

Abstract
Objective
During the course of rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS) are chronically exposed to an inflammatory milieu. In the current study we test the hypothesis that chronic exposure of FLS to TNFα augments inflammatory responses to secondary stimuli (priming effect).
Methods
FLS obtained from RA patients were chronically exposed to TNFα (3 days) and then were stimulated with interferons (IFNs). Expression of IFN-target genes was measured by real-time quantitative reverse transcription-polymerase chain reaction and enzyme-linked immunosorbent assay. Total STAT1 protein and IFN-mediated STAT1 activation were evaluated by Western blotting. Total histone levels, histone acetylation, NF-κB p65 and RNA polymerase II (pol II) recruitment were measured at the promoter of CXCL10 (encodes IP-10) by chromatin immunoprecipitation assays.
Results
Prolonged pre-exposure of FLS to TNFα enhanced the magnitude and extended the kinetics of CXCL10/IP-10, CXCL9/MIG and CXCL11/ITAC production upon subsequent IFN stimulation. This phenotype was retained over a period of days even after the removal of TNFα. Prolonged TNFα decreased histone levels, increased acetylation of the remaining histones, and heightened recruitment of NF-κB p65 and pol II to the CXCL10 promoter. In parallel, an increase in intracellular STAT1 led to amplification of IFN-induced STAT1 activation.
Conclusions
Our study reveals a novel pathogenic function of TNFα, namely prolonged and gene-specific priming of FLS for enhanced transcription of inflammatory chemokine genes due to priming of chromatin, sustained activation of NF-κB, and amplification of STAT1 activation downstream of IFNs. These data also suggest that FLS gain an “inflammatory memory” upon chronic exposure to TNFα.
Keywords: fibroblast-like synoviocytes, rheumatoid arthritis, signal transduction, TNFα, chromatin
Introduction
Scientific breakthroughs and technological advances have deepened our understanding of the pathogenesis of rheumatoid arthritis (RA). Recent evidence from human and animal studies supports the role of specific environmental factors, such as smoking and the microbiome, as disease triggers (1, 2). Genome-wide association studies (GWAS) have identified new genetic associations, uncovering novel pathogenic molecular pathways (3). The ENCODE project revealed a striking link of mutations in gene enhancers with disease predisposition, since it was shown that the majority of disease-associated single nucleotide polymorphisms (SNPs) lie within non-coding regions of DNA, which represent putative regulatory elements (4). In addition, the introduction of biologics and kinase inhibitors has revolutionized the treatment of RA patients (5, 6). Despite this progress, sustained disease remission and drug-free remission are still unmet needs for many RA patients (7). Thus, a challenge in the field is to discover and characterize novel treatment targets.
Fibroblast-like synoviocytes (FLS) have recently emerged as attractive therapeutic targets in RA. Targeting FLS, either with genetic deletion of cadherin-11 or by monoclonal antibodies against cadherin-11, ameliorates joint damage and inflammation in mouse models of arthritis (8). The concept that FLS in RA are not innocent bystanders, but function as pathogenic cells is supported by several studies. FLS derived from pannus exhibit a unique phenotype that shares morphologic features with tumor cells and thus RA FLS were described as “transformed” cells (9). RA FLS, but not FLS derived from osteoarthritis, gain and retain an invasive and destructive capacity against articular cartilage (10). FLS, during the chronic course of synovitis in RA, are a major source of inflammatory cytokines, chemokines, complement, growth factors and tissue destructive enzymes (11). Thus, RA FLS mediate synovial recruitment, retention, organization, activation and survival of inflammatory cells, enhance synovial neoangiogenesis, and induce osteoclastogenesis and cartilage degradation (9, 12).
In vivo evidence from DNaseII-null, TNF-Transgenic, and TNFΔARE mice suggests that systemically elevated levels of TNFα induce chronic synovitis, which was independent of adaptive immunity (13-17). Notably, in the TNFΔARE model, it was shown that FLS were the primary responder cells, sufficient for the full development of chronic synovitis (17), supporting the concept that FLS are hypersensitive to systemic TNFα. Along these lines, we have recently reported that TNFα induces in RA FLS a sustained and unremitting inflammatory response, characterized by sustained activation of the classical NF-κB pathway and continuous transcription of pathogenic mediators (18). Our observations support a model where FLS become hypersensitive to chronic TNFα potentially due to functional incompetence of signaling brakes and removal of chromatin barriers at key pathogenic gene loci, such as IL6.
The current study extends our previous investigation on the long term effects of TNFα on FLS. We have modeled, ex vivo, the chronic synovitis of RA by sustained exposure of FLS to TNFα. Using this system we have tested the hypothesis that chronic TNFα primes FLS for enhanced inflammatory responses to subsequent stimuli. Strikingly, we observed that, upon chronic exposure to TNFα, FLS gain and retain an “inflammatory memory”, manifested by sustained synergistic production of inflammatory chemokines upon subsequent interferon (IFN) stimulation, even after TNFα has been removed from the system. Our results uncover a priming mechanism whereby chronic pre-exposure of FLS to TNFα increases the intracellular STAT1 reservoir and primes chromatin at the CXCL10 promoter, allowing extended transcription upon secondary inflammatory stimulation due to the unopposed binding of transcription factors and RNA polymerase II (pol II).
Materials and Methods
Patients
Synovial tissues were obtained from RA patients who underwent total knee replacement or elbow synoviectomy (protocol approved by the Hospital for Special Surgery Institutional Review Board). The diagnosis of RA was based on the 1987 American College of Rheumatology criteria (19).
Cell purification
Synovial tissue fragments were incubated with liberase (Roche) for 90 min at 37 °C and cells were allowed to adhere to tissue culture dishes and passaged every 3-5 days. 4-5 passages yielded a relatively homogeneous population of FLS.
Cell Culture
FLS were cultured in alpha-MEM (plus 10% FBS, 1% Glutamine and 1% Penicillin/Streptomycin). The following reagents were used as indicated: TNFα (10 ng/ml) (Pepro Tech), IFNβ (1,000 U/ml) (Pepro Tech), IFNγ (0.1-100 U/ml) (Roche), infliximab (10 μg/ml) (Janssen Biotech), IKK Inhibitor II (50 μM) (Calbiochem), and dimethyl sulphoxide (DMSO) (Sigma Aldrich) was used as a vehicle control.
Real-time quantitative RT-PCR (qPCR)
RNA was extracted from 0.5×106 FLS using RNeasy mini kit (Qiagen) and 1 μg was reverse transcribed using a First Strand cDNA synthesis kit (Fermentas). qPCR was performed using SYBR Green supermix following the manufacturer's protocols, and triplicate reactions were run for each sample. The human oligonucleotide primers used were: GAPDH: 5-ATCAAGAAGGTGGTGAAGCA-3 and 5-GTCGCTGTTGAAGTCAGAGGA-3; CXCL10: 5- ATTTGCTGCCTTATCTTTCTG-3 and 5- TCTCACCCTTCTTTTTCATTGTAG-3; CXCL10 Primary Transcript (PT): 5- GAGGAGGAATGGATCACATCA-3 and 5-GAGGAGGAATGGATCACATCA-3; CXCL9: 5- ATCAGCACCAACCAAGGGACT-3 and 5- GCTTTTTCTTTTGGCTGACCTG-3; CXCL11: 5-GAAGGATGAAAGGTGGGTGA-3 and 5- AAGCACTTTGTAAACTCCGATG-3; STAT1: 5-TGGGTTTGACAAGGTTCTT-3 and 5- TATGCAGTGCCACGGAAAG-3.
Immunoblotting and ELISA
Lysates from 105 FLS were fractioned on polyacrylamide gels, transferred to polyvinylidene fluoride membranes and incubated with antibodies against pY-STAT1 (Cell Signaling), STAT1 (BD Transduction Laboratory) and p38 (Santa Cruz Biotechnology).
We measured CXCL10/IP-10 protein in culture supernatants (0.5×106 FLS in 3ml medium) with sandwich ELISA, using paired capture and detection antibodies against CXCL10/IP-10 (BD Pharmingen).
Chromatin immunoprecipitation assay (ChIP)
Cells were treated with 1% formaldehyde to crosslink chromatin. Fixed cells were incubated with lysis buffer and sonicated using a Bioruptor® device (Diagenode UCD400). 5% of sonicated cell lysates was saved as input. Chromatin was immunoprecipitated using antibodies to STAT1 (Abcam), Histone 4 (H4), acetylated H4 (Millipore), NF-κB p65 subunit (Abcam) and RNA Polymerase II (Santa Cruz Biotechnology). Crosslinks were reversed, DNA was purified and enrichment of the target DNA was measured by real time PCR. The human oligonucleotide primers used were:
CXCL10 Promoter: 5-GTGCTGAGACTGGAGGTTCC-3 and 5- GGGAGGGAAAATGGCTTTGC -3; Hemoglobin b (HBB) Promoter: 5- GAGGGCTGAGGGTTTGAAGT-3 and 5-TGCTCCTGGGAGTAGATTGG-3.
Statistical Analysis
Results are expressed as mean ± SEM and GraphPad Prism Analyical Software Version 5.93 for Windows was used. For statistical analysis the one-tailed, paired Student t test was used.
Results
Prolonged exposure to TNFα primes FLS for enhanced and sustained production of inflammatory chemokines upon IFN-stimulation
Our goal was to test the hypothesis that chronic exposure of FLS to TNFα augments inflammatory responses to secondary stimuli. We cultured RA FLS in the presence or absence of TNFα (10 ng/ml) for 3 days, and then cells were stimulated with either IFNβ (1,000 U/ml) or IFNγ (100 U/ml) for 3 hours. TNFα was added on the first day and was not replenished. We found modest induction of CXCL10/IP-10 mRNA by TNFα alone (Figure 1A left panel, second bar). Since CXCL10/IP-10 is NF-κB-dependent and IFN-inducible (20), the observed up-regulation, potentially results from the combination of sustained NF-κB signaling and autocrine IFN function downstream of chronic TNFα (18, 21, 22). As expected, stimulation of FLS with IFNs also induced the expression of CXCL10/IP-10 mRNA (Figure 1A left panel, third and fifth bars). FLS chronically exposed to TNFα displayed a substantially higher expression of CXCL10/IP-10 mRNA upon IFNs stimulation (Figure 1A left panel, fourth and sixth bars). Notably, the levels of CXCL10/IP-10 mRNA induced by IFN stimulation in FLS pre-exposed to chronic TNFα exceeded by far the sum of induction by TNFα alone and IFNs alone (Figure 1A left panel: compare second and third bars to fourth bar, as well as second and fifth bars to sixth bar). A similar pattern of synergistic induction was observed when CXCL10/IP-10 protein was measured by ELISA in culture supernatants (Figure 1A right panel). In addition, we measured levels of active transcription using primers specific for the first intronic region of CXCL10 gene. As shown in Figure 1B, there was a synergistic induction of CXCL10/IP-10 primary transcripts by IFNs and chronic TNFα, suggesting that synergy occurs at the level of transcription. A similar pattern of super-induction by IFNs in FLS chronically exposed to TNFα was observed for CXCL9/MIG and CXCL11/ITAC (Supplementary Figure 1). Notably, the priming effect of TNFα on IFN responses was gene specific, since IFN-mediated induction of other classic IFN-target genes, such as SOCS1, STAT1, IRF1and IFIT2/ISG54, was not affected by chronic pre-exposure to TNFα (Supplementary Figure 2).
Figure 1. Prolonged TNFα synergizes with interferons (IFN) for induction of CXCL10 transcription in RA FLS.
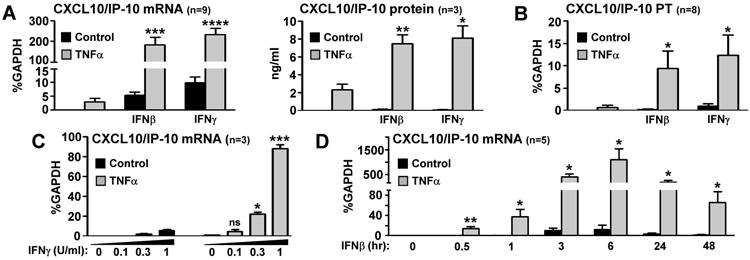
RA FLS were cultured for 3 days in the presence or absence of TNFα (10 ng/ml), which was added on the first day of culture and was not replenished. On day 3 the cells were stimulated with IFNβ (1,000 U/ml) or IFNγ (100 U/ml) for either 3 hours (A-B) or 0.5-48 hours (D). (C), A dose response to 3 hours IFNγ (0, 0.1, 0.3 and 1 U/ml) was performed. The expression of CXCL10/IP-10 mRNA and primary transcripts (PT) were measured by qPCR and CXCL10/IP-10 protein was measured with ELISA. For PT (B), primers specific for the first intronic region of CXCL10 gene were used. Values are the mean ±SEM and were normalized relative to mRNA for GAPDH. *= p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001 and ns=not significant, by one-tailed paired Student t test.
A classic paradox in RA pathogenesis is that, despite the abundance of activated M1-like cells (classically activated macrophages that express an IFN signature), there is very low amount of IFNγ within the inflamed synovium (23, 24). Thus, it has been debated whether and how these low concentrations of IFNγ have any biological effect during the course of synovitis. We have addressed this question with dose response experiments using IFNγ ranging from 0.1-100 U/ml. Doses of IFNγ ≤ 3 U/ml were suboptimal for induction of target-genes (data not shown). Interestingly we have seen substantial induction of CXCL10/IP-10, CXCL9/MIG and CXCL11/ITAC even by concentrations of IFNγ as low as 0.3 U/ml in FLS pre-treated with TNFα (Figure 1C and data not shown). These data suggest that chronic exposure to TNFα sensitizes FLS to the pro-inflammatory effects of suboptimal amounts of IFNγ.
Since IFNβ has been reported to be highly expressed in synovial lining of RA patients (25), we wondered whether chronic exposure to TNFα modifies the kinetics of FLS pro-inflammatory responses to saturating doses of IFNβ (1,000 U/ml). RA FLS were exposed to TNFα for 3 days as described above, and then stimulated with IFNβ in time course experiments (0-48 hours of IFNβ stimulation). IFNβ-induced CXCL10/IP-10, CXCL9/MIG and CXCL11/ITAC mRNA peaked between 3 and 6 hours and returned back to baseline by 24 hours (Figure 1D and data not shown). Notably, in cells pre-exposed to chronic TNFα there was robust expression of these chemokines even after 48 hours of IFN stimulation (Figure 1D and data not shown). These data suggest that chronic TNFα not only enhances the magnitude, but also prolongs the kinetics of IFN-induced pro-inflammatory chemokines in RA FLS.
FLS gain an “inflammatory memory” upon chronic exposure to TNFα
It has recently been proposed that, during the chronic course of synovitis, RA FLS gain and retain a pathogenic phenotype, due to prolonged exposure to inflammatory stimuli (9). In this context, we cultured FLS in the presence or absence of TNFα for 3 days and then inflammatory input was removed by washing cells, adding new medium and blocking any residual TNFα with infliximab (10 μg/ml). Following a weaning period of 24 hours, we measured inflammatory response to subsequent stimulation with IFNβ (1,000 U/ml) or IFNγ (100 U/ml) (the experimental set up is described in Figure 2A). Notably, FLS pre-exposed to TNFα display a more robust induction of CXCL10/IP-10 mRNA by IFNs, compared to control FLS, despite the removal of the priming stimulus (Figure 2B). The same phenotype was retained even when the weaning period was extended up to three days (data not shown), suggesting that FLS gain and retain an “inflammatory memory” upon chronic exposure to TNFα.
Figure 2. Chronic exposure to TNFα induces an “inflammatory memory” in RA FLS.
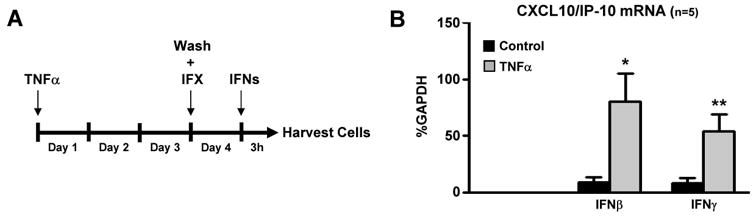
(A), RA FLS were cultured for 3 days in the presence or absence of TNFα (10 ng/ml), which was added on the first day of culture and was not replenished. Then inflammatory input was removed by washing cells, adding new medium and blocking any residual TNFα with infliximab (IFX, 10 μg/ml). Following a weaning period of 24 hours, cells were stimulated with IFNβ (1,000 U/ml) or IFNγ (100 U/ml) for 3 hours. (B), The expression of CXCL10/IP-10 mRNA was measured by qPCR. Values are the mean ±SEM and were normalized relative to mRNA for GAPDH. *= p<0.05 and **=p<0.01, by one-tailed paired Student t test.
Prolonged TNFα functions as STAT1-amplifier in FLS
Interferons signal mainly through the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) intracellular signal transduction pathway to achieve transcriptional activation of INF-inducible genes (26, 27). Since CXCL10, CXCL9 and CXCL11 are classic STAT1-targets, we tested the hypothesis that chronic exposure to TNFα might modify STAT1 activation downstream of IFNs. Stimulation of FLS with 1000 U/ml of IFNβ (Figure 3A, left panel) or 100 U/ml of IFNγ (Figure 3A, right panel) resulted in the expected tyrosine 701 phosphorylation of STAT1 (pY-STAT1). Verifying our hypothesis, in FLS pre-exposed to chronic TNFα, IFN-induced activation of STAT1 was remarkably higher in all time points tested (10-60 minutes, Figure 3A). We have also found that TNFα induces expression of total STAT1 protein and mRNA with delayed but sustained kinetics (24-72 hours of TNFα stimulation, Figure 3B-C). Overall, these data suggest that chronic TNFα augments the magnitude and prolongs the kinetics of STAT1 activation downstream of IFN stimulation, due to an increase of the intracellular STAT1 reservoir. Then, we measured STAT1 levels at CXCL10 promoter using ChIP assay. We observed that pre-exposure to TNFα for 72 hours augmented STAT1 recruitment at CXCL10 promoter upon subsequent IFN stimulation (Figure 3D), supporting the hypothesis that the TNFα-induced increase in STAT1 reservoir may contributes to the observed super-induction of CXCL10/IP-10.
Figure 3. STAT1 amplifier function of chronic TNFα in RA FLS.
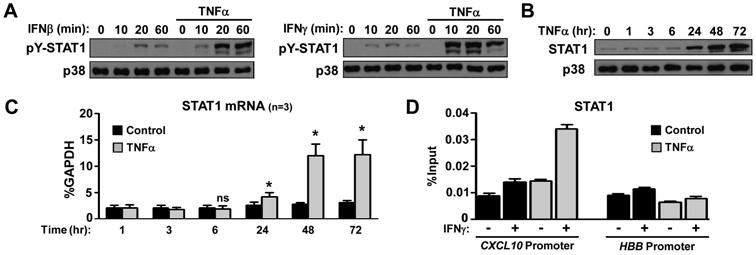
RA FLS were cultured for 3 days in the presence or absence of TNFα (10 ng/ml), which was added on the first day of culture and was not replenished (A, D). (A), On day 3 the cells were stimulated with IFNβ (1,000 U/ml) or IFNγ (100 U/ml) for 10-60 minutes and STAT1 tyrosine phosphorylation (pY) was measured by immunoblotting. The expression of STAT1 protein (B) and mRNA (C), upon TNFα time course stimulation, was measured by immunoblotting and qPCR. Values are the mean ±SEM and were normalized relative to mRNA for GAPDH. *= p<0.05 and ns=not significant, by one-tailed paired Student t test. (D), Chromatin immunoprecipitation was performed with anti-STAT1 antibody. The enrichment of immunoprecipitated STAT1 at the CXCL10 and hemoglobin B (HBB; negative control) promoters is shown as the percentage of input.
The priming effect of TNFα is dependent on sustained NF-κB signaling in FLS
A key finding of our recent report was that TNFα induces sustained activation of the classical NF-κB pathway in RA FLS, manifested by prolonged nuclear localization of p65 (18). Since CXCL10, CXCL9 and CXCL11 contain κB sites at their promoters (28), we have used ChIP assays to test the hypothesis that pre-exposure of FLS to TNFα leads to prolonged promoter binding of p65. Control FLS display very low occupancy of p65 at the CXCL10 promoter (Figure 4A, first bar). Upon TNFα stimulation there is induction of p65 binding within 3 hours, that is further increased at 24 and 72 hours (Figure 4A), suggestive of either continuous upstream signaling input or nuclear retention of p65. IKK inhibitor II (iIKK II, 50 μM) was added to the cultures on day 3 of TNFα stimulation to block nuclear influx of p65, and then FLS were stimulated with IFNs. Pharmacologic inhibition of NF-κB abrogated the synergistic induction of CXCL10/IP-10 (Figure 4B), indicating that the sustained signaling input by the classic NF-κB pathway contributes to the observed super-induction of CXCL10/IP-10 by IFNs.
Figure 4. Priming effects of TNFα is dependent on sustained NF-κB activity.
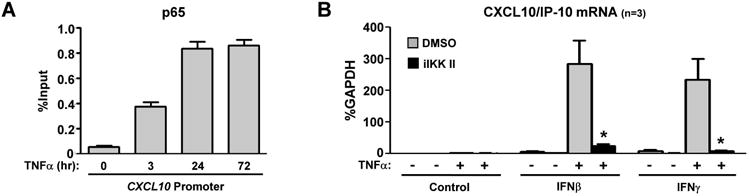
(A), A time course (0-72 hours) stimulation of FLS with TNFα (10 ng/ml) was performed and chromatin immunoprecipitation was done with anti-p65 antibody. The enrichment of immunoprecipitated p65 at the CXCL10 promoter is shown as the percentage of input. Results are representative of three independent experiments. (B), FLS were cultured for 3 days in the presence or absence of TNFα (10 ng/ml), which was added on the first day of culture and was not replenished. On day 3, DMSO or IKK inhibitor II (iIKK II, 50μM) was added for 30 minutes and then cells were stimulated for 3 hours with IFNβ (1,000 U/ml) or IFNγ (100 U/ml). The expression of CXCL10/IP10 mRNA was measured by qPCR. Values are the mean ±SEM and were normalized to mRNA for GAPDH. *= p<0.05, by one-tailed paired Student t test.
TNFα removes the chromatin barrier from CXCL10/IP-10 promoter
NF-κB is considered a “signaling transcription factor”, meaning that it is recruited to chromatin which is already open (29, 30). Thus, we have tested the hypothesis that, upon chronic exposure of FLS to TNFα, an open chromatin state is established at CXCL10 promoter. Chromatin accessibility can be increased by depletion of histones/nucleosomes, which exposes DNA to transcription factors, or by histone acetylation, which weakens DNA-histone interactions. We thus examined the effects of TNFα on histone occupancy and acetylation at the promoter of CXCL10. Exposure of FLS to TNFα decreased histone 4 (H4) levels at CXCL10 promoter within 3 hours (Figure 5A, left panel, first and second bars). Histone depletion at CXCL10 promoter was more dramatic at 24 hours and was retained at 48 and 72 hours post TNFα stimulation (Figure 5A). Depletion of H4 was minor if any at HBB promoter, which served as negative control (Figure 5A, right panel). Notably, the remaining histones at CXCL10 promoter showed increased acetylation, as measured by the ratio of acetylated H4 to total H4 (Figure 5B). Induction of a more accessible chromatin state and recruitment of p65 would facilitate binding of RNA polymerase II (pol II); indeed, we observed a striking increase of pol II recruitment at CXCL10 promoter 72 hours after TNFα stimulation (Figure 5C). Overall, these observations suggest that TNFα removes the chromatin barrier from the promoter of CXCL10 allowing abundant binding of transcription factors and unopposed recruitment of the transcriptional machinery.
Figure 5. TNFα induces sustained chromatin opening and prolonged recruitment of RNA polymerase II (pol II) at CXCL10 promoter of RA FLS.
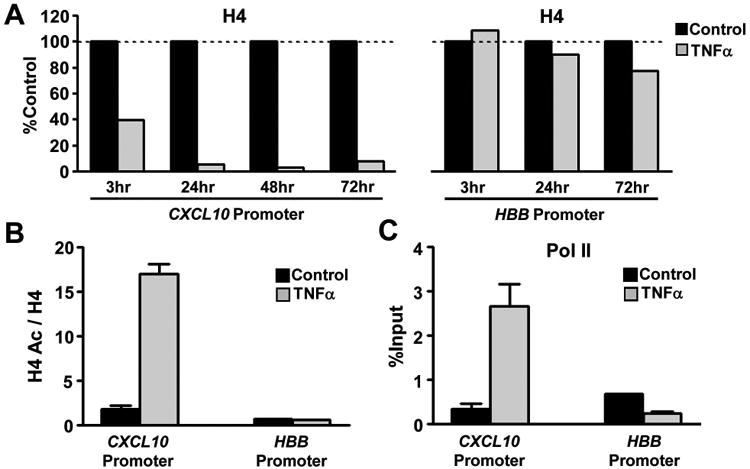
(A), FLS were cultured in the presence or absence of TNFα (10 ng/ml) in time course experiments (3-72 hours) and chromatin immunoprecipitation was performed with anti-histone 4 (H4) antibody. The occupancy of H4 at CXCL10 and hemoglobin B (HBB; negative control) promoters in control cells was set as 100 and occupancy of H4 in TNFα stimulated cells is shown as percentage of occupancy in control cells. (B-C), At 72 hours after TNFα stimulation FLS were harvested, and chromatin immunoprecipitation was performed with anti- H4 and anti-acetylated H4 (B) or anti-pol II (C) antibodies. Levels of acetylated H4 (H4 Ac) were normalized to those of total H4 (B). The enrichment of pol II at the CXCL10 and HBB promoters is shown as the percentage of input (C). Results are representative of three independent experiments.
Following the above observations, the key question becomes what is the molecular mechanism downstream of TNFα that opens the chromatin at CXCL10 promoter of RA FLS. To investigate the potential role of NF-κB pathway, the IKK inhibitor II (iIKK II, 50 μM) was added in the culture either before (Figure 6A) or 24 hours after (Figure 6B) TNFα. H4 depletion was neither prevented nor diminished by pharmacologic inhibition of IKK (Figure 6A, B), suggesting that NF-κB activation downstream of TNFα is not critical for the regulation of histone levels at CXCL10 promoter. These data are consistent with published literature suggesting that p65 itself is not able to open the chromatin (29, 30).
Figure 6. NF-κB pathway is not critical for TNFα-induced histone depletion in RA FLS.
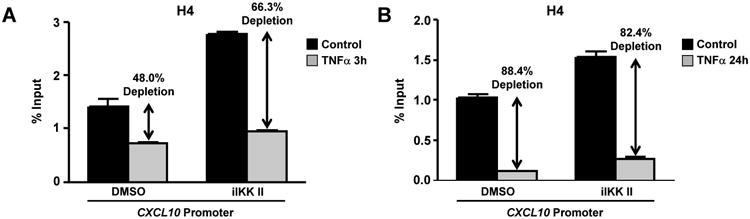
(A), RA FLS were exposed to IKK inhibitor II (iIKK II, 50μM) or DMSO for 30 minutes and then TNFα (10 ng/ml) was added for 3 hours. (B), RA FLS were stimulated with TNFα (10 ng/ml) for 24 hours and then iIKK II (50μM) or DMSO were added for 3 hours. Chromatin immunoprecipitation was performed with anti-H4 antibody. The occupancy of H4 at the CXCL10 promoter is shown as the percentage of input. Results are representative of three independent experiments.
Discussion
Our study reveals a novel pathogenic function of TNFα, namely priming of FLS for enhanced inflammatory responses by overcoming the chromatin barrier in inflammatory chemokine genes, activating NF-κB in a sustained pattern, and augmenting STAT1 activation downstream of IFNs. Synergy between TNFα and IFNs was initially described in cancer and viral biology, where TNFα was found to augment the anti-tumor and anti-viral effects of IFNs (31, 32). A series of subsequent studies revealed synergistic induction of various genes in a cell-specific manner (summarized in (33)) and shaped a paradigm for the molecular basis of synergy. For genes that contain κB motifs and STAT-binding sites, TNFα-induced NF-κB and IFN-induced STAT work cooperatively by binding to promoters at the same time (20, 28).
Our findings provide further insights into this model and the current study is the first one to investigate synergy in the context of chronic TNFα, which is more relevant to RA pathogenesis. In agreement with our recent report (18), we have showed sustained NF-κB activation downstream of TNFα, which was critical for the super-induction of inflammatory chemokines by IFNs. IFN-activated STAT1 and TNFα-activated p65 are both “signaling transcription factors”, which means they are recruited to chromatin that is already open (30, 34). Our results suggest that chronic exposure of FLS to TNFα depletes histones and hyperacetylates remaining histones, leading to loosening of chromatin at the locus of chromosome 4, where CXCL10, CXCL9 and CXCL11 genes are located next to each other (Supplementary Figure 1C). Open chromatin in this specific location allows unopposed recruitment of p65, STAT1 and pol II and provides the molecular basis for the gene-specific synergy. This model explains the increased magnitude and the extended kinetics of chemokine production upon IFN stimulation in FLS pre-exposed to TNFα.
Our observation that TNFα mediates induction of STAT1 in RA FLS indicates that, in the course of synovial inflammation, TNFα might operate as an amplifier of STAT1 activation. Since pharmacologic blockade of Jak-STAT pathway was proven effective in RA patients (6), understanding the molecular mechanism of the STAT1-amplifier function of TNFα is biologically significant. The existence of a TNFα-induced IFNβ loop has previously been identified by our group and others in several cell types, such as macrophages and FLS (21, 22, 35, 36). In macrophages, TNFα mediates IFNβ production through interferon response factor 1 (IRF1) (35). Secreted IFNβ then acts in an autocrine and paracrine manner, and modulates cellular responses. In this context, we believe that the observed increase of intracellular STAT1 reservoir in TNFα-stimulated FLS was the result of a TNFα-induced IFN loop. In patients with RA we have found increased STAT1 expression and activation within the synovial intima, where FLS are abundant (37). Our previous studies have shown a priming capacity of IFNs due to induction of STAT1 (37). Thereby we propose that a TNFα-IFNβ-STAT1 axis is activated during the course of RA synovitis and primes FLS for enhanced responses to cytokines with STAT1-activating potential. This model identifies RA FLS, in addition to lymphocytes and macrophages, as biologically important targets of Jak inhibitors.
A striking finding of our study is that, despite the removal of TNFα, FLS acquire and retain for days an “inflammatory memory”. Since the priming effect of chronic TNFα decays slowly over a period of a few days after washing and blocking TNFα (data not shown), it can be considered “short-term memory”. The current model describing the pathogenic functions of FLS during the course of RA proposes that FLS can behave as passive responders and imprinted aggressors (9). The passive responder behavior of FLS corresponds to a pathogenic phenotype induced by an exogenous stimulus that is rapidly reversed upon removal of the triggering factor (no memory). The imprinted aggressor phenotype refers to an autonomous pathogenic behavior, due to somatic mutations or epigenetic alterations, which is maintained irrespective of external stimulation (long-term memory) (9, 10). Our observations add to the model a third modus of FLS behavior with short-term memory, where the pathogenic phenotype is retained despite the removal of the primary stimulus and reverses with slow kinetics. An attractive molecular explanation for “short-term memory” could be the induction by chronic TNFα of molecules and chromatin modifications that are not rapidly reversed or are turned over with slow kinetics (38). Along these lines, we have seen that the intracellular STAT1 content remains high and the increased STAT1 activation upon IFN-stimulation is retained, even after the removal of TNFα (Supplementary Figure 3).
IFNs are pleiotropic cytokines and depending on the context can either suppress or promote immune and inflammatory responses (26). IFNs exert homeostatic functions by increasing production of anti-inflammatory factors, decreasing pro-inflammatory mediators, suppressing Th17 differentiation, and inhibiting osteoclastogenesis (26). Thereby, IFNβ and IFNγ have been used therapeutically in RA patients, but interestingly results were disappointing (39, 40). On the other hand IFNs boost immunity and inflammation by mediating activation and priming of macrophages and dendritic cells, enhancing Th1 differentiation, augmenting antibody responses, stimulating germinal center formation, and inducing inflammatory chemokine production (including CXCL10/IP-10, CXCL9/MIG and CXCL11/ITAC) (26). Our results suggest that in the context of RA synovitis, gene-specific priming of FLS, due to chronic exposure to TNFα, might switch the functions of endogenous or exogenous IFNs to a more pro-inflammatory direction characterized by excessive production of CXCL10/IP-10, CXCL9/MIG and CXCL11/ITAC.
CXCL10/IP-10 has been found highly expressed in the synovium and serum of RA patients (41, 42). Our results propose that, during the course of RA, TNFα-primed FLS respond even to very low amounts of endogenous IFNs, and become a major cellular source of CXCL10/IP-10. Several studies in animal models and humans support a pathogenic role of CXCL10/IP-10 and its receptor CXCR3 in RA (43). The proposed mechanisms are synovial recruitment and homing of inflammatory cells, increased invasiveness of FLS to cartilage, enhanced osteoclastogenesis, and atherogenic potential (43, 44). Along these lines, pharmacologic blockade of CXCL10/IP-10 or CXCR3 inhibited arthritis in animal models (45-48), and administration of a monoclonal antibody against CXCL10/IP-10 improved symptoms and signs in RA patients (49). Since CXCL10/IP-10-CXCR3 pathway is pathogenic and represents an attractive therapeutic target for RA, a challenge in the field is to understand the molecular mechanisms that regulate its activation in the context of synovitis. Our study provides a molecular explanation for the production of CXCL10/IP-10, CXCL9/MIG and CXCL11/ITAC by FLS during the course of synovial inflammation, where TNFα and IFNs are co-expressed.
Supplementary Material
Acknowledgments
We thank the patients and Dr. Mark Figgie of the Hospital for Special Surgery for providing synovial tissues. We thank Laura Donlin for critical review of the manuscript.
This work was supported by grants from the NIH (L.B.I), Arthritis National Research Foundation (G.D.K) and Sontag Foundation (G.D.K)
Footnotes
Author contributions: C.S., A.L., Y.Q. and K.L. designed and performed experiments, interpreted data and wrote the manuscript. G.D.K. and L.B.I oversaw the project, designed and supervised experiments, conceptualized the project, interpreted data and wrote the manuscript. G.D.K. had access to all primary data and is responsible for its integrity.
Conflict of interest: The authors declare no financial conflicts of interest.
References
- 1.Klareskog L, Malmstrom V, Lundberg K, Padyukov L, Alfredsson L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin Immunol. 2011;23(2):92–8. doi: 10.1016/j.smim.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Brusca SB, Abramson SB, Scher JU. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol. 2014;26(1):101–7. doi: 10.1097/BOR.0000000000000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(3):141–53. doi: 10.1038/nrrheum.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smolen JS, Aletaha D, Koeller M, Weisman MH, Emery P. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370(9602):1861–74. doi: 10.1016/S0140-6736(07)60784-3. [DOI] [PubMed] [Google Scholar]
- 6.Chakravarty SD, Poulikakos PI, Ivashkiv LB, Salmon JE, Kalliolias GD. Kinase inhibitors: a new tool for the treatment of rheumatoid arthritis. Clin Immunol. 2013;148(1):66–78. doi: 10.1016/j.clim.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 7.Prince FH, Bykerk VP, Shadick NA, Lu B, Cui J, Frits M, et al. Sustained rheumatoid arthritis remission is uncommon in clinical practice. Arthritis Res Ther. 2012;14(2):R68. doi: 10.1186/ar3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315(5814):1006–10. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 9.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2012;9(1):24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149(5):1607–15. [PMC free article] [PubMed] [Google Scholar]
- 11.Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–70. doi: 10.1111/j.1600-065X.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- 12.Buckley CD. Why does chronic inflammation persist: An unexpected role for fibroblasts. Immunol Lett. 2011;138(1):12–4. doi: 10.1016/j.imlet.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443(7114):998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 14.Kawane K, Tanaka H, Kitahara Y, Shimaoka S, Nagata S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proc Natl Acad Sci U S A. 2010;107(45):19432–7. doi: 10.1073/pnas.1010603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J. 1991;10(13):4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–98. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 17.Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205(2):331–7. doi: 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee A, Qiao Y, Grigoriev G, Chen J, Park-Min KH, Park SH, et al. Tumor necrosis factor alpha induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2013;65(4):928–38. doi: 10.1002/art.37853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 20.Ohmori Y, Hamilton TA. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J Immunol. 1995;154(10):5235–44. [PubMed] [Google Scholar]
- 21.Imaizumi T, Matsumiya T, Yoshida H, Naraoka T, Uesato R, Ishibashi Y, et al. Tumor-necrosis factor-alpha induces retinoic acid-inducible gene-I in rheumatoid fibroblast-like synoviocytes. Immunol Lett. 2009;122(1):89–93. doi: 10.1016/j.imlet.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Rosengren S, Corr M, Firestein GS, Boyle DL. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis. 2012;71(3):440–7. doi: 10.1136/ard.2011.150284. [DOI] [PubMed] [Google Scholar]
- 23.Firestein GS, Alvaro-Gracia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144(9):3347–53. [PubMed] [Google Scholar]
- 24.Sengupta TK, Chen A, Zhong Z, Darnell JE, Jr, Ivashkiv LB. Activation of monocyte effector genes and STAT family transcription factors by inflammatory synovial fluid is independent of interferon gamma. J Exp Med. 1995;181(3):1015–25. doi: 10.1084/jem.181.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tak PP. IFN-beta in rheumatoid arthritis. Front Biosci. 2004;9:3242–7. doi: 10.2741/1475. [DOI] [PubMed] [Google Scholar]
- 26.Kalliolias GD, Ivashkiv LB. Overview of the biology of type I interferons. Arthritis Res Ther. 2010;12(Suppl 1):S1. doi: 10.1186/ar2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31(4):539–50. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohmori Y, Schreiber RD, Hamilton TA. Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J Biol Chem. 1997;272(23):14899–907. doi: 10.1074/jbc.272.23.14899. [DOI] [PubMed] [Google Scholar]
- 29.Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3(1):69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 30.Natoli G. NF-kappaB and chromatin: ten years on the path from basic mechanisms to candidate drugs. Immunol Rev. 2012;246(1):183–92. doi: 10.1111/j.1600-065X.2012.01103.x. [DOI] [PubMed] [Google Scholar]
- 31.Fransen L, Van der Heyden J, Ruysschaert R, Fiers W. Recombinant tumor necrosis factor: its effect and its synergism with interferon-gamma on a variety of normal and transformed human cell lines. Eur J Cancer Clin Oncol. 1986;22(4):419–26. doi: 10.1016/0277-5379(86)90107-0. [DOI] [PubMed] [Google Scholar]
- 32.Wong GH, Goeddel DV. Tumour necrosis factors alpha and beta inhibit virus replication and synergize with interferons. Nature. 1986;323(6091):819–22. doi: 10.1038/323819a0. [DOI] [PubMed] [Google Scholar]
- 33.Bartee E, Mohamed MR, McFadden G. Tumor necrosis factor and interferon: cytokines in harmony. Curr Opin Microbiol. 2008;11(4):378–83. doi: 10.1016/j.mib.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiao Y, Giannopoulou EG, Chan CH, Park SH, Gong S, Chen J, et al. Synergistic activation of inflammatory cytokine genes by interferon-gamma-induced chromatin remodeling and toll-like receptor signaling. Immunity. 2013;39(3):454–69. doi: 10.1016/j.immuni.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9(4):378–87. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 36.Gordon RA, Grigoriev G, Lee A, Kalliolias GD, Ivashkiv LB. The interferon signature and STAT1 expression in rheumatoid arthritis synovial fluid macrophages are induced by tumor necrosis factor alpha and counter-regulated by the synovial fluid microenvironment. Arthritis Rheum. 2012;64(10):3119–28. doi: 10.1002/art.34544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, et al. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat Immunol. 2002;3(9):859–66. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- 38.Monticelli S, Natoli G. Short-term memory of danger signals and environmental stimuli in immune cells. Nat Immunol. 2013;14(8):777–84. doi: 10.1038/ni.2636. [DOI] [PubMed] [Google Scholar]
- 39.Genovese MC, Chakravarty EF, Krishnan E, Moreland LW. A randomized, controlled trial of interferon-beta-1a (Avonex(R)) in patients with rheumatoid arthritis: a pilot study [ISRCTN03626626] Arthritis Res Ther. 2004;6(1):R73–R77. doi: 10.1186/ar1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veys EM, Menkes CJ, Emery P. A randomized, double-blind study comparing twenty-four-week treatment with recombinant interferon-gamma versus placebo in the treatment of rheumatoid arthritis. Arthritis Rheum. 1997;40(1):62–8. doi: 10.1002/art.1780400110. [DOI] [PubMed] [Google Scholar]
- 41.Kuan WP, Tam LS, Wong CK, Ko FW, Li T, Zhu T, et al. CXCL 9 and CXCL 10 as Sensitive markers of disease activity in patients with rheumatoid arthritis. J Rheumatol. 2010;37(2):257–64. doi: 10.3899/jrheum.090769. [DOI] [PubMed] [Google Scholar]
- 42.Hanaoka R, Kasama T, Muramatsu M, Yajima N, Shiozawa F, Miwa Y, et al. A novel mechanism for the regulation of IFN-gamma inducible protein-10 expression in rheumatoid arthritis. Arthritis Res Ther. 2003;5(2):R74–81. doi: 10.1186/ar616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee EY, Lee ZH, Song YW. The interaction between CXCL10 and cytokines in chronic inflammatory arthritis. Autoimmun Rev. 2012;12(5):554–7. doi: 10.1016/j.autrev.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Zernecke A, Shagdarsuren E, Weber C. Chemokines in atherosclerosis: an update. Arterioscler Thromb Vasc Biol. 2008;28(11):1897–908. doi: 10.1161/ATVBAHA.107.161174. [DOI] [PubMed] [Google Scholar]
- 45.Salomon I, Netzer N, Wildbaum G, Schif-Zuck S, Maor G, Karin N. Targeting the function of IFN-gamma-inducible protein 10 suppresses ongoing adjuvant arthritis. J Immunol. 2002;169(5):2685–93. doi: 10.4049/jimmunol.169.5.2685. [DOI] [PubMed] [Google Scholar]
- 46.Mohan K, Issekutz TB. Blockade of chemokine receptor CXCR3 inhibits T cell recruitment to inflamed joints and decreases the severity of adjuvant arthritis. J Immunol. 2007;179(12):8463–9. doi: 10.4049/jimmunol.179.12.8463. [DOI] [PubMed] [Google Scholar]
- 47.Kwak HB, Ha H, Kim HN, Lee JH, Kim HS, Lee S, et al. Reciprocal cross-talk between RANKL and interferon-gamma-inducible protein 10 is responsible for bone-erosive experimental arthritis. Arthritis Rheum. 2008;58(5):1332–42. doi: 10.1002/art.23372. [DOI] [PubMed] [Google Scholar]
- 48.O'Boyle G, Fox CR, Walden HR, Willet JD, Mavin ER, Hine DW, et al. Chemokine receptor CXCR3 agonist prevents human T-cell migration in a humanized model of arthritic inflammation. Proc Natl Acad Sci U S A. 2012;109(12):4598–603. doi: 10.1073/pnas.1118104109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yellin M, Paliienko I, Balanescu A, Ter-Vartanian S, Tseluyko V, Xu LA, et al. A phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64(6):1730–9. doi: 10.1002/art.34330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.