

Abstract
The proteasome is a multi-subunit complex responsible for most non-lysosomal turnover of proteins in eukaryotic cells. Proteasome inhibitors are of great interest clinically, particularly for the treatment of multiple myeloma (MM). Unfortunately, resistance arises almost inevitably to these active site-targeted drugs. One strategy to overcome this resistance is to inhibit other steps in the protein turnover cascade mediated by the proteasome. Previously, Anchoori et al. identified Rpn13 as the target of an electrophilic compound (RA-190) that was selectively toxic to MM cells (Cancer Cell 24, 791–805 (2013)), suggesting that this sub-unit of the proteasome is also a viable cancer drug target. Here we describe the discovery of the first highly selective, reversible Rpn13 ligands and show that they are also selectively toxic to MM cells. These data strongly support the hypothesis that Rpn13 is a viable target for the development of drugs to treat MM and other cancers.
Keywords: Multiple myeloma, proteasome, Rpn13, peptoid, combinatorial chemistry, peptoid
Introduction
The 26S proteasome is responsible for most non-lysosomal protein degradation in eukaryotic cells and, as such, is involved in many different cellular pathways.1 The proteasome is comprised of two major sub-structures, the 20S core particle (CP) and the 19S regulatory particle (RP)2,3 (Figure 1). The former is a barrel-shaped structure4,5 comprised of four stacked heptameric rings (two rings of β proteins 1–7 sandwiched between two rings of α proteins 1–7). The three proteolytic active sites are found inside of the cavity. The openings at the top and bottom are so narrow that polypeptides must be completely unstructured to pass through. Most proteasome substrates are modified with K48-linked poly-ubiquitin (Ub) chains. Rpn106 and Rpn13,7 which reside in the 19S RP, mediate catalyst-substrate recognition by acting as ubiquitin receptors. Once bound to the proteasome, the ubiquitin chains are removed by dedicated ubiquitin-specific proteases (USPs) associated with the complex and the substrate is unfolded so that the unstructured peptide chain can be fed into the 20S CP.8 Substrate unfolding, as well as gate opening, is carried out by six homologous AAA class ATPases called Rpt1-6.9
Figure 1.

Structure of the 26S proteasome (right) with the subunits of the 19S RP labeled and strctures of compounds that inhibit proteasomal activity (left). RA-190 (ref 28) and KDT-11 (described in this publication) target Rpn13 (labeled N13 as this is a non-ATPase subunit) within the 19S RP, while Bortezomib covalently reacts with the β5 subunit of the 20S CP.
Pharmacological inhibitors of proteasome function are invaluable tools in both the laboratory and the clinic.10 Peptide aldehydes, such as MG-132, are moderately selective proteasome inhibitors that have been used widely to probe the role of proteasome-mediated proteolysis in a plethora of cellular processes. The peptide boronic acid Bortezomib (Velcade) (Figure 1), which hits primarily the chymotryptic β5 sub-unit of the 20S core complex, has revolutionized the treatment of multiple myeloma (MM) and is being investigated for the treatment of other cancers, along with second-generation inhibitors that lack some of Bortezomib’s off-target toxicities.11,12 Bortezomib displays an acceptable therapeutic window in MM, perhaps because myeloma is a malignancy of antibody-secreting plasma cells, which produce enormous amounts of immunoglobulin proteins. Many of these proteins are likely mis-folded and require destruction by the proteasome in order avoid cellular toxicity, placing an unusually high burden on the unfolded protein response (UPR) pathway in this cell type.13
Unfortunately, Bortezomib resistance arises rapidly and almost inevitably14 highlighting the need for the development of new inhibitors to overcome this resistance. Most work along these lines has focused on second generation proteasome inhibitors15,16 that also target the active site(s) but bind to the proteasome in a fashion that is not blocked by mutations in the β5 sub-unit that weaken Bortezomib association.14,17–22
An alternative strategy to overcome Bortezomib resistance in MM would be to inhibit one or more functions of the 19S RP. However, the highly developed pharmacology for blocking the proteolytic function of the proteasome stands in sharp contrast to the underdeveloped state of manipulating the various steps in substrate processing mediated by the 19S RP. Inhibitors of the ubiquitin-specific proteases USP14 and USP7 have been reported23,24,25 as has a peptoid inhibitor of the Rpt4 AAA ATPase,26,27 but none of these compounds are potent nor has their selectivity been characterized thoroughly.
An important contribution to this area came recently with the discovery by Roden and colleagues of an amino acid-chalcone conjugate called RA-190 (Figure 1) that inhibits the ubiquitin-proteasome pathway in various cancer cells, but does not target the 20S active sites.28,29 Aided by the fact that RA-190 is a covalent proteasome inhibitor, Rpn13, one of the ubiquitin receptors, was identified as the target of this compound.28 Interestingly, Rpn13 is not essential in mice30 and Δrpn13 yeast strains only show a phenotype in a Δrpn10 background7 or when subjected to cellular stress.31 Rpn13 is overexpressed in a variety of cancers,32 including MM, ovarian, cervical, pancreatic and colorectal. This has led to the suggestion that Rpn13 may be a stress-induced accessory factor for the proteasome in cells that must turn over abnormally high levels of mis-folded proteins, but that it is mostly dispensable in unstressed cells,28,33 presumably because the other ubiquitin receptor, Rpn10 is able to handle the load under these conditions. This raises the possibility that Rpn13 inhibitors may have an expanded therapeutic window compared to active site proteasome inhibitors or compounds targeting other components of the complex that are essential in all cells. Therefore, there is a need for the discovery of new chemical matter targeting Rpn13.
In this study we report the discovery of a peptoid ligand for Rpn13 (called KDT-11, Figure 1) that displays modest affinity (KD = 2 μM) but very high selectivity for Rpn13. It is shown that this compound is toxic to MM cells but has little effect on HEK-293T cells. Moreover, KDT-11 acts synergistically with Bortezomib. The peptoid is shown to bind a surface of Rpn13 that is different than that recognized by RA-190. The fact that these two Rpn13-binding compounds effect similar phenotypes in cellular assays, despite having completely different chemical structures and recognizing different surfaces of the protein, argues strongly that Rpn13 inhibition is indeed the source of selective toxicity to MM cells, not some off-target effect.34
Results
Library Synthesis and Screening
A one bead one compound peptoid library was created by split and pool synthesis35,36 using the “sub- monomer” method37,38 on 90 μm TentaGel beads.39 The library was separated from the bead via the linker shown in Figure 2A. Each compound contained five variable residues surrounding a central, conserved unit displaying an amine side chain (Figure 2B). Ten amines were employed as diversity elements, yielding a library of 100,000 molecules with molecular weights ranging from 512 g/mol to 1380 g/mol. In the first four variable positions, many of the amines employed were α-branched (Figure 2C; for a full listing of all of the amines used, see Supporting Information Figure S1). This favors the cis amide bond rotomer40–42 over the trans (Figure 2D) thus reducing the “floppiness” of the main chain. At the N-terminal position, secondary amines were employed (Figure 2C). 36 beads were chosen at random from the library, the molecules were released from the bead and analyzed by tandem mass spectrometry. 33 gave mass spectra that allowed unequivocal determination of the structure of the compound, so the library was deemed of sufficiently high quality to carry forward (Supporting Information Figure S2).
Figure 2.

General structure of the library used to screen for Rpn13 ligands. Each N-substituted glycine unit is derived from bromoacetic acid and an amine. (A) Linker structure that incorporates moieties needed for peptoid cleavage or to aid in MS identification. (B) Backbone structure of the library, which contains five variable positions. (C) Branched amines utilized in the “sub-monomer” library synthesis that provided a steric constraint to the backbone, preventing some of the “floppiness” associated with peptoids because of the preference for the cis-amide conformation (illustrated in (D)).
For screening, approximately 5–6 copies of the peptoid library was incubated with recombinant, His6-tagged human Rpn13 in the presence of a large excess of non-specific competitor proteins (Figure 3, i; also see Supporting Information Figure S3). After washing away unbound proteins, the beads were incubated with anti-Rpn13 antibodies (Figure 3, ii). After another wash, the beads were incubated with anti-rabbit-IgG antibody-coated iron oxide particles (Figure 3, iii) and the magnetized TentaGel beads were collected using a strong magnet.43 275 beads were isolated in this fashion. These “hits” were washed with a denaturing buffer to remove any bound proteins and placed into wells of a microtiter plate (one bead per well). The peptoid was liberated from the bead by reaction with cyanogen bromide and sequenced by tandem mass spectrometry.
Figure 3.
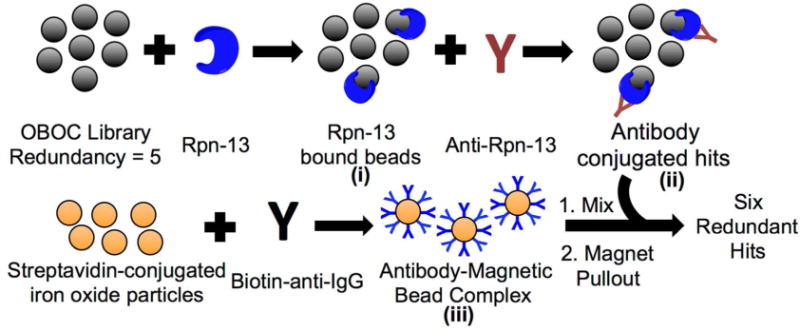
Library screening scheme utilizing recombinant His6-tagged Rpn13 (blue) as the target. The OBOC library was first exposed to Rpn13 in the presence of an excess of competitor proteins, followed by a rabbit anti-Rpn13 IgG antibody (red Y-shaped molecule). After thorough washing, the compound-displaying beads were mixed with magnetic particles linked to streptavidin and coated with biotin-conjugated anti-rabbit IgG. Only hit beads will bind the iron oxide particles and be pulled out by a magnet because of the network of peptoid-Rpn13-anti-Rpn13-secondary antibody interactions.
False positives are terribly common in OBOC library screening experiments44,45 but we have shown that compounds isolated more than once from redundant libraries are almost always bona fide ligands.45 Six molecules (Figure 4) were found more than once in the hit pool (three were found three times and three were found four times) so further efforts were focused on these compounds exclusively. All six of these primary hits were re-synthesized to include a fluorescein for fluorescent polarization (FP) analysis. Titration with Rpn13 revealed all of the repeat hits to be weak ligands, with compounds 1 and 2 showing the highest affinities (Supporting Information Figure S4).
Figure 4.

Structures of the six first-generation hits. Because a five-fold redundant library was employed in the screen, hits were considered as bona fide only if they were isolated on at least three different beads. These hit structures were then elucidated with tandem MALDI mass spectrometry after release from the bead. Fluorescent derivatives of these molecules were synthesized to facilitate binding studies.
Improvement of the Primary Screening Hits
As a first step towards the identification of higher affinity compounds and to identify the elements of the molecule critical for binding Rpn13, a small “derivative library” was synthesized. At each variable position, three amines were employed in the split and pool synthesis: methylamine and the two amines present at the corresponding position in hits 1 and 2. The goal of this “methyl scan” was determine the amine-derived side chains necessary for binding to Rpn13. In addition, the central ethylene diamine was acetylated or left as the free amine. The theoretical diversity of this library was 486 compounds. Fluorescein was appended to the alkyne in the linker region of each molecule by click chemistry.46,47 1,500 beads were picked and distributed into individual wells of microtiter plates, providing about three-fold coverage of the library. The peptoids were cleaved from the resin and the solutions were split between two 384-well plates, one for FP analysis and the other for structure determination by MALDI, Figure 5A. For FP analysis, 5 μM of Rpn13 was added to each well and the change in fluorescent polarization was measured. Compounds 1 and 2 gave a signal of 45 mP with 5 μM of Rpn13 under these conditions, so any compounds from the derivative library that gave a signal higher than 45 mP was considered a potentially improved derivative (Supporting Information Figure S5). The structures of these molecules were determined by tandem mass spectrometry. Only sequences found at least twice in the >45 mp category were considered further. Figure 5B summarizes the amine monomers present at each position of the hit compounds.
Figure 5.

Identification of an improved Rpn13 ligand. (A) Synthesis of a second-generation library based upon the primary hits. After synthesis, the peptoids are removed from resin, analyzed for their ability to bind Rpn-13, and hit structures were determined by MALDI. (B) Analysis of each amine monomer at every position of hit compounds with a mP greater than 45.
In this pool of putatively improved Rpn13 ligands, all of the molecules had an amide-containing side chain at position four and the N-methyl-fluorobenzyl unit at the N-terminus. There was a significant, but not absolute, preference for a particular side chain at positions one, three, and five. No significant bias was observed at position two. The nearly complete absence of methylamine from position five was interesting when considered in concert with the quite modest preference for one of the branched side chains over the other. This suggests that a bulky alkyl group at this position favors binding through a hydrophobic interaction.
Based on this information, fluorescein-conjugated derivatives of compounds 7–9 (Figure 6A) were re-synthesized, purified, and their affinity for Rpn13 was determined. These compounds include the consensus sequence inferred from the Figure 5B data and differ only in the position two residue. These molecules possessed essentially identical Kd values of approximately 4 μM (Figure 6B), confirming that the nature of the side chain at position 2 indeed has no effect on binding.
Figure 6.

Finally, since analysis of the small derivative library indicated a strong preference for an acetyl group on the central amine side chain, several peptoids were synthesized that tested the effect of different amide groups at this position. Most of them had the same affinity for Rpn13 as did compound 9 (Supporting Information Figure S6), except for extremely bulky groups that showed a slightly poorer affinity. We concluded that the Rpn13 binding is favored by including a amide moiety rather than an amine at this position, but the nature of the amide is not critical.
Rpn13-binding peptoids are selectively toxic to multiple myeloma cells
To determine if Rpn13-binding peptoids (Figure 7A) affect the function of the protein, and thus proteasome-mediated proteolysis, their effect on the viability of MM and HEK-293T cells was determined (Figure 7B). HEK-293T cells were chosen as a negative control cell line because, unlike MM cells, HEK-293T cells are not plasma cells and do not require unusually high proteasome activity to survive. Peptoids 7, 8, and 9 were all toxic to MM.1R cells with EC50s ranging from approximately 5 μM to 50 μM. All of these peptoids also displayed toxicity towards HEK-293T cells. Peptoids 8 and 9 showed a modest therapeutic window (about 10-fold for 9) though compound 7 appeared to be equally toxic to both cell types. Compound 10, with an amine side chain, was about 10-fold less potent. Strikingly, however, the presence of an aromatic ring in the side chain of position four (11 and 12) essentially eliminated toxicity to the HEK-293T, with no significant cell death observed even at a concentration of 100 μM similar to the results observed with MG-132. Since the nature of the amide side chain has no effect on the affinity of the peptoid for Rpn13 (Fig. 7A), this result suggests that the particular amide in 11 ablates one or more off-target interactions that are the cause of toxicity in HEK-293 T cells.
Figure 7.

(A) Summary of the best compounds tested for binding to Rpn13. (B) Cell viability testing of MM and HEK-293 cells (B) with 2nd (7–9) and 3rd generation hits (10–12). Compounds 9, 11, and 12 possessed the most cytotoxic activity against MM. The largest therapeutic window was seen with compounds 11 and 12, which are not toxic to HEK-293 cells even at 100 μM concentration.
Peptoid 11 was also tested against two other types of MM, two sarcoma cell lines, and an ovarian cancer cell line. Similar EC50 values were obtained for the other MM cell lines and slightly higher values for the sarcoma and ovarian, Supporting Information Figure S7.
Is inhibition of Rpn13 the cause of selective toxicity to MM cells?
A concern in any pharmacological experiment is whether off-target effects might contribute to the phenotype observed. This is especially acute when the phenotype is something as general as cellular toxicity. Indeed, presumably because Rpn13 is non-essential in normal cells, there has been a call for further evidence that the cellular toxicity of RA-190 is indeed due to Rpn13 inhibition and not an off-target effect.34 As a first step towards addressing this question in the context of the peptoid ligands, the selectivity of 11 for Rpn13 was assessed. In vitro, 11 bound Rpn13 at least 100-fold more tightly than several other proteins tested (Supporting Information Figure S8).
More importantly, an affinity chromatography experiment was performed to assess the selectivity of peptoid binding to Rpn13 in the context of the MM cellular proteome. A derivative of 11 was coupled to iodoacetamide-modified agarose beads (Thermo Scientific SulfoLink Resin). The beads were packed into a small column onto which lysate from MM cells was loaded. After a one-hour incubation, the column was washed with loading buffer, followed by washing with a high salt (1 M NaCl) buffer. Finally, any tightly bound proteins were eluted with a denaturing wash (8M Guanidinium HCl). Both the crude lysate and the fraction eluted with Guanidinium HCl were subjected to SDS-PAGE analysis and stained with Coomassie Brilliant Blue. Remarkably, the eluent from the peptoid 11 affinity column was comprised of predominantly a single band (Figure 8A). Western blotting (Figure 8B) with an anti-Rpn13 antibody confirmed that this band was Rpn13.
Figure 8.
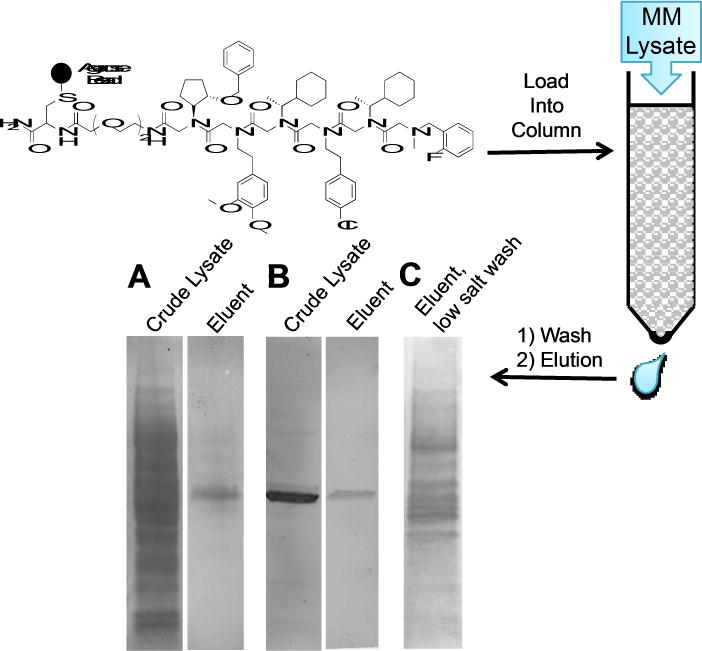
Analysis of the proteins that bind to peptoid 11 in a crude lysate prepared from MM cells. 11 was linked to an agarose matrix and exposed to an MM lysate. After washing with loading buffer, a second wash was done with a high salt buffer (1 M NaCl). Finally, tightly bound proteins were eluted with a denaturing buffer (8M Guanidinium HCl) A. SDS-PAGE analysis, followed Coomassie staining, of the lysate loaded onto the column and of the eluent released by the denaturing wash. B. A western blot with anti-Rpn13 using the same gel shown in A. C. Coomassie-stained gel of the eluent from the denaturing wash in an experiment in which the high salt wash had been omitted to preserve Rpn13-protein interactions. The same experiments with a control peptoid are presented in Supporting Information Figure S8.
This experiment was repeated, except this time, the 1M NaCl wash was excluded in order to preserve any moderate affinity interactions between Rpn13 and other cellular proteins. After washing with loading buffer, all of the bound protein was eluted with denaturing buffer. Under these conditions, Rpn13 eluted along with several associated protein (Fig. 8C), many of which were shown to be components of the 19S RP by LC-MS/MS proteomic analysis (Supporting Information Table S1). These data indicate that peptoid 11 can engage Rpn13 when it is assembled into the proteasome. This conclusion is also supported by a glycerol gradient analysis (Supporting Information Figure S11). Rpn13 was detected in the same fractions as Rpn2 and the 20S β5 subunit. Taken together, these data argue that whatever the mechanism of inhibition of proteasome-mediated proteolysis by 11, it is not due to inhibition of Rpn13 loading into the proteasome.
Finally, returning to the question of whether Rpn13 inhibition is indeed the source of cellular toxicity, the relative toxicities of peptoids 11 and 2 to MM cells were compared. If inhibition of Rpn13 is indeed the physiologically relevant target, then one would expect that the measured EC50s of these compounds should correlate roughly with their affinities for Rpn13, which differ by about 13-fold. As shown by the dose response curves in Figure 9, this was indeed the case. Moreover, peptoid 13, a scrambled version of 9, showed no binding to Rpn13 and was not toxic to MM.
Figure 9.
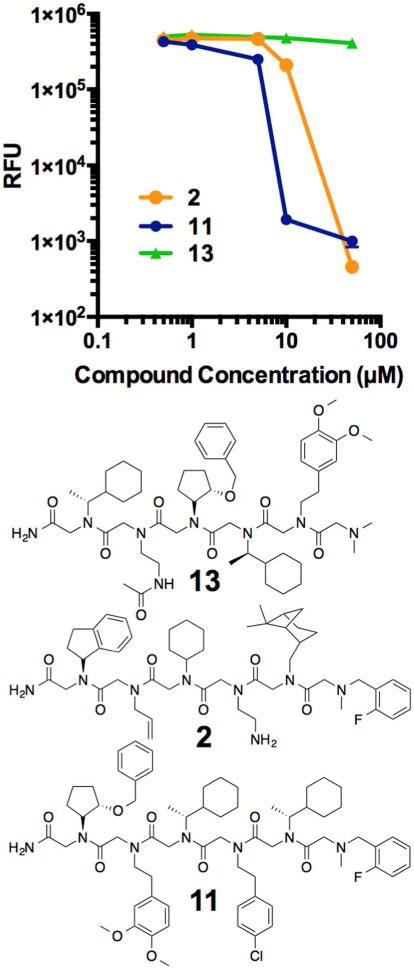
Evaluation of the toxicity of compounds 2, 11, and 13 to MM cells. RFU is a measure of cell viability. 13, which does not bind to purified Rpn13, has no effect on MM cells. As the binding affinity to Rpn13 increases (13-fold difference between 2 and 11) so does the efficacy against MM.
KDT-11 is a proteasome inhibitor in MM cells that causes ubiquitin accumulation
The data described above argues that Rpn13 is the physiologically relevant target of peptoid 11. Is this effect mediated through inhibition of proteasome-mediated proteolysis? If so, then one would expect that treatment of MM cells with 11 would result in a buildup of poly-ubiquitylated proteins at levels well above that of the normal steady state. As shown in Figure 10A, this is indeed the case. MM cells incubated with 11 show approximately the same level of these intermediates as cells incubated with the classical proteasome inhibitor MG-132 (also see Supporting Information Figure S9). HEK293-T cells were also treated with 11 and MG-132. As seen in Figure 10B, accumulation of ubiquitylated proteins does occur relative to the vehicle-treated cells, but cell death does not follow, as expected from the fact HEK cells are not so exquisitely dependent on high level proteasome activity. It is also interesting that the level of poly-Ub protein buildup in 11-treated HEK293-T cells is much lower than seen in 11-treated MM cells (compare Figs. 10A and 10B), consistent with the idea that protein turnover is more dependent on Rpn13 activity in the MM cells.
Figure 10.
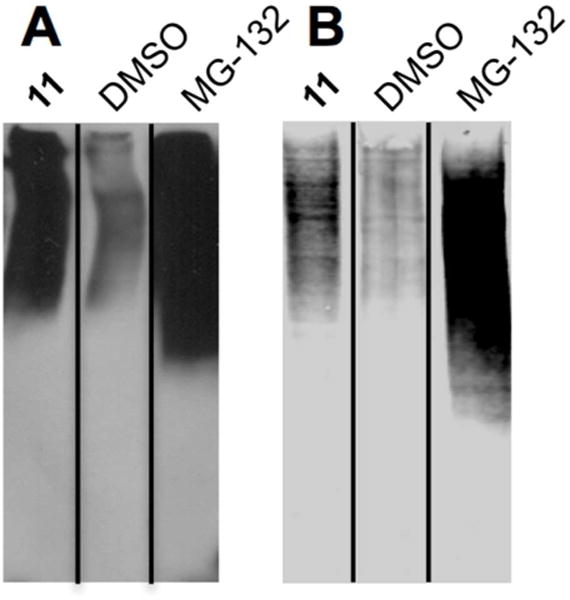
SDS-PAGE analysis of lysates prepared from cells treated with the compounds indicated. The gels were Western blotted using an anti-Ub antibody to visualize poly-ubiquitin conjugates. (A) MM cells and (B) HEK293-T cells. These experiments were performed in triplicate (Supporting Information Figure S10).
Peptoid 11 and Bortezomib act synergistically
Our mechanistic hypothesis that peptoid is an inhibitor of Rpn13 predicts two things. First, that 11 and Bortezomib should act synergistically in MM cells if they indeed inhibit different steps in the proteasome-catalyzed protein degradation pathway. Second, 11 should not inhibit proteasome-mediated peptidolysis, since in this case the substrate is not ubiquitylated and need not be processed by the 19S RP. As shown in Fig. 11, both of these hypotheses proved to be true.
Figure 11. Peptoid 11 act synergistically on different steps in the proteasome-mediated protein degradation pathway.

(A) The dose-dependent toxicity of Bortezomib to MM cells was assessed in the presence and absence of a sub-lethal dose of peptoid 11 (5 μM). (B) Purified 26S proteasome was tested for its ability to cleave Suc-LLVY-AMC in the presence of Bortezomib or peptoid 11. Hydrolysis of the fluorescent peptide is inhibited only in the presence of Bortezomib.
The sensitivity of MM cells to Bortezomib was analyzed in the presence of a sub-lethal dose (5 μM) of peptoid 11 or in its absence. As shown in Figure 11A (also see Supporting Information Figure S12), there was indeed clear synergy between the two compounds, with far more cell killing observed at 1 nM or 5 nM Bortezomib in the presence of 11 than in its absence.
Figure 11B shows an experiment in which purified 26S proteasome was incubated with a trimeric, fluorogenic peptide substrate containing a chymotrypsin cleavage site. A dose-sensitive decrease of fluorescence when the 26S was treated with Bortezomib. No change in fluorescence was observed when treated the solution was treated with 11.
Taken together, the data shown in Fig. 11 provide further evidence that peptoid 11 is an inhibitor of the 19S RP (via Rpn13) and targets a different step in the turnover of poly-ubiquitylated proteins than does Bortezomib.
Peptoid 11 and RA-190 recognize different surfaces of Rpn13
RA-190, the only previously reported Rpn13 inhibitor, links covalently to Cys88 near the ubiquitin-binding domain.29 To ask if peptoid 11 binds to the same surface, though reversibly, a competition experiment was conducted in which Rpn13 was incubated with fluorescently labeled 11, then the complex was challenged with increasing amounts of RA-190. As shown in Figure 12, the level of fluorescence polarization was insensitive to the amount of RA-190 present, even at concentrations far above its IC50 of 100 nM.28 Challenge of the labeled peptoid-Rpn13 complex with K48-linked ubiquitin chains also did not compete binding. In contrast, when the fluorescently labeled 11-Rpn13 complex was challenged with increasing amount of unlabeled 11, dose-dependent reduction of the FP signal was observed, as expected. These data show that peptoid 11 binds to a surface of Rpn13 distinct from those recognized by either RA-190 or ubiquitin chains.
Figure 12.
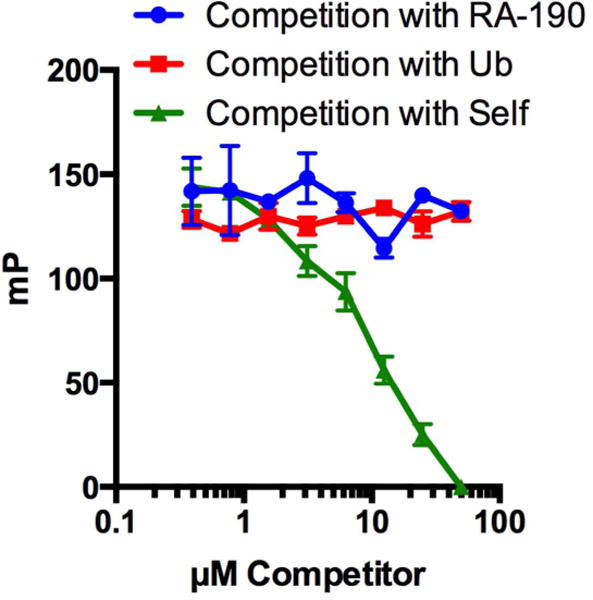
Competition of fluorescently-labeled 11 with an increasing concentration of RA-190, K48-linked penta-ubiquitin chains (Ub), or unlabeled 11. RA-190 and ubiquitin do not compete binding of compound 11 to Rpn13, providing evidence that 11 does not bind in the Ub or RA-190 pocket.
Discussion
The pioneering work of Roden and colleagues has suggested that Rpn13 may be an outstanding target for inhibition of the proteasome in MM cells and other cancers.29 However, there is always the possibility that the phenotype elicited by a bioactive compound may be due to off-target effects, particularly gross effects such as cellular toxicity. One of the best ways to support the idea that an observed phenotype is due to drugging a particular target protein is to isolate two structurally distinct inhibitors of that target and demonstrate that they elicit the same phenotype.
Towards this goal, a library of peptoids was screened against Rpn13. Six “hits” were identified (Figure 4), all of which proved to be weak ligands for Rpn13. An improved second-generation Rpn13 ligand, peptoid 11, was isolated by screening a small derivative library. While this peptoid, which will be referred to as KDT-11 hereafter (Figure 1), has only a modest EC50 value (5 μM), it is highly selective for Rpn13, as shown by the affinity chromatography experiment presented in Figure 8. Competition binding experiments demonstrated that KDT-11 bound a site on Rpn13 that is distinct from that recognized by RA-190 or, for that matter, K48-linked ubiquitin chains (Figure 12).
Conclusion
In conclusion, we have discovered the first reversible inhibitor of Rpn13, one of two ubiquitin receptors of the 19S RP. The peptoid 11 (henceforth called KDT-11) binds Rpn13 with modest affinity, but high selectivity. KDT-11 bears no resemblance to RA-190 and binds a distinct site on Rpn13. Yet both compounds elicit similar biological activity, including blockade of proteasome-mediated proteolysis of poly-ubiquitylated proteins and selective toxicity to MM cells. This argues strongly that Rpn13 is the relevant target for mediating the cytotoxic effects and, together with the work of Anchoori et. al., confirms Rpn13 as an attractive target for the development of novel drugs for MM and perhaps other cancers.28 Finally, the availability of these two very different reagents for inhibition of Rpn13 function should greatly aid further research into understanding why Rpn13 appears to have a critical role in many cancer cells but is largely dispensable in normal cells.
Supplementary Material
Acknowledgments
We thank Prof. Alfred Goldberg for advice and helpful discussions at the outset of this project. D.J.T. was supported by a Ruth Kirschstein post-doctoral fellowship from the NIH (5F32CA186612-02). This work was supported by a contract from the NHLBI (N01-HV-00242).
Footnotes
Supporting Information Available: Detailed procedures for the synthesis of compounds, binding assays, cytotoxic assays and all other experimental protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Baumeister W, Walz J, Zuhl F, Seemuller E. Cell. 1998;92:367. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 2.Glickman MH, Rubin DM, Fried VA, Finley D. Mol Cell Biol. 1998;18:3149. doi: 10.1128/mcb.18.6.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lander GC, Martin AG, Nogales E. Curr Op Struc Biol. 2013;23:243. doi: 10.1016/j.sbi.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groll M, Ditzel L, Lowe J, Stock D, Bochtier M, Bartunik HD, Huber R. Nature. 1997;386:463. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 5.Groll M, Kim KB, Kairies N, Huber R, Crews CM. J Amer Chem Soc. 2000;122:1237. [Google Scholar]
- 6.Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. J Biol Chem. 1994;269:7059. [PubMed] [Google Scholar]
- 7.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. Nature. 2008;453:481. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verma R, Aravind L, Oania R, McDonald WH, Yates JRI, Koonin EV, Deshaies RJ. Science. 2002;298:611. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 9.Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ESC, Siddiqui SM, Wah DA, Baker TA. Cell. 2004;119:9. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kisselev AF, Goldberg AL. Chemistry & Biology. 2001;8:739. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 11.Paramore A, Frantz S. Nature reviews. Drug discovery. 2003;2:611. doi: 10.1038/nrd1159. [DOI] [PubMed] [Google Scholar]
- 12.Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP. Curr Pharm Des. 2013;19:4025. doi: 10.2174/1381612811319220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cvek B, Dvorak Z. Curr Pharm Des. 2011;17:1483. doi: 10.2174/138161211796197124. [DOI] [PubMed] [Google Scholar]
- 14.Kale AJ, Moore BS. J Med Chem. 2012;55:10317. doi: 10.1021/jm300434z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steele JM. Journal of oncology pharmacy practice. 2013;19:348. doi: 10.1177/1078155212470388. [DOI] [PubMed] [Google Scholar]
- 16.Thompson JL. The Annals of pharmacotherapy. 2013;47:56. doi: 10.1345/aph.1R561. [DOI] [PubMed] [Google Scholar]
- 17.Lu S, Chen Z, Yang J, Chen L, Gong S, Zhou H, Guo L, Wang J. Exp Hematol. 2008;36:1278. doi: 10.1016/j.exphem.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Lu S, Yang J, Chen Z, Gong S, Zhou H, Xu X, Wang J. Exp Hematol. 2009;37:831. doi: 10.1016/j.exphem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Lu S, Yang J, Song X, Gong S, Zhou H, Guo L, Song N, Bao X, Chen P, Wang J. J Pharmacol Exp Ther. 2008;326:423. doi: 10.1124/jpet.108.138131. [DOI] [PubMed] [Google Scholar]
- 20.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G. Blood. 2008;112:2489. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 21.Ri M, Iida S, Nakashima T, Miyazaki H, Mori F, Ito A, Inagaki A, Kusumoto S, Ishida T, Komatsu H, Shiotsu Y, Ueda R. Leukemia. 2010;24:1506. doi: 10.1038/leu.2010.137. [DOI] [PubMed] [Google Scholar]
- 22.Harshbarger W, Miller C, Diedrich C, Sacchettini J. Structure. 2015;23:418. doi: 10.1016/j.str.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Lee BH, Finley D, King RW. Curr Protoc Chem Biol. 2012;4:311. doi: 10.1002/9780470559277.ch120078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D. Nature. 2010;467:179. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, Weinstock J, Kingsbury WD, Hideshima T, Shah PK, Minvielle S, Altun M, Kessler BM, Orlowski R, Richardson P, Munshi N, Anderson KC. Cancer Cell. 2012;22:345. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim HS, Archer CT, Kodadek T. J Am Chem Soc. 2007;129:7750. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim HS, Cai D, Archer CT, Kodadek T. J Am Chem Soc. 2007;129:12936. doi: 10.1021/ja075469+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anchoori RK, Karanam B, Peng S, Wang JW, Jiang R, Tanno T, Orlowski RZ, Matsui W, Zhao M, Rudek MA, Hung CF, Chen X, Walters KJ, Roden RB. Cancer Cell. 2013;24:791. doi: 10.1016/j.ccr.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anchoori RK, Khan SR, Sueblinvong T, Felthauser A, Iizuka Y, Gavioli R, Destro F, Isaksson Vogel R, Peng S, Roden RB, Bazzaro M. PloS one. 2011;6:e23888. doi: 10.1371/journal.pone.0023888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Shami A, Jhaver KG, Vogel P, Wilkins C, Humphries J, Davis JJ, Xu N, Potter DG, Gerhardt B, Mullinax R, Shirley CR, Anderson SJ, Oravecz T. PloS one. 2010;5:e13654. doi: 10.1371/journal.pone.0013654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tkach JM, Yimit A, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, Davis TN, Nislow C, Brown GW. Nat Cell Biol. 2012;14:966. doi: 10.1038/ncb2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Neoplasia. 2004;6:744. doi: 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kisselev AF. Cancer Cell. 2013;24:691. doi: 10.1016/j.ccr.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 34.Boettner B. SciBX. 2014;7 [Google Scholar]
- 35.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 36.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Nature. 1991;354:84. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 37.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Amer Chem Soc. 1992;114:10646. [Google Scholar]
- 38.Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. Methods Enzymol. 1996;267:437. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- 39.Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. J Am Chem Soc. 2003;125:13995. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- 40.Roy O, Caumes C, Esvan Y, Didierjean C, Faure S, Taillefumier C. Org Lett. 2013 doi: 10.1021/ol400820y. [DOI] [PubMed] [Google Scholar]
- 41.Yoo B, Kirshenbaum K. Current opinion in chemical biology. 2008;12:714. doi: 10.1016/j.cbpa.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 42.Stringer JR, Crapster JA, Guzei IA, Blackwell HE. J Am Chem Soc. 2011 doi: 10.1021/ja204755p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Astle JM, Simpson LS, Huang Y, Reddy MM, Wilson R, Connell S, Wilson J, Kodadek T. Chem & Biol. 2010;17:38. doi: 10.1016/j.chembiol.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lian W, Upadhyaya P, Rhodes CA, Liu Y, Pei D. J Am Chem Soc. 2013;135:11990. doi: 10.1021/ja405106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doran TM, Gao Y, Mendes K, Dean S, Simanski S, Kodadek T. ACS Comb Sci. 2014;16:259. doi: 10.1021/co500030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 47.Hintersteiner M, Kimmerlin T, Kalthoff F, Stoeckli M, Garavel G, Seifert JM, Meisner NC, Uhl V, Buehler C, Weidemann T, Auer M. Chemistry & Biology. 2009;16:724. doi: 10.1016/j.chembiol.2009.06.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.