]. Six Panx1 subunits oligomerize [25] to form a pannexon channel in the non-junctional plasma membrane [26]. The channel properties are unusual, and, as shown below, vary with the mode of stimulation.
Innexins are bifunctional, as they can form gap junction channels as well as non-junctional membrane channels, innexons. The properties of innexons as far as tested are identical to pannexon properties [42–45].
4. Evidence for an ATP release function of Panx1
The pannexon channel fulfils all criteria listed above for a legitimate ATP release channel.
(a) Reversal potential measurements in a potassium ATP gradient suggest high ATP permeability of pannexons in cells expressing Panx1 exogenously [31] or endogenously [21]. Expression of Panx1 also confers stimulus-dependent ATP release to cells [31].
(b) Cells for which channel-mediated ATP release has been documented express Panx1, including erythrocytes, airway epithelial cells, astrocytes, macrophages, hepatocytes, lymphocytes, kidney tubule cells and pituitary cells. Inversely, knockdown or knockout of Panx1 expression leads to attenuation or loss of ATP release in erythrocytes [46], astrocytes [47], airway epithelial cells [37,48], macrophages [49] and pituitary cells [50].
(c) In polarized cells, including airway epithelial cells and kidney tubulus cells, Panx1 co-localizes with the ATP release site [37,38].
(d) Pannexon channels can open under physiological conditions, including regular extracellular calcium concentration [34] in response to stimulation by mechanical stress, low oxygen environment, the ligands ATP, glutamate and angiotensin II binding to their respective receptors, and increased intracellular calcium ion concentration.
(e) Activation of pannexons by several of the aforementioned stimuli can occur at the resting membrane potential. This property allows for efficient release of ATP downstream of both the concentration and voltage gradients.
(f) ATP release has been determined in many cells to correlate with the uptake of extracellular tracer molecules (typically fluorescent dyes) that are either negatively or positively charged. The pannexon channel is permeable to both negatively charged and positively charged dyes [21,24,51].
(g) ATP acting through either metabotropic P2Y [52] or ionotropic P2X [53] receptors can activate pannexon channels, enabling the phenomenon of ATP-induced ATP release and thereby amplifying the ATP signal.
(h) The positive feedback loop provided by ATP-induced ATP release is counteracted by a negative feedback. At high concentrations, extracellular ATP inhibits currents through pannexons [54]. This effect is mediated by amino acids in both extracellular loops of Panx1 [55]. In several cell types, including astrocytes, neurons and macrophages, Panx1 is closely associated with the purinergic receptor P2X7, and pannexons can be opened by ATP through this association. Several ligands to the P2X7 receptor, irrespective of whether they act as agonist or antagonist at the receptor, inhibit pannexon function. The affinity of the ATP binding site on Panx1 is lower than that on the P2X7 receptor. This constellation allows for initial amplification of the ATP signal by the positive feedback loop between pannexon and receptor. Under physiological conditions, the amplification is limited by the negative feedback on the pannexon. This represents an important mechanism to preserve cell integrity, because without it, apoptotic pathways would be activated even with minute elevations of extracellular ATP.
(i) The pharmacology of pannexons matches well with that of non-vesicular ATP release. Pannexons are inhibited by a variety of compounds belonging to different chemical groups and pharmacological classes [35]. Prime examples are probenecid, glyburide and artemesinin. Only one compound, dipyridamole, known to affect ATP release does not inhibit pannexons [46]. Thus, in some cells, including mouse erythrocytes, another non-vesicular ATP release pathway must exist.
(j) Mutations and chemical modification of Panx1 affect ATP release, consistent with an ATP permeation pathway provided by pannexons [56].
5. Release of compounds other than ATP
The pannexon's maximal single-channel conductance is on the order of 500 pS [31,57,58]. Flux of tracer molecules through pannexons does not exhibit charge selectivity, as both negatively and positively charged fluorescent dyes applied at the extracellular surface enter cells when pannexons are open. The exact size exclusion limit for pannexons has not been determined. However, polyethyleneglycol molecules up to 1.5 kDa [41] enter the channel to a sufficient depth to affect channel conductance. Whether this is a measure of the pore itself or that of an extended channel vestibulum needs further investigation.
Nevertheless, based on the large pore size and the apparent lack of selectivity, it can be expected that molecules in the size range of ATP will pass through pannexons without hindrance. The signalling molecules uridine 5′-triphosphate (UTP) and glutamate fall into this category. Furthermore, it appears that epoxyeicosatrienoic acids permeate pannexons [59].
6. Expression and function of pannexons in the central nervous system
At present, it is uncertain whether the release of ATP and similar-sized molecules, such as glutamate and other transmitters, is the exclusive function of pannexons. Nevertheless, there is accumulating evidence that pannexons are involved in multiple functions of the central nervous system (CNS). This is indicated by the apparent involvement of pannexons in pathological states such as epilepsy [60], migraine [61], hypoxic depolarization [57], Crohn's disease with its neuronal loss in the enteric nervous system [62] and neuroinflammation [63]. Panx1 is widely expressed in the body, including the brain. Panx2, which is essentially uncharacterized in terms of channel properties and functional roles [64], is almost exclusively expressed in the CNS. Expression of Panx1 includes astrocytes and neurons [65]. In neurons, it appears that Panx1 may exist in different conformations or associations with other proteins. Immunohistochemical analysis of Panx1 expression has shown that different validated anti-Panx1 antibodies stain different parts of neurons [66]. The most likely interpretation of this phenomenon is epitope masking or unmasking by either differential protein folding, differential post-translational modification or differential interaction with other proteins in cell bodies and dendrites.
Several intimate interactions between Panx1 and other proteins in astrocytes and neurons have been identified. A direct physical interaction is indicated by co-immunoprecipitation for Panx1 and the P2X7 receptor [63] or the potassium channel subunit Kvβ3 [67]. Furthermore, Panx1 can be activated by glutamate binding to NMDA receptors, by noradrenaline through alpha adrenergic receptors, or by angiotensin II binding to AT1 receptors [68–70]. The interaction of Panx1 with Kvβ3 is intriguing, as the pharmacology of the pannexon is changed by this interaction. The inhibitory effect of carbenoxolone, probenecid and reducing agents was attenuated in co-expressing cells when compared with the effect in cells expressing Panx1 alone. In the CNS, Panx1 and Kvβ3 co-localize in pyramidal neurons in the hippocampus and in Purkinje cells in the cerebellum [67]. The specific functional role of this co-expression is not known.
7. Activation of pannexons in physiological and pathological conditions
An experimentally convenient way to activate pannexons is by depolarizing voltage steps applied to cells. However, the voltages required for channel activation are in the positive range and thus do not occur physiologically except in excitatory cells for a brief period during the peak of an action potential. Nevertheless, a number of physiological stimuli for pannexons have been identified that open the channel at the resting membrane potential.
(a) Pannexons are mechanosensitive and can be opened at −50 mV in excised membrane patches by application of negative pressure applied to the patch pipette [31]. This channel property could be the basis for the observed swelling-induced ATP release from erythrocytes [21] and airway epithelial cells in response to hypotonic stress [37,48]. Activation of pannexons in response to cell swelling has also been observed in other cell types, including lens [71], neurons [72], astrocytes [73], bovine ciliary epithelial cells [74] and fibrosarcoma cells [75].
(b) Low oxygen elicits pannexon-mediated ATP release from erythrocytes [21,76] and neurons [57]. In neurons, low oxygen could be sensed via the orthodox mitochondrial pathway, which in turn activates pannexons. In the organelle-free erythrocyte without mitochondria, a more direct activation of pannexons must exist. Because the activity of the potassium channel slo1 BK can be modulated by haem [77], a similar mechanism may control pannexon activity in erythrocytes in an (oxygen)-dependent way.
(c) Increase in cytoplasmic calcium ion concentration ([Ca2+]i) has been shown to activate pannexons in excised inside–out membrane patches [52]. Consistent with this activation mechanism, pannexons are activated by ligands binding to metabotropic receptors signalling through IP3 and [Ca2+]i. Receptor-mediated pannexon activation has been shown for P2Y receptors [52,78] and angiotensin II receptors [70]. Buffering intracellular Ca2+ with 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA) prevents activation of pannexons by angiotensin II and ATP through their respective receptors [70,78]. This observation lends further support to the notion that increases in [Ca2+]i can activate pannexons. Because each point has its counterpoint, it should be noted that induction of pannexon currents by increased [Ca2+]i can also be missed [79].
(d) Pannexons can be opened by ATP binding to purinergic receptors. In addition to the aforementioned activation through the metabotropic P2Y receptors, binding of ATP to ionotropic P2X7 receptors also leads to pannexon activity [51,53]. It appears that some other P2X receptors have the same capability [80]. While it is conceivable that the Ca2+ influx through a P2X receptor can trigger pannexon activation, this process is not needed, because removal of extracellular calcium does not attenuate the ATP-induced pannexon current [53]. Thus, other signalling events must be involved. Because Panx1 can be immunoprecipitated with P2X protein by antibodies directed to either Panx1 or P2X [63,80,81], an activation of pannexons by direct protein–protein interaction is a distinct possibility. In addition (or instead), another signalling pathway appears to be involved the activation mechanism. Pannexon currents in cells co-expressing Panx1 and P2X7R are attenuated by inhibition of Src tyrosine kinases by a TAT-P2X7 peptide representing the death signal domain in the COOH terminus of P2X7R, by the P2X7R blocker KN-62 or by the Src tyrosine inhibitor PP2 [82].
While the identification of the molecular components involved in ATP-induced ATP release is recent, the phenomenon as such has long been known [83–85]. Considering that ATP at high concentrations in the extracellular medium leads to cell death, this positive feedback loop for ATP release is potentially dangerous. Under physiological conditions, the negative feedback in form of pannexon inhibition by extracellular ATP counteracts the ATP-induced ATP release and thereby limits overstimulation of apoptotic/pyroptotic pathways [86].
(e) Glutamate binding to NMDA receptors activates pannexons [68]. This process is thought to be responsible for anoxic depolarization of pyramidal neurons, as occurring in stroke. Intriguingly, the signalling between receptor and pannexon is similar to that described for the P2X7/pannexon interaction. While Ca2+ influx through the NMDA receptors in principle could be sufficient for the activation process, experimental evidence here also points to an important role of Src kinases in the activation of pannexons [87].
(f) Pannexons can be activated by agonists of the alpha 1D adrenergic receptor [69]. This amplifies the adrenergic signalling cascade enhancing the vasoconstrictive effect of adrenergic agonists in resistance vessels. The mechanism of activation of pannexons through alpha adrenergic receptors is not yet known. Because alpha adrenergic receptors typically signal through phosphatidylinositol–IP3–Ca2+, pannexons might be directly stimulated by increased [Ca2+]i, although other possibilities remain [88].
(g) Thrombin binding to its receptor increases [Ca2+]i and stimulates release of ATP [89]. Pre-loading the cells with BAPTA attenuates this, as if the increase in [Ca2+]i were directly responsible for pannexon opening and ATP release. While pharmacological evidence indicates that the RhoA/ROCK pathway is also activated by the thrombin receptor activation [48], it is not known if this pathway is obligatory.
(h) Bradykinin-induced increase in [Ca2+]i involves pannexon activity and P2Y receptor activation, as indicated by pharmacological evidence. Whether the initial increase in [Ca2+]i is the primary stimulus for pannexons, which then activate P2Y receptors to further amplify the signal, is not clear [90].
(i) Histamine-induced ATP release seems to follow the same pattern as described for bradykinin [91].
(j) Angiotensin II-induced ATP release is inhibited by intracellular calcium chelation with BAPTA and thus pannexon activation could be a direct consequence of the rise in intracellular [Ca2+] [70].
(k) Thromboxane receptor-mediated activation of pannexons, in contrast, involves the cAMP → protein kinase A pathway [92].
(l) Regulation of catecholamine release from adrenal chromaffin cells apparently involves pannexon activity and ATP release, which in turn amplifies the calcium signalling triggered by nicotinic receptors [93].
(m) Increases in extracellular K+ concentration ([K+]o) stimulates pannexon-mediated ATP release [63]. The dose–response curve shows a steady increase in ATP release from 10 mM [K+]o to at least 150 mM [K+]o [47,58,63]. Several independent observations indicate that [K+]o acts on pannexons. Xenopus oocytes expressing Panx1 de novo acquire K+-stimulated ATP release. KO/knockdown of Panx1 abolishes K+-induced ATP release in astrocytes. The K+-induced pannexon activation is not an effect of depolarization as judged by the voltage dependence of pannexons. Indeed, [K+]o activates pannexons directly at any membrane potential in the range −100 to +100 mV under voltage clamp conditions (see below).
Activation of pannexons by all the aforementioned stimuli is rapidly reversible. For many physiological functions involving pannexons, such as platelet activation, oxygen supply, hearing, airway cilia beat regulation, quick reversibility is indispensable. Considering pannexons' large conductance and the permeability to all molecules up to 1.5 kDa, prolonged activation of pannexons would lead to the rundown of ionic gradients and loss of energy necessary for cell survival. Indeed, excessive or irreversible activation of pannexons leads to cell death [53,63,94,95]. The self-inhibition of pannexons by ATP is a component of fine tuning of a potentially dangerous channel by preventing prolonged opening.
(n) Cleavage of Panx1 by caspase 3 at its cognizant site in the carboxyterminal sequence of the protein irreversibly activates pannexons, invariably killing the cells [95]. This type of activation occurs in only a select group of cells and only after these cells are committed to apoptotic cell death. The expected ensuing ATP release mobilizes macrophages to the site of cell death. ATP in this setting has been termed the ‘find me signal’. A similar phenomenon also occurs in brain lesions, where ATP initiates the response of microglia to an injury [96]. In a leech model for nerve injury, which is amenable to a rigorous analysis of molecular events, the signalling responsible for migration of microglia to a lesion is complex. Directional movement and accumulation of microglia involve at least three signals: the ATP released at the lesion site is the ‘go’ signal, NO is the ‘where’ signal [97] and ArA is a ‘stop’ signal [44].
Caspase cleavage of Panx1, however, is not an obligatory event in the anoxic depolarization that precedes death of neurons [87]. Western blots of hippocampal slice preparations show only the uncleaved form of Panx1 under conditions that open pannexons. In addition, pharmacology shows that anoxia-induced pannexon activation is caspase-independent.
Published data do not show whether the caspase 3 cleaved pannexon is permeable to ATP. Single-channel recordings of pannexons under conditions where the caspase-cleaved form should prevail show a unitary conductance of 75 pS [98] or even as low as 20 pS [99]. As shown below, the low conductance pannexon is not permeable to ATP. Either the patches with the low conductance pannexon did not happen to contain the fully activated, ATP-permeable configuration or the caspase-cleaved Panx1 does not form pannexons with ATP permeability. The latter interpretation is also suggested by a lack of reversal potential shift associated with caspase 3 cleavage of Panx1 [95,99,100]. In contrast, the K+-stimulated pannexon is ATP permeable [31,58] and exhibits a large shift in the reversal potential [47].
8. Two channel conformations of Panx1
A preponderance of evidence designates pannexons as the main conduit for efflux of ATP from many cell types (reviewed in references [26,86,101]). Nonetheless, recent independent studies have shown the pannexon to be a highly selective chloride channel with no ATP permeability [102,103]. Furthermore, in these studies, the pannexon exhibited a unitary conductance of 74 [103] or 68 pS [102] rather than exhibiting the complex gating behaviour and maximal conductance of 500 pS reported earlier [21,31,52,57].
Chloride selectivity of pannexons in the Ma et al. [102] and Romanov et al. [103] studies is indicated by two basic observations. Replacement of extracellular Na+ by NMDG+ had no effect on pannexon currents. Replacement of extracellular Cl− by gluconate strongly attenuated the currents and replacement by glutamate and aspartate abolished pannexon currents altogether. Furthermore, no ATP release from cells overexpressing Panx1 was observed despite robust pannexon currents.
Several explanations could account for the discrepancies in the reported channel conductances and permeabilities of pannexons. For example, different cell types with differing modulators of pannexons could be responsible for the differing channel properties [101,103]. Alternatively, Panx1 may serve as a regulator of the actual ATP release conduit.
Scrutiny of the experimental conditions used in the various studies, however, reveals marked discrepancies. Notably, the low conductance, ATP-impermeable pannexons were observed with the channels activated exclusively by voltage. In contrast, the high conductance, ATP-permeable pannexons were observed with different physiological/pathological channel stimuli. A systematic analysis of this phenomenon revealed that, depending on the stimulus modality, pannexons assume different conformations with different conductances and permeabilities [58].
The exclusively voltage-gated pannexon is not permeable to ATP. Measuring ATP release from voltage-clamped oocytes with a luciferase assay revealed that no pannexon-mediated release occurred despite robust voltage-induced pannexon currents [58]. Increasing [K+]o under otherwise identical conditions resulted in an ATP release of the same magnitude as observed in K+-stimulated unclamped oocytes. The K+-stimulated ATP release was independent of the membrane holding potential in the range from −60 to +40 mV.
The two permeability states of pannexons correlate with the two conductance states of the channel. The previously published differences in conductance states were found in different cell types, opening the possibility for cell-type-specific properties of pannexons. However, the two conductance states can be seen in the same cell and even in the same channel contained in an excised outside–out membrane patch [58]. A simple change of the extracellular ion composition induces the change in conductance. With Na+ in the extracellular solution, the channel opens only at positive potentials as also seen for whole cell currents. The unitary conductance under this condition is about 50 pS and thus in the same range as reported for other cell types under comparable experimental conditions [102,103]. Replacing extracellular Na+ with K+ opens pannexons in the voltage range from −100 to +100 mV, also consistent with macroscopic currents. In high [K+]o, the pannexon currents are complex, indicating the existence of several subconductance states and a maximal conductance of ca 500 pS (figure 1). The large pannexon conductance and/or ATP permeability have been observed in several cell types with different physiological or pathological stimuli. This includes K+-activated pannexons in oocytes expressing Panx1, erythrocytes and astrocytes, [31], low-oxygen-stimulated neurons or erythrocytes [21,57], mechanically stressed pannexons in oocytes or erythrocytes [21,31], intracellular calcium-induced pannexon activity in oocytes [52], ligand–receptor activation mechanisms of pannexons listed above, some possibly by elevated [Ca2+]i or other signalling chains. Thus, K+-activation of pannexons mimics that by physiological activators of the channel. K+ can therefore be used as a surrogate for the physiological activators. For experiments, K+-stimulation is considerably more convenient than some of the physiological stimuli (figure 2).
Figure 1.
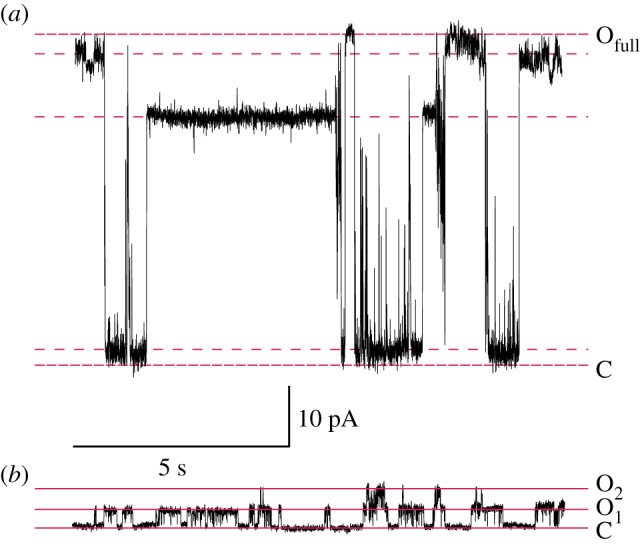
Single-channel currents through Panx1 channels with K+ or Na+ in the extracellular solution. (a) A pannexon in an inside–out membrane patch exposed to high extracellular [K+] and clamped at –100 mV exhibited a maximal conductance of approximately 500 pS. The fully open and fully closed states are indicated by red lines; the dashed lines indicate levels of three major subconductances. Both the bath and pipette solutions contained 140 mM KGlu, 10 mM KCl and 5 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES; pH 7.5). (b) An outside–out patch exposed to a low extracellular [K+] (Ringer solution) and clamped at +50 mV containing two channels that opened to the current levels O1 and O2. Both the pipette and bath solutions contained 140 mM NaCl, 10 mM KCl and 5 mM TES (pH 7.5). As shown previously, the conversion between small and large channel conductance can be observed in the same channel in an outside–out patch by switching between normal Ringer and high K+ in the extracellular solutions [58]. (Online version in colour.)
Figure 2.

Permeability and conductances of pannexons vary with the activation mode. The pannexon has high conductance and is permeable to ATP, when activated by various physiological stimuli or by elevated extracellular [K+]. Activation of pannexons only by stepping the membrane potential to positive values results in a small channel conductance and selective permeability to Cl−. It is unclear which channel figuration is assumed by the caspase 3 cleaved pannexon. No direct measurements of ATP permeability of the caspase-cleaved pannexon are available, and the single-channel conductance has been reported to be 20 or 75 pS. (Online version in colour.)
In retrospect, it is clear that the search for ATP permeability in the exclusively voltage-gated pannexon would be futile. An unfavourable voltage gradient at high positive membrane potentials limits or even prevents ATP efflux from cells following the concentration gradient. For example, no ATP efflux through connexin hemichannels, which open only at extreme positive voltages, is measurable at +80 mV, but ATP release occurs briefly during the tail currents during repolarization when some of the channels are still open and both concentration and voltage gradients are directed outwards [104]. For the same reasons, one cannot expect a robust influx of positively charged YoPro (4-[(3-methyl-1,3-benzoxazol-2(3H)-ylidene)methyl]-1-[3-(trimethylammonio)propyl]quinolinium diiodide) against the strong electrical gradient of positive voltages required to open pannexons by voltage alone [105].
That different stimuli trigger two conformations of pannexons is also indicated by differential thiol reagent accessibility. Panx1 has a carboxyterminal cysteine, which can be reacted with thiol reagents in the voltage-activated state of the channel [56]. In contrast, pannexons opened with extracellular K+ cannot be modified by thiol reagents [58]. These observations suggest that the inner (cytoplasmic) aspect of the pannexon assumes drastically different conformations with different stimuli. Cysteines replacing autochthonous amino acids in the outer (extracellular) portion of pannexon channel permeation pathway can be reacted with thiol reagents with either stimulation modality. Thus, the pore structure at the extracellular side of the channel is similar or identical with different stimuli. Figure 3 illustrates schematically the two conductance/permeability states of pannexons.
Figure 3.

Cartoon of pannexon activation by different stimuli. In the unstimulated pannexon (centre), the channel pore is occluded and the terminal cysteine is not accessible to thiol reagents. Activation by stepping the membrane potential to positive values renders the terminal cysteine reactive to extracellularly applied thiol reagents. The single-channel conductance under this condition is circa 50 pS (left). Activation of pannexons by elevated extracellular [K+] at any membrane potential yields a channel with high conductance (500 pS; right). In this state, the terminal cysteine (yellow dot) is not sensitive to thiol reagents, while an engineered cysteine at the extracellular vestibulum is. In high extracellular [K+], the pannexon is permeable to ATP and likely has the same configuration as pannexons activated by various physiological stimuli. (Online version in colour.)
9. Allosteric effect of K+ on Panx1
Several lines of evidence indicate that the opening of pannexons by extracellular K+ is due to a direct allosteric effect. Under voltage clamp conditions, K+ activates pannexons over a wide voltage range (−100 to +100 mV). Various ion substitutions do not interfere with the K+ effect. K+ stimulates pannexons in diverse cell types, including oocytes, erythrocytes and astrocytes, making it unlikely that another protein is required. The K+-mediated activation is observed in excised membrane patches containing a pannexon. The most compelling evidence for a direct action of K+ on Panx1 comes from electron microscopic (EM) studies. With a soluble factor (K+), it is possible to activate pannexons assembled from purified Panx1 protein in vitro and visualize the effects with negative stain EM techniques. The Sosinsky laboratory showed that K+ changes the diameters of the extra- and intracellular vestibula of pannexons by 30% and 300%, respectively [58].
10. ATP and K+ entangle on pannexons
The initial determination of ATP permeability of pannexons has been performed on excised membrane patches exposed to a 10 : 1 gradient of potassium ATP. ATP permeability in this experiment was indicated by the reversal potential being ca 20 mV shifted from the potassium equilibrium potential. Considering the inhibitory effect of ATP on pannexons and the use of high concentrations of ATP in the extracellular aspect of the membrane patch, this experiment should not have worked. A reassessment of the effect of ATP on pannexons in different ionic conditions, however, revealed that the inhibitory effect of ATP on pannexons is attenuated in a dose-dependent way by increased extracellular K+.
The interplay of K+ and ATP can be observed directly in Panx1-expressing oocytes [94]. Membrane currents induced by depolarization are inhibited by ATP and more effectively by BzATP (benzoyladenosinetriphosphate). Increasing extracellular [K+] attenuates or abolishes the inhibitory effect of ATP and BzATP depending on the [K+] and [BzATP] used, as if the two compounds compete at overlapping binding sites. Consistent with this, K+ also attenuates the inhibitory effect on pannexons of BB FCF (brilliant blue for colouring food) and Fast Green, which have an overlapping binding site on Panx1 with that of ATP [106].
11. Pannexons can mediate cell death
The entanglement between ATP and K+ on pannexons can also be observed in astrocytes and in brain slices as an approximation of in vivo conditions [94]. Astrocytes exposed to high [K+]o (50 mM) release lactate dehydrogenase (LDH), indicating cell death. LDH release is abrogated by either blocking pannexon activity pharmacologically or by Panx1 knockout. Knockout of the P2X7 receptor does not affect K+-induced LDH release. At a moderate [K+]o (25 mM), low BzATP (a potent ATP analogue for pannexon inhibition) enhances (through P2X7) and high BzATP attenuates LDH release from astrocytes, consistent with the K+/ATP interaction at the pannexon observed in oocytes.
Similarly, activation of the ‘executioner’ caspase 3 by K+ is inhibited at low [K+]o, but not at high [K+]o, by BzATP. Caspase 3 activation is absent in Panx1 KO cells but is unaltered in P2X7 KO cells. This shows that Panx1 is upstream of caspase 3 in the apoptotic signalling chain. Thus, Panx1 is both an activator and a downstream substrate of caspase 3, because as pointed out above, cleavage of Panx1 by caspase 3 can activate pannexons.
Yet another indicator of apoptotic cell death, phosphatidylserine (PS) exposure on the external leaflet of the plasma membrane is stimulated by K+ in a Panx1-dependent way. Annexin binding as a measure of PS exposure is stimulated by K+ in wt astrocytes, but not in Panx1 KO astrocytes. With low K+ stimulation, BzATP attenuates annexin binding, as predicted by the interplay between K+ and ATP on pannexons observed in oocytes.
Additionally, in situ blockade of pannexons by ATP as determined by dye uptake in brain slices is present in wt mice and P2X7 KO mice, but not in Panx1 KO mice. In the absence of the oocyte data, one would be hard pressed to interpret the data obtained in cultured astrocytes and brain slices.
The interplay between ATP and K+ at pannexons has profound consequences in pathological conditions where damaged cells spill a series of pannexon activators onto adjacent healthy neighbouring cells [86,94]. Among these activators is K+, which is a direct pannexon stimulant and also abolishes the negative feedback control on pannexon-mediated ATP release. In this constellation, the unopposed positive feedback for ATP release will result in overstimulation of P2X purinergic receptors and, owing to their linkage to the inflammasome, cause apoptotic/pyroptotic death of cells not damaged by the original insult. This phenomenon has long been known and is termed secondary cell death. Typically, secondary cell death makes the lesion volume several fold larger than the primary lesion. Because this process extends over several hours, there is a therapeutic window to limit secondary cell death and Panx1 is a logical target for such intervention.
12. What does it all mean?
Since the discovery of the pannexins in 2000 [36], 460 papers (as of December 2014) have been dedicated to this small family of ‘gap junction’ proteins comprising three members. Rather than forming the cell-to-cell channels of authentic gap junction proteins that the innexins and connexins do, pannexins apparently form exclusively non-junctional membrane channels. These pannexons allow the exchange of small molecules between the intra- and extracellular spaces. While the repertoire of potential permeants is large, only the permeation of ATP through pannexons has been studied in detail.
In several physiological settings, ATP release through pannexons is the primary event, i.e. a stimulus activates pannexons. Direct pannexon-mediated ATP release is found for example in erythrocytes exposed to low oxygen or mechanical stress, airway epithelial cells stimulated mechanically and also tubular cells in the kidney.
In several activation schemes of pannexons, the ensuing ATP release is a secondary event with exclusive amplification function for the signalling cascade initiated by the initial receptor ligand. This applies to all presently known ligand-mediated pannexon activations. In the case of ATP, it is a self-amplification of purinergic signalling. For the other ligands, pannexon-mediated ATP release amplifies other signalling cascades, as shown for alpha-adrenergic, histamine, thrombin and angiotensin II signalling.
It is possible that pannexons exert other functions unrelated to the release of ATP, glutamate and other compounds in the same size range. Alternative roles of Panx1 may even include non-channel functions of the protein.
The function of the other two pannexins (Panx2 and Panx3) has remained elusive. Panx2 expression is almost exclusively found in the nervous system. While the Panx2 protein is capable of forming a channel [64], no physiological stimuli have been identified so far. Furthermore, its predominant localization in cytoplasmic compartments [107] suggests an alternative function to that of Panx1.
Panx3 is similarly mysterious, because its localization in some studies is also mainly intracellular [108,109]. Furthermore, the expression of Panx3 is restricted to skin and bone [110], excluding a general function comparable to that of Panx1. Thus, the challenge to identify physiological functions for Panx2 and Panx3 remains and some inspired approaches will be required.
Acknowledgements
The critical reading of the manuscript by Dr Kenneth Muller is very much appreciated.
Funding statement
The author thanks the Craig Nielsen Foundation for support. All applications related to the work by the author described here were triaged by NIH study sections.
Competing interests
The author has no competing interests.
References
- 1.Burnstock G. 1990. Overview. Purinergic mechanisms. Ann. N.Y. Acad. Sci. 603, 1–17; discussion 8 ( 10.1111/j.1749-6632.1990.tb37657.x) [DOI] [PubMed] [Google Scholar]
- 2.Burnstock G. 1995. Current state of purinoceptor research. Pharm. Acta Helv. 69, 231–242. ( 10.1016/0031-6865(94)00043-U) [DOI] [PubMed] [Google Scholar]
- 3.Burnstock G. 2008. Purinergic signalling and disorders of the central nervous system. Nat. Rev. Drug Discov. 7, 575–590. ( 10.1038/nrd2605) [DOI] [PubMed] [Google Scholar]
- 4.Drury AN, Szent-Gyorgyi A. 1929. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J. Physiol. 68, 213–237. ( 10.1113/jphysiol.1929.sp002608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holton FA, Holton P. 1953. The possibility that ATP is a transmitter at sensory nerve endings. J. Physiol. 119, 50P–51P. [PubMed] [Google Scholar]
- 6.Holton FA, Holton P. 1954. The capillary dilator substances in dry powders of spinal roots; a possible role of adenosine triphosphate in chemical transmission from nerve endings. J. Physiol. 126, 124–140. ( 10.1113/jphysiol.1954.sp005198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnstock G, Campbell G, Satchell D, Smythe A. 1970. Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. Br. J. Pharmacol. 40, 668–688. ( 10.1111/j.1476-5381.1970.tb10646.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnstock G. 1971. Neural nomenclature. Nature 229, 282–283. ( 10.1038/229282d0) [DOI] [PubMed] [Google Scholar]
- 9.Su C, Bevan JA, Burnstock G. 1971. [3H]adenosine triphosphate: release during stimulation of enteric nerves. Science 173, 336–338. ( 10.1126/science.173.3994.336) [DOI] [PubMed] [Google Scholar]
- 10.MacKenzie I, Burnstock G, Dolly JO. 1982. The effects of purified botulinum neurotoxin type A on cholinergic, adrenergic and non-adrenergic, atropine-resistant autonomic neuromuscular transmission. Neuroscience 7, 997–1006. ( 10.1016/0306-4522(82)90056-2) [DOI] [PubMed] [Google Scholar]
- 11.Wagner JA, Carlson SS, Kelly RB. 1978. Chemical and physical characterization of cholinergic synaptic vesicles. Biochemistry 17, 1199–1206. ( 10.1021/bi00600a010) [DOI] [PubMed] [Google Scholar]
- 12.Silinsky EM. 1975. On the association between transmitter secretion and the release of adenine nucleotides from mammalian motor nerve terminals. J. Physiol. 247, 145–162. ( 10.1113/jphysiol.1975.sp010925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Unsworth CD, Johnson RG. 1990. Acetylcholine and ATP are coreleased from the electromotor nerve terminals of Narcine brasiliensis by an exocytotic mechanism. Proc. Natl Acad. Sci. USA 87, 553–557. ( 10.1073/pnas.87.2.553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y. 2008. Identification of a vesicular nucleotide transporter. Proc. Natl Acad. Sci. USA 105, 5683–5686. ( 10.1073/pnas.0800141105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsson M, Sawada K, Morland C, Hiasa M, Ormel L, Moriyama Y, Gundersen V. 2011. Functional and anatomical identification of a vesicular transporter mediating neuronal ATP release. Cereb. Cortex 22, 1203–1214. ( 10.1093/cercor/bhr203) [DOI] [PubMed] [Google Scholar]
- 16.Sperlágh B, Vizi SE. 1996. Neuronal synthesis, storage and release of ATP. The Neurosciences 8, 175–186. ( 10.1006/smns.1996.0023) [DOI] [Google Scholar]
- 17.Edwards FA, Gibb AJ. 1993. ATP: a fast neurotransmitter. FEBS Lett. 325, 86–89. ( 10.1016/0014-5793(93)81419-Z) [DOI] [PubMed] [Google Scholar]
- 18.Bergfeld GR, Forrester T. 1992. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc. Res. 26, 40–47. ( 10.1093/cvr/26.1.40) [DOI] [PubMed] [Google Scholar]
- 19.Hamann M, Attwell D. 1996. Non-synaptic release of ATP by electrical stimulation in slices of rat hippocampus, cerebellum and habenula. Eur. J. Neurosci. 8, 1510–1515. ( 10.1111/j.1460-9568.1996.tb01613.x) [DOI] [PubMed] [Google Scholar]
- 20.Leybaert L, Braet K, Vandamme W, Cabooter L, Martin PE, Evans WH. 2003. Connexin channels, connexin mimetic peptides and ATP release. Cell. Commun. Adhes. 10, 251–257. ( 10.1080/cac.10.4-6.251.257) [DOI] [PubMed] [Google Scholar]
- 21.Locovei S, Bao L, Dahl G. 2006. Pannexin 1 in erythrocytes: function without a gap. Proc. Natl Acad. Sci. USA 103, 7655–7659. ( 10.1073/pnas.0601037103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verkhratsky A, Burnstock G. 2014. Biology of purinergic signalling: its ancient evolutionary roots, its omnipresence and its multiple functional significance. Bioessays 36, 697–705. ( 10.1002/bies.201400024) [DOI] [PubMed] [Google Scholar]
- 23.Ballerini P, Di Iorio P, Ciccarelli R, Nargi E, D'Alimonte I, Traversa U, Rathbone MP, Caciagli F. 2002. Glial cells express multiple ATP binding cassette proteins which are involved in ATP release. Neuroreport 13, 1789–1792. ( 10.1097/00001756-200210070-00019) [DOI] [PubMed] [Google Scholar]
- 24.Silverman W, Locovei S, Dahl G. 2008. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am. J. Physiol. Cell Physiol. 295, C761–C767. ( 10.1152/ajpcell.00227.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boassa D, Ambrosi C, Qiu F, Dahl G, Gaietta G, Sosinsky G. 2007. Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J. Biol. Chem. 282, 31 733–31 743. ( 10.1074/jbc.M702422200) [DOI] [PubMed] [Google Scholar]
- 26.Dahl G, Locovei S. 2006. Pannexin: to gap or not to gap, is that a question? IUBMB Life 58, 409–419. ( 10.1080/15216540600794526) [DOI] [PubMed] [Google Scholar]
- 27.Reisin IL, et al. 1994. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J. Biol. Chem. 269, 20 584–20 591. [PubMed] [Google Scholar]
- 28.Liu HT, Toychiev AH, Takahashi N, Sabirov RZ, Okada Y. 2008. Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res. 18, 558–565. ( 10.1038/cr.2008.49) [DOI] [PubMed] [Google Scholar]
- 29.Cotrina ML, Lin JH, Lopez-Garcia JC, Naus CC, Nedergaard M. 2000. ATP-mediated glia signaling. J. Neurosci. 20, 2835–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taruno A, et al. 2013. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 495, 223–226. ( 10.1038/nature11906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bao L, Locovei S, Dahl G. 2004. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 572, 65–68. ( 10.1016/j.febslet.2004.07.009) [DOI] [PubMed] [Google Scholar]
- 32.Grygorczyk R, Tabcharani JA, Hanrahan JW. 1996. CFTR channels expressed in CHO cells do not have detectable ATP conductance. J. Membr. Biol. 151, 139–148. ( 10.1007/s002329900065) [DOI] [PubMed] [Google Scholar]
- 33.Wiszniewski L, Sanz J, Scerri I, Gasparotto E, Dudez T, Lacroix JS, Suter S, Gallati S, Chanson M. 2007. Functional expression of connexin30 and connexin31 in the polarized human airway epithelium. Differentiation 75, 382–392. ( 10.1111/j.1432-0436.2007.00157.x) [DOI] [PubMed] [Google Scholar]
- 34.Bruzzone R, Barbe MT, Jakob NJ, Monyer H. 2005. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J. Neurochem. 92, 1033–1043. ( 10.1111/j.1471-4159.2004.02947.x) [DOI] [PubMed] [Google Scholar]
- 35.Dahl G, Qiu F, Wang J. 2013. The bizarre pharmacology of the ATP release channel pannexin1. Neuropharmacology 75, 150–159. ( 10.1016/j.neuropharm.2013.02.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panchin Y, Kelmanson I, Matz M, Lukyanov K, Usman N, Lukyanov S. 2000. A ubiquitous family of putative gap junction molecules. Curr. Biol. 10, R473–R474. ( 10.1016/S0960-9822(00)00576-5) [DOI] [PubMed] [Google Scholar]
- 37.Ransford GA, Fregien N, Qiu F, Dahl G, Conner GE, Salathe M. 2009. Pannexin 1 contributes to ATP release in airway epithelia. Am. J. Respir. Cell Mol. Biol. 41, 525–534. ( 10.1165/rcmb.2008-0367OC) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanner F, Lam L, Nguyen MT, Yu A, Peti-Peterdi J. 2012. Intrarenal localization of the plasma membrane ATP channel pannexin1. Am. J. Physiol. Renal Physiol. 303, F1454–F1459. ( 10.1152/ajprenal.00206.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penuela S, Bhalla R, Gong XQ, Cowan KN, Celetti SJ, Cowan BJ, Bai D, Shao Q, Laird DW. 2007. Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J. Cell Sci. 120, 3772–3783. ( 10.1242/jcs.009514) [DOI] [PubMed] [Google Scholar]
- 40.Bruzzone R, Hormuzdi SG, Barbe MT, Herb A, Monyer H. 2003. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl Acad. Sci. USA 100, 13 644–13 649. ( 10.1073/pnas.2233464100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Ma M, Locovei S, Keane RW, Dahl G. 2007. Modulation of membrane channel currents by gap junction protein mimetic peptides: size matters. Am. J. Physiol. Cell Physiol. 293, C1112–C1119. ( 10.1152/ajpcell.00097.2007) [DOI] [PubMed] [Google Scholar]
- 42.Bao L, Samuels S, Locovei S, Macagno ER, Muller KJ, Dahl G. 2007. Innexins form two types of channels. FEBS Lett. 581, 5703–5708. ( 10.1016/j.febslet.2007.11.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuels SE, Lipitz JB, Dahl G, Muller KJ. 2010. Neuroglial ATP release through innexin channels controls microglial cell movement to a nerve injury. J. Gen. Physiol. 136, 425–442. ( 10.1085/jgp.201010476) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samuels SE, Lipitz JB, Wang J, Dahl G, Muller KJ. 2013. Arachidonic acid closes innexin/pannexin channels and thereby inhibits microglia cell movement to a nerve injury. Dev. Neurobiol. 73, 621–631. ( 10.1002/dneu.22088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dahl G, Muller KJ. 2014. Innexin and pannexin channels and their signaling. FEBS Lett. 588, 1396–1402. ( 10.1016/j.febslet.2014.03.007) [DOI] [PubMed] [Google Scholar]
- 46.Qiu F, Wang J, Spray DC, Scemes E, Dahl G. 2011. Two non-vesicular ATP release pathways in the mouse erythrocyte membrane. FEBS Lett. 585, 3430–3435. ( 10.1016/j.febslet.2011.09.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suadicani SO, Iglesias R, Wang J, Dahl G, Spray DC, Scemes E. 2012. ATP signaling is deficient in cultured pannexin1-null mouse astrocytes. Glia 60, 1106–1116. ( 10.1002/glia.22338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seminario-Vidal L, et al. 2011. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J. Biol. Chem. 286, 26 277–26 286. ( 10.1074/jbc.M111.260562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, Dubyak GR, Hackos D, Dixit VM. 2011. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J. Immunol. 186, 6553–6561. ( 10.4049/jimmunol.1100478) [DOI] [PubMed] [Google Scholar]
- 50.Li S, Bjelobaba I, Yan Z, Kucka M, Tomic M, Stojilkovic SS. 2011. Expression and roles of pannexins in ATP release in the pituitary gland. Endocrinology 152, 2342–2352. ( 10.1210/en.2010-1216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pelegrin P, Surprenant A. 2006. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 25, 5071–5082. ( 10.1038/sj.emboj.7601378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Locovei S, Wang J, Dahl G. 2006. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 580, 239–244. ( 10.1016/j.febslet.2005.12.004) [DOI] [PubMed] [Google Scholar]
- 53.Locovei S, Scemes E, Qiu F, Spray DC, Dahl G. 2007. Pannexin1 is part of the pore forming unit of the P2X receptor death complex. FEBS Lett. 581, 483–488. ( 10.1016/j.febslet.2006.12.056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu F, Dahl G. 2009. A permeant regulating its permeation pore: inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Cell Physiol. 296, C250–C255. ( 10.1152/ajpcell.00433.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qiu F, Wang J, Dahl G. 2012. Alanine substitution scanning of pannexin1 reveals amino acid residues mediating ATP sensitivity. Purinergic Signal. 8, 81–90. ( 10.1007/s11302-011-9263-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Dahl G. 2010. SCAM analysis of Panx1 suggests a peculiar pore structure. J. Gen. Physiol. 136, 515–527. ( 10.1085/jgp.201010440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thompson RJ, Zhou N, MacVicar BA. 2006. Ischemia opens neuronal gap junction hemichannels. Science 312, 924–927. ( 10.1126/science.1126241) [DOI] [PubMed] [Google Scholar]
- 58.Wang J, Ambrosi C, Qiu F, Jackson DG, Sosinsky G, Dahl G. 2014. The membrane protein Pannexin1 forms two open-channel conformations depending on the mode of activation. Sci. Signal. 7, ra69 ( 10.1126/scisignal.2005431) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang H, Zhu AG, Mamczur M, Falck JR, Lerea KM, McGiff JC. 2007. Stimulation of rat erythrocyte P2X7 receptor induces the release of epoxyeicosatrienoic acids. Br. J. Pharmacol. 151, 1033–1040. ( 10.1038/sj.bjp.0707311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santiago MF VJ, Patel NK, Lutz SE, Caille D, Charollais A, Meda P, Scemes E. 2011. Targeting Pannexin1 improves seizure outcome. PLoS ONE 6, e25178 ( 10.1371/journal.pone.0025178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD, Dalkara T. 2013. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339, 1092–1095. ( 10.1126/science.1231897) [DOI] [PubMed] [Google Scholar]
- 62.Gulbransen BD, et al. 2012. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 18, 600–604. ( 10.1038/nm.2679) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, Keane RW, Dahl G. 2009. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 284, 18 143–18 151. ( 10.1074/jbc.M109.004804) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ambrosi C, Gassmann O, Pranskevich JN, Boassa D, Smock A, Wang J, Dahl G, Steinem C, Sosinsky GE. 2010. Pannexin1 and Pannexin2 channels show quaternary similarities to connexons and different oligomerization numbers from each other. J. Biol. Chem. 285, 24 420–24 431. ( 10.1074/jbc.M110.115444) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang Y, Grinspan JB, Abrams CK, Scherer SS. 2007. Pannexin1 is expressed by neurons and glia but does not form functional gap junctions. Glia 55, 46–56. ( 10.1002/glia.20435) [DOI] [PubMed] [Google Scholar]
- 66.Cone AC, Ambrosi C, Scemes E, Martone ME, Sosinsky GE. 2013. A comparative antibody analysis of pannexin1 expression in four rat brain regions reveals varying subcellular localizations. Front. Pharmacol. 4, 6 ( 10.3389/fphar.2013.00006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bunse S, Locovei S, Schmidt M, Qiu F, Zoidl G, Dahl G, Dermietzel R. 2009. The potassium channel subunit Kvbeta3 interacts with pannexin 1 and attenuates its sensitivity to changes in redox potentials. FEBS J. 276, 6258–6270. ( 10.1111/j.1742-4658.2009.07334.x) [DOI] [PubMed] [Google Scholar]
- 68.Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA. 2008. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 322, 1555–1559. ( 10.1126/science.1165209) [DOI] [PubMed] [Google Scholar]
- 69.Billaud M, Sandilos JK, Isakson BE. 2012. Pannexin 1 in the regulation of vascular tone. Trends Cardiovasc. Med. 22, 68–72. ( 10.1016/j.tcm.2012.06.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murali S, Zhang M, Nurse CA. 2014. Angiotensin II mobilizes intracellular calcium and activates pannexin-1 channels in rat carotid body type II cells via AT1 receptors. J. Physiol. 592, 4747–4762. ( 10.1113/jphysiol.2014.279299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shahidullah M, Mandal A, Beimgraben C, Delamere NA. 2011. Hyposmotic stress causes ATP release and stimulates Na,K-ATPase activity in porcine lens. J. Cell Physiol. 227, 1428–1437. ( 10.1002/jcp.22858) [DOI] [PubMed] [Google Scholar]
- 72.Xia J, Lim JC, Lu W, Beckel JM, Macarak EJ, Laties AM, Mitchell CH. 2012. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J. Physiol. 590, 2285–2304. ( 10.1113/jphysiol.2012.227983) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beckel JM, et al. 2014. Mechanosensitive release of adenosine 5′-triphosphate through pannexin channels and mechanosensitive upregulation of pannexin channels in optic nerve head astrocytes: a mechanism for purinergic involvement in chronic strain. Glia 62, 1486–1501. ( 10.1002/glia.22695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li A, Leung CT, Peterson-Yantorno K, Mitchell CH, Civan MM. 2010. Pathways for ATP release by bovine ciliary epithelial cells, the initial step in purinergic regulation of aqueous humor inflow. Am. J. Physiol. Cell Physiol. 299, C1308–C1317. ( 10.1152/ajpcell.00333.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Islam MR, Uramoto H, Okada T, Sabirov RZ, Okada Y. 2012. Maxi-anion channel and pannexin 1 hemichannel constitute separate pathways for swelling-induced ATP release in murine L929 fibrosarcoma cells. Am. J. Physiol. Cell Physiol. 303, C924–C935. ( 10.1152/ajpcell.00459.2011) [DOI] [PubMed] [Google Scholar]
- 76.Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. 2010. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am. J. Physiol. Heart Circ. Physiol. 299, H1146–H1152. ( 10.1152/ajpheart.00301.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang XD, Xu R, Reynolds MF, Garcia ML, Heinemann SH, Hoshi T. 2003. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 425, 531–535. ( 10.1038/nature02003) [DOI] [PubMed] [Google Scholar]
- 78.Zhang M, Piskuric NA, Vollmer C, Nurse CA. 2012. P2Y2 receptor activation opens pannexin-1 channels in rat carotid body type II cells: potential role in amplifying the neurotransmitter ATP. J. Physiol. 590, 4335–4350. ( 10.1113/jphysiol.2012.236265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma W, Hui H, Pelegrin P, Surprenant A. 2009. Pharmacological characterization of pannexin-1 currents expressed in mammalian cells. J. Pharmacol. Exp. Ther. 328, 409–418. ( 10.1124/jpet.108.146365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hung SC, Choi CH, Said-Sadier N, Johnson L, Atanasova KR, Sellami H, Yilmaz Ö, Ojcius DM. 2013. P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS ONE 8, e70210 ( 10.1371/journal.pone.0070210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li S, Tomic M, Stojilkovic SS. 2011. Characterization of novel Pannexin 1 isoforms from rat pituitary cells and their association with ATP-gated P2X channels. Gen. Comp. Endocrinol. 174, 202–210. ( 10.1016/j.ygcen.2011.08.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iglesias R, Locovei S, Roque A, Alberto AP, Dahl G, Spray DC, Scemes E. 2008. P2X7 receptor–Pannexin1 complex: pharmacology and signaling. Am. J. Physiol. Cell Physiol. 295, C752–C760. ( 10.1152/ajpcell.00228.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Di Virgilio F, et al. 2001. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood 97, 587–600. ( 10.1182/blood.V97.3.587) [DOI] [PubMed] [Google Scholar]
- 84.Ballerini P, et al. 1996. Rat astroglial P2Z (P2X7) receptors regulate intracellular calcium and purine release. Neuroreport 7, 2533–2537. ( 10.1097/00001756-199611040-00026) [DOI] [PubMed] [Google Scholar]
- 85.Bodin P, Burnstock G. 1996. ATP-stimulated release of ATP by human endothelial cells. J. Cardiovasc. Pharmacol. 27, 872–875. ( 10.1097/00005344-199606000-00015) [DOI] [PubMed] [Google Scholar]
- 86.Dahl G, Keane RW. 2012. Pannexin: from discovery to bedside in 11±4 years? Brain Res. 1487, 150–159. ( 10.1016/j.brainres.2012.04.058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weilinger NL, Tang PL, Thompson RJ. 2012. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 32, 12 579–12 588. ( 10.1523/JNEUROSCI.1267-12.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Isakson BE, Thompson RJ. 2014. Pannexin-1 as a potentiator of ligand-gated receptor signaling. Channels (Austin) 8, 118–123. ( 10.4161/chan.27978) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seminario-Vidal L, Kreda S, Jones L, O'Neal W, Trejo J, Boucher RC, Lazarowski ER. 2009. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of Rho- and Ca2+-dependent signaling pathways. J. Biol. Chem. 284, 20 638–20 648. ( 10.1074/jbc.M109.004762) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pinheiro AR, Paramos-de-Carvalho D, Certal M, Costa C, Magalhaes-Cardoso MT, Ferreirinha F, Costa M, Correia-de-Sá P. 2013. Bradykinin-induced Ca2+ signaling in human subcutaneous fibroblasts involves ATP release via hemichannels leading to P2Y12 receptors activation. Cell Commun. Signal. 11, 70 ( 10.1186/1478-811X-11-70) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pinheiro AR, et al. 2013. Histamine induces ATP release from human subcutaneous fibroblasts, via pannexin-1 hemichannels, leading to Ca2+ mobilization and cell proliferation. J. Biol. Chem. 288, 27 571–27 583. ( 10.1074/jbc.M113.460865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.da Silva-Souza HA, de Lira MN, Patel NK, Spray DC, Persechini PM, Scemes E. 2014. Inhibitors of the 5-lipoxygenase pathway activate pannexin1 channels in macrophages via the thromboxane receptor. Am. J. Physiol. Cell Physiol. 307, C571–C579. ( 10.1152/ajpcell.00087.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Momboisse F, et al. 2014. Pannexin 1 channels: new actors in the regulation of catecholamine release from adrenal chromaffin cells. Front. Cell Neurosci. 8, 270 ( 10.3389/fncel.2014.00270) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jackson DG, Wang J, Keane RW, Scemes E, Dahl G. 2014. ATP and potassium ions: a deadly combination for astrocytes. Sci. Rep. 4, 4576 ( 10.1038/srep04576) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chekeni FB, et al. 2010. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 467, 863–867. ( 10.1038/nature09413) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan W-B. 2005. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. ( 10.1038/nn1472) [DOI] [PubMed] [Google Scholar]
- 97.Duan Y, Sahley CL, Muller KJ. 2009. ATP and NO dually control migration of microglia to nerve lesions. Dev. Neurobiol. 69, 60–72. ( 10.1002/dneu.20689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chiu YH, Ravichandran KS, Bayliss DA. 2014. Intrinsic properties and regulation of Pannexin 1 channel. Channels (Austin) 8, 103–109. ( 10.4161/chan.27545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Poon IK, Chiu YH, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, Ravichandran KS. 2014. Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature 507, 329–334. ( 10.1038/nature13147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. 2012. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J. Biol. Chem. 287, 11 303–11 311. ( 10.1074/jbc.M111.323378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.MacVicar BA, Thompson RJ. 2010. Non-junction functions of pannexin-1 channels. Trends Neurosci. 33, 93–102. ( 10.1016/j.tins.2009.11.007) [DOI] [PubMed] [Google Scholar]
- 102.Ma W, Compan V, Zheng W, Martin E, North RA, Verkhratsky A, Surprenant A. 2012. Pannexin 1 forms an anion-selective channel. Pflugers Arch. 463, 585–592. ( 10.1007/s00424-012-1077-z) [DOI] [PubMed] [Google Scholar]
- 103.Romanov RA, Bystrova MF, Rogachevskaya OA, Sadovnikov VB, Shestopalov VI, Kolesnikov SS. 2012. The ATP permeability of pannexin 1 channels in a heterologous system and in mammalian taste cells is dispensable. J. Cell Sci. 125, 5514–5523. ( 10.1242/jcs.111062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nualart-Marti A, et al. 2013. Role of connexin 32 hemichannels in the release of ATP from peripheral nerves. Glia 61, 1976–1989. ( 10.1002/glia.22568) [DOI] [PubMed] [Google Scholar]
- 105.Hansen DB, Ye ZC, Calloe K, Braunstein TH, Hofgaard JP, Ransom BR, Nielsen MS, MacAulay N. 2014. Activation, permeability, and inhibition of astrocytic and neuronal large pore (hemi)channels. J. Biol. Chem. 289, 26 058–26 073. ( 10.1074/jbc.M114.582155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang J, Jackson DG, Dahl G. 2013. The food dye FD&C Blue No. 1 is a selective inhibitor of the ATP release channel Panx1. J. Gen. Physiol. 141, 649–656. ( 10.1085/jgp.201310966) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Le Vasseur M, Lelowski J, Bechberger JF, Sin WC, Naus CC. 2014. Pannexin 2 protein expression is not restricted to the CNS. Front. Cell Neurosci. 8, 392 ( 10.3389/fncel.2014.00392) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iwamoto T, Nakamura T, Doyle A, Ishikawa M, de Vega S, Fukumoto S, Yamada Y. 2010. Pannexin 3 regulates intracellular ATP/cAMP levels and promotes chondrocyte differentiation. J. Biol. Chem. 285, 18 948–18 958. ( 10.1074/jbc.M110.127027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ishikawa M, Iwamoto T, Nakamura T, Doyle A, Fukumoto S, Yamada Y. 2011. Pannexin 3 functions as an ER Ca2+ channel, hemichannel, and gap junction to promote osteoblast differentiation. J. Cell Biol. 193, 1257–1274. ( 10.1083/jcb.201101050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baranova A, et al. 2004. The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics 83, 706–716. ( 10.1016/j.ygeno.2003.09.025) [DOI] [PubMed] [Google Scholar]