

Abstract
Parkinson's disease affects more than five million people worldwide, yet no therapeutic has been identified that can slow or halt the progression of this debilitating disease. A new study in tau knockout mice suggests that tau deficiency causes impaired ferroportin-coupled iron export, by retention of the amyloid precursor protein, a neuronal ferroxidase partner, in the endoplasmic reticulum. This leads to parkinsonism through intracellular iron accumulation and degeneration of dopamine neurons (pages X-Y).
Parkinson's disease (PD) is the most common neurodegenerative movement disorder, affecting more than five million people worldwide1. A combination of genetic and environmental factors may cause PD2. The defining neuropathological hallmarks of PD are the progressive and selective degeneration of dopaminergic (DA) neurons of the substantia nigra (SN), as well as the formation of intraneuronal, filamentous protein inclusions enriched in α-synuclein and ubiquitin known as Lewy bodies and Lewy neurites3. In addition, tau pathology in PD and genome wide association (GWA) studies suggest that tau is somehow involved in PD pathogenesis 4. These cellular and molecular changes ultimately result in the onset of the cardinal clinical motor features of PD, including rest tremor, rigidity, postural instability and bradykinesia, as well as cognitive dysfunction in advanced cases3.
In this issue of Nature Medicine, Lei et al5 provide an unexpected twist for tau in the pathogenesis of PD. They find that soluble tau is reduced in the SN and suggest a model in which the reduction of soluble tau leads to the intracellular accumulation of iron and a late-age non-progressive degeneration of DA neurons through inhibiting the neuronal ferroxidase partner for ferroportin-coupled iron export by decreasing surface trafficking of the amyloid precursor protein (APP) (Figure 1). The authors find that tau knockout leads to dopaminergic degeneration, increased iron levels, levodopa responsive locomotor deficits and cognitive deficiencies by 12 months of age, mimicking sporadic PD. Unlike sporadic PD the pathologic changes do not progress. Their data suggests that the reciprocal relationship between changes in tau levels and iron accumulation is specific to the SN, as these pathological alterations remain absent from other brain regions, including neocortex and cerebellum.
Figure 1.
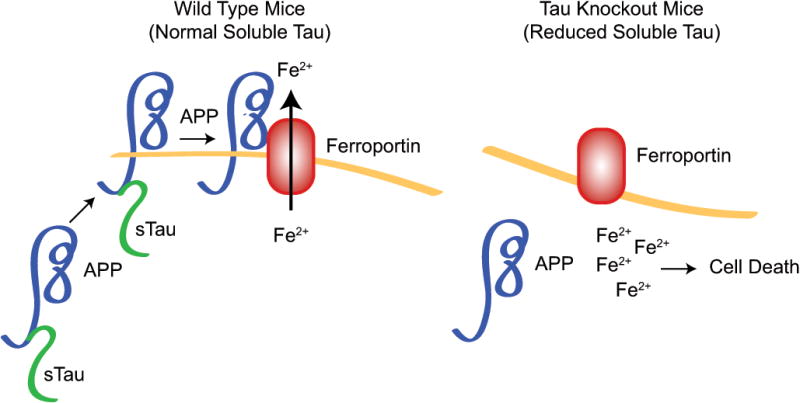
Effect of tau deficiency on dopaminergic degeneration. In the presence of functional tau (WT), neuronal iron (Fe2+) export is governed by tau's interaction with the amyloid precursor protein (APP). Binding of APP to tau results in the trafficking of the protein complex to the neuronal surface where APP interacts with ferroporin (Fnp), thereby allowing iron export from neurons. Tau deficiency in patients with PD or knock out (Tau KO), as shown by Lei et al., abolishes iron export from neurons. Lack of tau prevents dissociation of immature APP from the endoplasmic reticulum (ER), thereby abolishing the trafficking of APP to the neuronal surface where it matures and binds to Fnp. The absence of this interaction prevents Fe2+ from exiting neurons, leading to a toxic intracellular accumulation of Fe2+ and ultimately dopaminergic degeneration.
How might iron accumulate in tau deficient neurons and lead to degeneration of DA neurons? Using primary cortical cultures the authors investigated the primary route by which iron is exported from neurons; namely by means of the ferroportin protein which interacts with the amyloid precursor protein (APP) to govern iron export 6. Tau associates with APP protein and regulates APP trafficking in neurons 7. The authors found that overall levels of APP increase in tau deficient neurons, yet subcellular fractionation studies show that loss of tau results in an accumulation and retention of immature APP in the endoplasmic reticulum (ER). These data suggest that tau deficiency prevents APP from being trafficked to the neuronal surface where it matures and interacts with ferroportin to govern iron export. The subsequent accumulation of iron within neurons is toxic, ultimately leading to the observed neuronal death in PD (Figure 1). While the data investigating the mechanism underlying toxic iron accumulation in tau knockout neurons is intriguing, it remains to be determined if this mechanism translates to dopaminergic neurons and if differences in cortical and dopaminergic neurons may influence this mechanism differently.
The authors also found that administration of the moderate iron chelator clioquinol (CQ) to tau deficient mice prevented the onset of both motor dysfunction and cognitive decline5. Thus, CQ may represent a potential neurotherapeutic for the treatment of neurodegenerative disorders with reduced soluble tau and impaired iron export. Notably, liver iron levels as well as zinc and copper levels were not altered by treatment with CQ. The importance of this finding lies in the fact that CQ appears to specifically target iron levels in the SN without producing other, potentially untoward side effects. These results are interesting as they demonstrate that CQ may represent an effective neurotherapeutic for the treatment of disorders with reduced soluble tau such as PD. Since soluble tau is potentially reduced in AD and tauopathies and iron accumulates in these disorders6,8, CQ treatment may be potentially beneficial in these disorders as well.
The herein described model system is challenged by previous findings from several other groups where tau knockout mice present with a normal lifespan and do not demonstrate any substantial neurological or behavioral deficits (for review see Morris et al,9). Moreover, the absence of tau does not prevent motor deficits in two mouse models of Parkinson's disease10. Since the majority of the latter studies were conducted in younger mice, a careful reassessment of these models in 12-24 month old mice is a necessity to determine and validate the contribution of tau deficiency in the neurodegeneration of DA neurons.
Although, Lei et al5 find that soluble tau is selectively reduced in the SN of PD patients without a corresponding increase in the levels of insoluble tau, most other neuropathological studies find increased levels of insoluble hyperphosphorylated tau in PD11, even in PD without dementia12. Since there is no evidence that tau aggregation lowers the levels of soluble tau in vivo 9, the question of the mechanism underlying a potential decrease or loss of tau in PD or other neurodegenerative diseases remains unknown. One would need to presume that soluble tau that remains in PD is somehow not functioning properly, since soluble tau is not completely absent in PD brains as it is in the tau knockout animals and hemizygous tau knockout animals are normal9. Thus, future studies will need to carefully evaluate the physical state of tau in PD to confirm that soluble tau is reduced and dysfunctional. This is critical as reducing tau has been suggested as a potential therapy for AD related dementia13 by normalizing network activity and reducing amyoid beta and excitotoxin related injury (for review see, Morris et al., 9). It may be necessary to reduce the aggregated forms of Tau while maintaining the soluble form of tau.
While the loss of dopaminergic neurons along with a phenotype mimicking sporadic PD is interesting, it remains unclear if Lewy bodies and Lewy neurites form in this model system. Moreover, it is questionable if the observed parkinsonian symptoms along with the robust loss in brain mass are solely due to the inability of neurons to maintain iron homeostasis, or if other factors may contribute to these observations. Specifically, functional consequences associated with tau loss have been stressed in a study using hippocampal cultures from tau knockout mice demonstrating delayed maturation of neuronal extensions in these cultures14. These results may suggest that the pathological changes observed by Lei et al.,5 may have additional underlying causes other than the sole accumulation of iron.
Overall these findings suggest the involvement of a potentially new mechanism associated with the selective loss of dopaminergic neurons in PD and associated parkinsonism syndromes (Figure 1). The finding that loss of tau interferes with iron export by preventing proper trafficking of APP to the neuronal surface where it interacts with ferroportin (Figure 1), as well as the discovery that pharmacological intervention by means of the moderate iron chelator CQ can rescue this phenotype suggests a therapeutic potential of CQ for PD. Future work should aim at identifying any long term risks associated with the use of CQ as well as the question if this drug could be used as a preventative measure and could be administered to patients who are at risk of developing PD.
Acknowledgments
This work was supported by grants from the NIH NS38377. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Program No. M-1.
Footnotes
Competing Financial Interests: The authors declare no competing financial interests.
Bibliography
- 1.Aron L, Klein R. Repairing the parkinsonian brain with neurotrophic factors. Trends Neurosci. 2011;34:88–100. doi: 10.1016/j.tins.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 3.Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nalls MA, et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lei P, et al. Tau deficiency induces a parkinsonism with dementia phenotype by obtunding APP-mediated iron export. Nature Medicine. 2012 doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 6.Duce JA, et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhukareva V, et al. Selective reduction of soluble tau proteins in sporadic and familial frontotemporal dementias: an international follow-up study. Acta Neuropathol. 2003;105:469–476. doi: 10.1007/s00401-002-0668-8. [DOI] [PubMed] [Google Scholar]
- 9.Morris M, Koyama A, Masliah E, Mucke L. Tau reduction does not prevent motor deficits in two mouse models of Parkinson's diseas. PLoS One. 2011;6:e29257. doi: 10.1371/journal.pone.0029257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Compta Y, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain. 2011;134:1493–1505. doi: 10.1093/brain/awr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wills J, et al. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson's disease brains with and without dementia. Exp Neurol. 2010;225:210–218. doi: 10.1016/j.expneurol.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vossel KA, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson HN, et al. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]