

Summary
Background
Recurrent malignant brain tumors (RMBTs) carry a poor prognosis. Dichloroacetate (DCA) activates mitochondrial oxidative metabolism and has shown activity against several human cancers.
Design
We conducted an open-label study of oral DCA in 15 adults with recurrent WHO grade III – IV gliomas or metastases from a primary cancer outside the central nervous system. The primary objective was detection of a dose limiting toxicity for RMBTs at 4 weeks of treatment, defined as any grade 4 or 5 toxicity, or grade 3 toxicity directly attributable to DCA, based on the National Cancer Institute’s Common Toxicity Criteria for Adverse Events, version 4.0. Secondary objectives involved safety, tolerability and hypothesis-generating data on disease status. Dosing was based on haplotype variation in glutathione transferase zeta 1/maleylacetoacetate isomerase (GSTZ1/MAAI), which participates in DCA and tyrosine catabolism.
Results
Eight patients completed at least 1 four week cycle. During this time, no dose-limiting toxicities occurred. No patient withdrew because of lack of tolerance to DCA, although 2 subjects experienced grade 0–1 distal parasthesias that led to elective withdrawal and/or dose-adjustment. All subjects completing at least 1 four week cycle remained clinically stable during this time and remained on DCA for an average of 75.5 days (range 26–312).
Conclusions
Chronic, oral DCA is feasible and well-tolerated in patients with recurrent malignant gliomas and other tumors metastatic to the brain using the dose range established for metabolic diseases. The importance of genetic-based dosing is confirmed and should be incorporated into future trials of chronic DCA administration.
Keywords: Dichloroacetate, Malignant (high grade) glioma, Warburg effect, Pyruvate dehydrogenase complex, Pyruvate dehydrogenase kinase, Phase 1 trial
Introduction
Recurrent malignant brain tumors (RMBTs), whether from primary malignant gliomas, or from metastasis to the central nervous system (CNS), have a poor prognosis [1, 2]. They share an increasing incidence, clinical and radiographic characteristics and paucity of effective therapies. Thus, Phase 1/2 trials of RMBTs for the investigation of potentially promising agents have been considered by the international oncology community to be both efficient and scientifically valid [3, 4].
Despite their diverse histological phenotypes, RMBTs may share a common biochemical pathobiology. Aerobic glycolysis, or the Warburg effect, is a hallmark of tumor metabolism that reflects a disproportionate increase in cytoplasmic glycolysis relative to mitochondrial glucose oxidation, even under conditions of adequate oxygenation [5]. A molecular component of aerobic glycolysis is stable upregulation of hypoxia inducible factor-1alpha (HIF-1α), which results in increased expression of glucose transporters, most enzymes of glycolysis and the mitochondrial enzyme pyruvate dehydrogenase kinase (PDK)[6]. PDK phosphorylates the E1α subunit of the multi-component pyruvate dehydrogenase complex (PDC), inhibiting its catalytic activity and, hence, mitochondrial oxidation of glucose (Fig. 1). Reduced expression in many cancers of the tumor suppressor gene p53 also contributes to the Warburg effect because it normally inhibits PDK, dampening its inhibitory effect on PDC [7].
Fig. 1.

The PDC catalyzes the rate-limiting step in the aerobic oxidation of glucose and pyruvate and of alanine and lactate, which are in equilibrium with pyruvate. PDC also functionally links cytoplasmic glycolysis with the mitochondrial tricarboxylic acid (TCA) cycle and is thus integral to cellular energetics. Panel A – The 9.5 M Da eukaryotic complex is organized into multiple copies of 3 enzymatic components [19, 20]. The heterotetrameric (α2β2) pyruvate dehydrogenase (E1) decarboxylates pyruvate in the presence of thiamine pyrophosphate (TPP). Dihydrolipoamide acetyltransferase (E2) transfers the acetyl group to a lipoic acid moiety that synthesizes up to 60 molecules of acetyl CoA from reduced coenzyme A per macromolecular complex. Reduced lipoate is reoxidzed by dihydrolipoamide dehydrogenase (E3) in a coupled redox reaction in which NADH is generated. The PDC also utilizes an E3 binding protein (E3BP) to tether the E3 component to the complex’s core. The net reaction thus provides glucose-derived acetyl CoA for the tricarboxylic (TCA) cycle and reducing equivalents (NADH) for the respiratory chain or for anabolic reactions, such as lipid synthesis. Four requisite cofactors enable pyruvate oxidation (thiamine; B1) and the synthesis of coenzyme A (pantothenic acid; B5), acetyl CoA (lipoic acid) and NADH (riboflavin; B2 and niacin; B3). The gene for the E1α subunit is located on the X chromosome and all components of the complex are nuclear encoded. Panel B – Rapid regulation of the PDC is mediated primarily by reversible phosphorylation of up to three serine residues on the E1α subunit, rendering the complex inactive. Phosphorylation of E1α is facilitated by a family of four pyruvate dehydrogenase kinase isoforms (PDK 1–4), whereas two pyruvate dehydrogenase phosphatase isoforms (PDP 1 and 2) dephosphorylate, and activate, the PDC. PDKs themselves are activated by a rise in the intramitochondrial ratio of acetyl CoA:CoA and NADH:NAD+, as well as by an increase in cellular energy charge (ATP:ADP). Pyruvate and certain structurally-related halogenated analogs, such as dichloroacetate (DCA), inhibit PDK activity. PDPs are positively regulated by insulin or magnesium ions and PDP1 can be activated by calcium ions. PDK and PDP isoforms are differentially expressed in tissues and PDK isoforms exhibit variable sensitivity to pyruvate and DCA (Adapted from Stacpoole P. Aging Cell 11:371–377, 2012)
Dichloroacetate (DCA) is the prototypic small molecule inhibitor of PDK, an emerging therapeutic target in oncology [8]. By inhibiting PDK, the drug maintains PDC in its active form and facilitates mitochondrial oxidation of glucose-derived pyruvate and lactate via the tricarboxylic acid (TCA) cycle [reviewed in 9]. DCA readily crosses the blood–brain barrier, can be measured in cerebrospinal fluid and activates PDC in brain [9, 10]. DCA has a plasma half-life of ~1 h in drug-naïve individuals, but may increase several-fold with chronic administration [11, 12]. DCA has been used for many years as an investigational drug to treat children with inborn errors of mitochondrial metabolism, including PDC deficiency [13], a disease also characterized by increased aerobic glycolysis [14] and pathological upregulation of HIF-1α [15]. Studies of human glioma and other cancer cells in culture or after transplant into immunocompromised animals show that DCA activates PDC and reverses the Warburg effect in these cells, initiating complex changes in tumor cell metabolism that lead to decreased angiogenesis and increased apoptotic tumor cell death [16, 17].
Recently, Michelakis and coworkers reported an open label trial of oral DCA in five patients with recurrent glioblastoma, with promising results in three subjects in terms of tumor biochemistry and progression [18]. Drug safety was limited initially by peripheral neuropathy that was reversible upon DCA dose reduction. We have shown that DCA-induced neuropathy may be at least partly dependent on its in vivo rate of biotransformation which, in turn, is influenced by both subject age [21] and haplotype variability in glutathione transferase zeta 1 (GSTZ1), which biotransforms DCA to glyoxylate [12]. Thus, there remains uncertainty regarding the chronic safety of DCA in adults with cancer, particularly among individuals in whom variations in GSTZ1 haplotype may prolong the drug’s plasma half-life, thereby increasing risk of toxicity.
Therefore, we undertook an open label, phase 1 trial of chronic, oral DCA in adults with RBMT in whom drug safety and tolerability were assessed in relation to GSTZ1 genotype. GSTZ1 is a bifunctional enzyme that, as maleylacetoacetate isomerase (MAAI), catalyzes the penultimate step in tyrosine metabolism [22] (Fig. 2). Consequently, we also examined the effect of DCA on the accumulation of the tyrosine intermediate maleylacetone, a biomarker of tyrosine metabolism.
Fig. 2.

Bifunctionality of GSTZ1/MAAI. GSTZ1 dehalogenates DCA to the naturally occurring molecule glyoxylate. MAAI isomerizes maleylacetoacetate and maleylacetone, respectively, to fumarylacetoacetate and fumarylacetone
Methods
This study was approved by the University of Florida (UF) Institutional Review Board and the Scientific Advisory Committee of the Clinical Research Center (CRC) at Shands Hospital at UF. An internal Data and Safety Monitoring Board (DSMB) included clinical oncologists experienced in cancer clinical trials of investigational agents. All subjects gave written informed consent to participate and were recruited without selection bias from the oncology clinic of one of the investigators (ED). The study was registered with clinicaltrials.gov (No. NCT 01111097) and was conducted under the auspice of IND #107,893 issued to PWS.
Study objectives
The primary aim of this study was to determine if a dose-limiting toxicity (DLT) occurred in adult patients with RMBTs, within the well-established dose-range used in metabolic diseases, within the first 4 consecutive weeks (cycle 1, formal study endpoint) of oral DCA administration, employing drug doses within the established dose range used in patients with non-malignant metabolic disorders [9, 13]. In addition, patients were allowed to continue DCA until progression, intolerance, toxicity, withdrawal or death. Secondary objectives were to investigate the kinetics of DCA and the effects of GSTZ1/MAAI haplotype variability thereon, evaluate a novel, noninvasive test of DCA in vivo dynamics and mechanism of action and provide hypothesis-generating data as to whether DCA might influence disease progression.
Patient eligibility
Eligibility criteria were similar to those commonly employed in other trials of advanced CNS tumors [23–29]. We included adults aged 21 years or over with histologically proven RMBTs defined as either 1) malignant (WHO III–IV) gliomas progressive after a radiation-containing initial therapy or 2) stage IV parenchymal metastases from systemic malignancies. Any number of prior progressions and therapies were allowed. Patients were required to have radiographic progression determined by contrast-enhanced magnetic resonance imaging (MRI), normal hematologic, renal, and liver function, and must have otherwise recovered from toxicities of prior therapy. Eligibility was based on medical history, review of systems, and an absence of greater than NCI-CTCAEv4.0 grade 3 (or “moderate”) peripheral neuropathy (PN) on exam, except when it was considered attributable to the location of the tumor and its prior therapies. Eligibility criteria also included an interval of at least 4 weeks since prior cytotoxic chemotherapy, a Karnofsky performance status (KPS) of ≥60, an Eastern Cooperative Oncology Group (ECOG) performance status of ≤2, the ability to speak English, and the ability to provide written informed consent. Patients with the potential for pregnancy within themselves or a partner agreed to use reliable birth control. Women of childbearing potential were required to have a negative pregnancy test result and not be breastfeeding. Exclusion criteria included previous medical use of DCA, concurrent anti-cancer therapy and serious concurrent medical conditions.
Study design
The trial was prospective, open-label and single arm (non-randomized). Eligible patients were assigned a DCA dose per kilogram (kg) body weight, as employed in prior DCA clinical trials [27]. Patients self-administered DCA at 8 AM and 8 PM (+/− 1 h). At entry patients were admitted to the CRC for confirmation of eligibility by history, physical examination, repeat of the imaging and blood chemistries described above. Medication diaries were reviewed by the study coordinator.
We employed a “3+3” study design, commonly used in comparable adult and pediatric phase 1 oncology trials [28, 29] (Table 1). This design entails testing 3 subjects at a given dose and escalating to the next dose if no patient has a dose limiting toxicity (DLT). However, if 2 or all of the first 3 subjects had a DLT, the study would be terminated unless the subjects had received the starting dose, in which case a dose reduction would occur. If one of the initial 3 subjects had a DLT, then 3 more subjects would be tested, with dose escalation if none of these subjects had a DLT. However, the study would be terminated if more than 1 of these subjects experienced a DLT. In accordance with the primary objective, the cohort dose levels were set within the clinically established dose range of DCA in metabolic diseases [11, 27]. The initial starting dose was 8.0 mg/kg/12 h, which was increased to 12.5 mg/kg/12 h, or decreased to 5.0 mg/kg/12 h if the first dose was not tolerated, as determined by the DSMB. However, during enrollment, new data on the impact of GSTZ1/MAAI genotype on DCA metabolism [12] resulted in an alteration in the assignment of dose level for this trial. Because individuals who harbor at least one wild type (EGT) haplotype for GSTZ1/MAAI metabolize DCA more rapidly than those who lack this haplotype, the protocol was amended to a priori stratify (i.e., not randomize) patients into one of two cohorts: 1) EGT carriers; and 2) non-EGT carriers. Patients deemed “fast” metabolizers (EGT carriers) continued in the 3+3 design. Patients deemed “slow” metabolizers (non-EGT carriers), were administered 4.0 mg/kg/12 h and were not included in the 3+ 3 design. Thus, the “slow” metabolizers were not included in determining the primary objective, but were followed for analysis of secondary objectives.
Table 1.
Cohorts of three patients treated at each dose level
| Cohort “fast” | Dose | Number of patients experiencing DLT within 4 week | DSMB Determination |
|---|---|---|---|
| 1 (N =3 to 6*) | 8.0 mg/kg/12 h | 0 | 12.5 mg/kg/12 h for next cohort |
| 1 | 3 more patients* will be tested at 8.0 mg/kg/12 h
|
||
| 2 or 3 | 5.0 mg/kg/12 h for the rest of the study | ||
| 2 (N =3) | 12.5 mg/kg/12 h | 0 | 12.5 mg/kg/12 h for the rest of the study |
| 1 | 3 more tested.
|
A DLT was defined as any NCI-CTCAEv4.0 grade 4–5 toxicity, any grade 3 toxicity directly attributable to protocol therapy requiring hospitalization and/or therapy interruption, or any grade 3 toxicity that could not be improved after 3 weeks of medical intervention [29]. An exception was made for patients who required a hospitalization, procedure or other study delay of less than 14 days that was unrelated to the safety or tolerability of DCA (e.g., adjustment of anti-epileptic drug or medical device).
Drug formulation and testing
Clinical grade crystalline sodium DCA (TCI America, Portland, OR) was independently analyzed for purity and homogeneity by gas chromatography–mass spectrometry (GC-MS) [30] and was formulated as a liquid preparation, as previously described [31]. Each lot of product was tested for content and pyrogenicity and was distributed in 400 ml amber colored bottles that were refrigerated at 4 °C. Under these conditions, DCA has a shelf life of at least one year.
Biochemical analyses
Plasma and urine samples were analyzed, in duplicate, by GC-MS for trough (dosing interval) levels of DCA [30] and the tyrosine intermediate maleylacetone (MA), a substrate for MAAI [12].
DNA isolation, genotyping and haplotype analysis
DNA was isolated from blood using Qiagen Gentra Puregene Blood Kits (Qiagen Intl., Alameda, CA). We genotyped samples for three non-synonymous SNPs: G94>A (rs3177427) Glu → Lys at position 32; G124> A (rs7972) Gly → Arg at position 42; and C245> T (rs1046428) Thr → Met at position 82 in the GSTZ1/MAAI gene and for the promoter SNP-1002 G > A (rs7160195) by pyrosequencing [32]. We inferred haplotypes by computational methods using the Bayesian haplotype reconstruction program, PHASE version 2.1 [33]. The five reported haplotypes and their frequencies in the general population are: EGT (~50 %), KGT (~30 %), EGM (~10–20 %), KRT (~5–10 %) and KGM (<5 %)(12).
Pyruvate Breath Test (PBT)
We applied a novel method to noninvasively estimate endogenous PDC activity in vivo. The rationale is based on the unique ability of PDC to irreversibly decarboxylate 1-13C-pyruvate to carbon dioxide, according to the equation:
A proof-of-concept study of the PBT in healthy subjects has been reported [34]. A 25 mg dose of sodium 1-13C-pyruvate (Cambridge Isotopes, Andover, MA) was dissolved in 25 ml tap water and administered to the patient after an overnight fast. The container was rinsed with another 75 ml of water and drunk. Subjects sat quietly with minimal moving for the duration of the test.
To investigate the potential utility of the PBT as a biomark-er for a DCA response in RMBTs, we performed a PBT in four subjects at the start of their first four week therapy cycle. Immediately after the final baseline breath sample was obtained, each subject received a single dose of DCA by mouth and, 60 min later, the PBT was repeated. The DCA dose was either 8 mg/kg or 4 mg/kg and was determined by the GSTZ1/MAAI haplotype, with EGT non-carriers receiving the lower dose. The purpose of this pre-therapy study was to determine each subject’s baseline response to 1-13C-pyruvate administration and the effectiveness of DCA in modifying that response. Thereafter, subjects underwent a repeat PBT at the DCA dosing interval following the completion of each four week therapy cycle.
Breath samples were collected at −10, −5, 10, 20, 30, 40, 50 and 60 min, after which the subject received breakfast. Breath samples were collected using a common straw upon exhaling into a 10 ml screw-cap tube with rubber septum (Exetainers, Labco, Ltd, Houston, TX). Subjects took full breaths and the entire breath was exhaled through the straw into the tube, which was then capped. The amount of 13CO2 in the Exetainer breath storage tubes was measured as atom % 13C with a gas isotope ratio mass spectrometer [35]. Standards of carbon dioxide gas at three different levels of atom % 13C were run before and after each daily run to check instrument performance. The analytical precision of the instrument was 0.0001 atom % 13C. The atom % 13C values of each breath sample were used to calculate the percent of the dose recovered in the breath during each time period. The % 1-13C-pyruvate dose metabolized area under the curve (AUC) for each time period was calculated by the linear trapezoid method [35]. The percent of the dose metabolized at each time point was calculated as:
CO2 production was estimated as 5 mmol/CO2/minutem2 body surface area. The percent dose metabolized/h at each time point was calculated as:
Whole blood lactate concentrations were measured (YSI Instruments, Yellow Springs, OH) after an overnight fast at baseline (before starting DCA) and at the end of each four week therapy cycle to evaluate the PBT results against the known lactate-lowering actions of DCA [36].
Statistical considerations
The sample size was estimated to be sufficient to analyze endpoints. Standard medically-indicated intervention, dose-reduction or delay, adverse event (AE) assessment and response assessment occurred according to established guidelines [24, 37–40]. The primary objective of the study was met when no DLTs occurred, within the dose range well-established in metabolic diseases, within the initial 4 weeks (cycle 1) of DCA administration in the first 8 patients with evaluable data. Based on prior DCA studies, determination of DLT was anticipated to occur within 4 weeks and within 8.0–12.5 mg/kg/12 h. The probability of declaring a dose safe when the probability of DLT is 10 %, 20 %, and 30 % are 91 %, 71 %, and 49 % respectively. The DSMB oversaw the assignment of cohorts and determination of DLT.
Results
Patient outcomes
Fifteen patients were enrolled in this study between 2009 and 2013 (Table 2). One remains on study at the time of manuscript submission. Patient demographics were similar to comparable phase 1 brain tumor trials [3, 25, 26] and included seven females and eight males, with an average age of 52 years at initial diagnosis (range 34–74) and an average age of 57 years at enrollment. Two patients had metastatic disease and 13 had progressive malignant gliomas that had all undergone prior surgery, radiation and chemotherapy (nine WHO IV glioblastomas, two WHO III anaplastic atrocytomas, one WHO III anaplastic oligodendroglioma without co-deletion of 1p/19q). The average number of previous cytotoxic therapies was 2.8. No intra-patient dose escalation occurred; thus, no maximum tolerated dose was sought. All patients remained at their assigned dose level for the remainder of their participation, with the exception of one subject who was allowed to lower his dose to 6.25 mg/kg/12 h from 12.5 mg/kg/12 h because of grade 1 sensory PN manifesting at the sixth therapy cycle. Compliance was excellent among evaluable patients, as indicated by medical records, medication diaries and return of unused medication.
Table 2.
Patient characteristics
| Subject # | Age (yrs) | Sex (M/F) | Diagnosis | Prior Therapy |
|---|---|---|---|---|
| 1 | 75 | F | Papillary serous adenocarcinoma of the uterus | Surgery (3), radiation (2), chemotherapy (3) |
| 2 | 52 | M | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), trial (1) |
| 3 | 45 | F | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), irinotecan/bevacizumab (1) |
| 4 | 45 | M | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), irinotecan/bevacizumab (1) |
| 5 | 60 | M | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), trial |
| 6 | 68 | M | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), |
| 7 | Withdrew the day following consent before starting DCA (Anaplastic Astrocytoma) | TMX/XRT + TMZ (1), alternative alkylator (1), irinotecan/bevacizumab (1) | ||
| 8 | 63 | F | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), |
| 9 | 52 | F | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), |
| 10 | 61 | M | Adenocarcinoma of the lung | Surgery (1), radiosurgery (2), chemotherapy (1) |
| 11 | 41 | M | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), |
| 12 | Tumor unexpectedly progressed before starting DCA | TMX/XRT + TMZ (1), alternative alkylator (1), bevacizumab (1) | ||
| 13 | 67 | M | Anaplastic Astrocytoma | TMX/XRT + TMZ (1), alternative alkylator (1), |
| 14 | Tumor unexpectedly progressed before starting DCA (Anaplastic oligodendroglioma) | TMX/XRT+ TMZ (1), alkylator (1), metronomic temozolomide, bevacizumab (1) | ||
| 15 | 44 | F | Glioblastoma | TMX/XRT + TMZ (1), alternative alkylator (1), Avg. 2.8 |
| ——— | Mean Age: 57 | ——— | ||
Key: TMX/XRT + TMZ: (surgery or biopsy - > temozolomide/radiation - > temozolomide (TMX/XRT + TMZ; 50); alkylator (BCNU or CCNU)
At submission, the median number of days on DCA was 34 days (mean=75.5 days; range: 26–312 days) and one patient remains on study at 330+ days (twelfth cycle). Data on six other subjects were un-evaluable because of the inability to complete four weeks of DCA for reasons unrelated to the trial, including unexpected general medical decline (tooth abscess, VP shunt malfunction, noncompliance, non-progression-related hydrocephalus), unexpected rapid progression, and self-withdrawal (illness of caregiver, elective Hospice). Three of these individuals withdrew before commencing DCA. The remaining four subjects who did receive DCA had an average therapy duration of 4 days (range 0–10 days) and did not report adverse drug-related events.
Eight evaluable patients had clinically and radiographically stable disease at the end of the fourth week on DCA as defined by RANO guidelines [38, 39]. At the date of submission, three patients are alive and five have died. On those alive, one remains on DCA. Two received subsequent therapy and their average survival at the time of submission is 296 days (range 240–330 days). Of those who have died, none received subsequent anti-tumor therapy and remained alive an average of 88 days (range 5–186) after having withdrawn from DCA.
DLTs and other Adverse Events (AEs)
No patient was withdrawn because of toxicity related to DCA. Using the NCI-CTCAEv4.0, no DLTs occurred within the 4 weeks, when using the dose range well-established in metabolic diseases. Table 3 summarizes AEs with at least possible attribution to DCA in the eight evaluable patients and included grade 1–2 episodes of fatigue (5), gait abnormalities (4), hypersomnolence (3) and sensory peripheral neuropathy (2). Each of these has been attributed to DCA in prior studies.
Table 3.
Adverse events possibly attributable to DCA
| Patient | Adverse Event | Grade | Cycle |
|---|---|---|---|
| 1 | fatigue | 1 | 2 |
| 2 | fatigue | 1 | 3 |
| hypersomnolence | 1 | 3 | |
| 3 | fatigue | 1 | 2 |
| hypersomnolence | 2 | 2 | |
| gait abnormalities | 1 | 1 | |
| 4 | None | ||
| 5 | fatigue | 1 | 2 |
| gait abnormalities | 1 | 2 | |
| 6 | fatigue | 1 | 1 |
| hypersomnolence | 1 | 1 | |
| gait abnormalities | 2 | 1 | |
| 7 | sensory peripheral neuropathy | 1 | 2 |
| 8 | gait abnormalities | 1 | 6 |
| sensory peripheral neuropathy | 1 | 6 |
Reversible sensory and motor peripheral neuropathy (PN) has been directly associated with dose and duration of DCA administration in adults [11]. Two evaluable patients in this trial reported non-progressive PN, characterized as parasthesias of the tips of the fingers and toes without muscle weakness or ataxia. Symptoms began at cycle 2 in patient 7, a predictable “slow” metabolizer and cycle 6 in patient 8, a “fast metabolizer” of DCA. Both patients and investigators assigned PN a grade 1; both patients denied adverse changes in their activities of daily living and desired to stay on DCA therapy. No other obvious causes of PN could be ascertained in either subject. Supportive care included physical therapy, vitamin and mineral supplementation, skin care and offers of gabapentin and/or a DCA drug holiday. For unrelated reasons, patient 7 elected to discontinue DCA at the end of the second cycle and reported complete resolution of his neuropathic symptoms within 60 days with the use of gabapentin. Patient 8 managed his PN with supportive care for several months before electing to take a drug holiday of less than one month, during which time he reported minor improvement. Additionally, and in contrast to the patient 8’s own self-assessment, his spouse and some coworkers have noted he exhibits a flatter affect while receiving DCA, without altering his capacity to continue functioning in an intellectually demanding profession. Because this patient elected to remain on study, and was past the DLT determination time-period, the IRB and DSMB allowed a single-patient dose adjustment to allow him to continue DCA at 6.25 mg/kg/12 h, half his original dose. The patient remains on this dose at the time of reporting (cycle12, 312 days same as above).
Asymptomatic, mild, reversible elevation of alanine aminotransferase and/or aspartate aminotransferase is also occasionally associated with chronic DCA administration [11]. Figure 3 illustrates the correlation between serum transaminase levels and DCA trough concentrations in patient 3, a “slow” DCA metabolizer. This 68 year old individual had gait abnormalities and normal serum transaminases at baseline and was begun on DCA (8 mg/kg/12 h). Initial DCA trough levels were undetectable in plasma and urine. After 26 days of therapy, the patient presented with worsening gait abnormalities and DCA was stopped. The patient’s GSTZ1/MAAI genotype was subsequently found to be EGM/EGM, which would predict a very slow rate of DCA plasma clearance [12]. However, at the time, the trial’s study design allowed GSTZ1/MAAI haplotype analysis to be reported only after DCA treatment had commenced. Three days after DCA was discontinued the patient’s plasma trough DCA concentration was 280 μg/ml and became undetectable 30 days after drug withdrawal. Serum aspartate aminotransferase rose from 19 U/L to a maximum of 46 U/L and returned to pre-therapy baseline levels within approximately 45 days after drug withdrawal. The patient’s gait abnormalities were improving until he died of unrelated general medical events 116 days after discontinuing DCA.
Fig. 3.
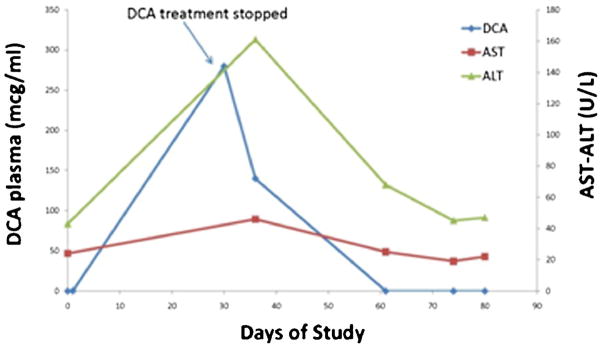
Relationship between plasma trough DCA and serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) concentrations in patient 6, a slow metabolizer of DCA (haplotype: EGM/EGM)
DCA and tyrosine metabolism
Table 4 shows plasma DCA trough and urinary MA concentrations in relation to drug dose and GSTZ1/MAAI genotype in patients who completed the first four week therapy cycle. In this small sample, there were no statistically significant corrections between DCA and MA levels or between the levels of either of these compounds and GSTZ1/MAAI genotype. After the first six patients had entered the study, the effect of GSTZ1/MAAI genotype on DCA kinetic became known [12]. Thus, patient 6, whose genotype would be predicted to confer a slow rate of DCA metabolism, was assigned a higher starting drug dose than were subsequent patients who lacked an EGT allele. Nevertheless, measurement of plasma trough DCA and urinary MA over time revealed no appreciable accumulation of either molecule. Mean and median plasma DCA levels exceeded the Ki of DCA of ~25 μg/ml (~0.2 mmol/l) for the PDK 2, the most ubiquitously expressed PDK isoform and the most sensitive to DCA inhibition [40].
Table 4.
Plasma trough DCA and Urinary MA levels at 30 Days
| Patient | Genotype* | DCA dose (mg/kg/12 h) | Plasma DCA (μg/ml) | Urinary MA (mg/g C) |
|---|---|---|---|---|
| 1 | EGT/EGT* | 8 | 55 | 4.3 |
| 2 | EGT/KRT* | 8 | 2.0 | <0.1 |
| 6 | EGM/EGM | 8 | 280 | 20.1 |
| 8 | KGT/KGT | 4 | 9.7 | <0.1 |
| 9 | EGT/EGT* | 12.5 | 113 | 11.8 |
| 10 | KGT/KGT | 12.5 | 85 | 45 |
| 11 | EGM/KGT | 4 | 64 | 11.1 |
| 13 | EGT/KRT* | 12.5 | 48 | 16.6 |
Fast metabolizers
Patient 13 Plasma DCA trough levels ranged from 44–65 μg/ml for cycles 1–3 (dose 12.5 mg/kg/12 h) and from 11.2–11.5 mcg/ml for cycles 5 and 6 (dose 6.25 mg/kg/12 h). He received no DCA during cycle 4 (DCA trough level <0.1 μg/ml), as described in the text
Pyruvate breath test
During the latter part of this trial the PBT became available and was applied to four patients (patients 10, 11, 13 and 15 of Table 5). These subjects underwent at least a baseline±DCA PBT in the overnight fasted state, together with measurement of blood lactate before receiving DCA. Figure 4 illustrates 13CO2 production in the pre-DCA and post-DCA state in patient 10 as a percent of the 1-13C-pyruvate dose administered per unit time and as the cumulative percent 13CO2, which reflects the area under the 13CO2 concentration-time curve. Peak 13CO2 production pre-DCA occurred 40 min after 1-13C-pyruvate administration. However, in patient 10 the peak 13CO2 production post-DCA occurred at 30 min with higher intensity. The cumulative percent 13CO2 production of the administered dose after 60 min was 28.70 % post-DCA and 22.26 % pre-DCA. Similar results were observed with patient 15. However, PBTs performed in patients 11 and 13 revealed that 13CO2 production was decreased after DCA. The time to peak 13CO2 was delayed with DCA and the cumulative percent 13CO2 was less (Fig. 5). Despite the inter-subject variability of the PBT, DCA administration was associated with a significant lactate-lowering effect (baseline lactate 1.1±0.45 mmol/l vs. DCA lactate 0.54 ±0.27 mmol/l; p<0.001) in these four subjects.
Table 5.
Plasma DCA trough and urinary maleylacetone concentrations
| Value | Plasma DCA (μg/ml) | Urinary MA (mg/g C) |
|---|---|---|
| Mean | 62.9 | 9.5 |
| Median | 48.0 | 4.3 |
| Stdev | 65.5 | 10.0 |
| Cmin | 2.9 | 1 |
| Cmax | 281 | 45 |
| N | 21 | 21 |
Samples were obtained at the dosing interval in the eight patients who completed at least one DCA treatment cycle and were analyzed as described in the Methods. MA concentrations expressed as mg/g creatinine. Cmin and Cmax denote the minimum or maximum plasma or urinary concentration, respectively, measured for each analyte. N denotes the number of samples
Fig. 4.

Pre- and post-DCA (baseline) Pyruvate Breath Test in patient 10, a 61 year old man with GSTZ1/MAAI genotype KGT/KGT
Fig. 5.

Pre- and post-DCA (baseline) Pyruvate Breath Tests in patient 11, a 41 year old man with a GSTZ1/MAAI genotype EGM/KGT
Discussion
These data indicate that oral DCA, administered within the dose range well-established in metabolic disease, is safe, well-tolerated and feasible for use in adults with RMBTs. DCA’s safety and tolerance are supported by protocol compliance of the eight evaluable patients and the average duration of survival of the five patients who died, with a mean survival duration of 140 days (range 75–146) from the start of DCA and 88 days (range 30–186) from withdrawal of DCA.
Nevertheless, safety is an important concern with DCA, based on toxicological findings in rodents (9), and a randomized controlled trial of the drug in adults with a primary mitochondrial disease due to mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) that was stopped prematurely because of new onset or exacerbation of peripheral neuropathy [41]. However, other studies have demonstrated excellent long-term safety in children and adolescents administered DCA for several years for therapy of primary mitochondrial diseases [42, 43]. Moreover, the earlier trials in adults with MELAS and the more recent trial in GBM patients in which neuropathy was a limiting AE did not account for the effect of GSTZ1/MAAI haplotype on drug kinetics and biotransformation and on the perturbation of the phenylalanine/tyrosine catabolic pathway in individuals who metabolize DCA slowly [12].
Although this phase I trial cannot provide definitive information on efficacy, it is noteworthy that DCA therapy was associated with clinical and radiographic evidence of disease stabilization through the first 4 weeks of DCA administration in all 8 evaluable patients whose average number of prior treatments was 2.8. This is at least non-inferior, if not possibly favorable, compared to other advanced cancer phase 1 trials [3, 4, 25, 26] and is consistent with early data in glioblastoma patients [18] and with experimental evidence for a metabolic modulation and selective killing of CNS cancers [17, 44]. In this regard, it is interesting that perturbation of the PDK-PDC axis by DCA is also the basis for the drug’s therapeutic effect in patients with congenital PDC deficiency [13] and in experimental and clinical models of myocardial ischemia and failure [45], pulmonary arterial hypertension [46] and certain other pathological states [47] in which the efficient mitochondrial conversion of substrate fuels to energy is perturbed. All of these conditions are associated with genetic or acquired suppression of PDC activity, leading to accelerated glycolysis and lactate production and decreased mitochondrial glucose oxidation.
Animal and clinical experiments indicate that a single oral dose of DCA has excellent bioavailability and can induce a lactate-lowering effect within 30 min of administration [37], a finding temporally associated in rats with activation of hepatic PDC [9]. To gain insight into the effect of DCA on endogenous PDC activity in humans, we developed and applied here a novel breath test that quantitates 13CO2 production from the PDC-catalyzed decarboxylation of orally administered 1-13C-pyruvate. We applied this method to some of the participants of this trial at enrollment and, subsequently, after the initial DCA dosing. We found that the PBT was easy to administer and well-tolerated by all subjects. In two patients (10 and 15), a single dose of DCA had an appreciable effect on the rate or magnitude of 13CO2 production. In two other patients (11 and 13), the opposite effect of DCA was observed, suggesting inhibition of pyruvate oxidation, despite evidence consistent with a blood lactate-lowering effect of the drug. Thus, there were notable inter-patient differences in the pre-therapy kinetics of 13CO2 as well as intra-subject variability during drug therapy. It is likely that the PBT measures mainly hepatic PDC activity, owing to the small amount of pyruvate administered and the first-pass effect by the liver, which has a high content of PDC. It is possible that PDC activity in the livers of some patients was suppressed with initial DCA treatment, as might be expected to occur in their tumor cells, but responded to chronic exposure to DCA with a return to or towards baseline 13CO2 production (data not shown). Further research will be required to confirm and extend our preliminary findings and determine the ultimate utility of the PBT as a useful index of endogenous PDC activity and as a biomarker of DCA dynamics.
Conclusion
DCA appears to be safe, tolerable and feasible for chronic administration in adults with RMBTs, when it is administered within the dose range commonly used in the chronic treatment of childhood metabolic diseases. Personalized dosing of patients, based on GSTZ1/MAAI genotype, is likely to enhance chronic safety and tolerability of the drug without diminishing efficacy. Specifically, patients who lack an EGT allele should probably be treated with a starting oral dose of 5 mg/kg/12 h that can be increased, based on absence of peripheral neuropathy, whereas subjects who possess at least one EGT allele should tolerate doses at least 6.25 mg/kg/12 h. Despite decades of clinical use, DCA exposure so far has not been associated with adverse interactions with other drugs, which is relevant to its consideration as combination therapy for cancer [e.g., 10, 47–52]. DCA’s unique site and mechanism of action interdicts several fundamental processes important to tumor survival and growth [9, 14, 44, 48–52]. Furthermore, recent animal and human studies supports its activity within the CNS parenchyma and anti-tumor effects in tumors occurring in the brain. Finally, the PBT holds promise as a noninvasive serial measurement of DCA’s in vivo pharmacodynamics. Further clinical trials of DCA in adult cancers occurring in the brain cancer are warranted.
Acknowledgments
We are grateful to the DSMB members for their dedication to this trial and to Ms. Candace Caputo for editorial assistance.
Funding This study was funded by Reliable Cancer Therapies, Brussels, Belgium, the Ocala Royal Dames Foundation, Ocala, FL, the Preston A. Wells, Jr., Center for Brain Tumor Therapy and a National Institutes of Health Clinical and Translational Science Award UL1 TR000064.
Footnotes
Conflict of interest disclosures PWS holds investigator INDs for DCA. DAW is President, Metabolic Solutions, Inc.
Authors’ contributions EMD, JJS and PWS developed the study design; EMD and BSC screened and enrolled subjects; EMD, PWS and BSC evaluated and treated patients; TL performed and interpreted the genotyping; ALS conducted mass spectrometric analyses; EMD, BSC and PWS analyzed routine clinical and imaging data; JRF and EMD analyzed exploratory imaging data; JJS reviewed interim safety data, PWS and DAW designed the breath test procedures; BSC conducted the pyruvate breath test; DAW analyzed 13CO2 breath samples; EMD, PWS and DAW wrote the manuscript.
Contributor Information
E. M. Dunbar, Department of Neurosurgery, JHMHC, College of Medicine, University of Florida, PO Box 100265, Gainesville, FL 32610-0265, USA
B. S. Coats, Department of Medicine, JHMHC, College of Medicine, University of Florida, PO Box 100226, Gainesville, FL 32610-0226, USA
A. L. Shroads, Department of Medicine, JHMHC, College of Medicine, University of Florida, PO Box 100226, Gainesville, FL 32610-0226, USA
T. Langaee, Department of Pharmacotherapy and Translational Research, Center for Pharmacogenomics, JHMHC, College of Pharmacy, University of Florida, PO Box 100486, Gainesville, FL 32610-0486, USA
A. Lew, Department of Medicine, JHMHC, College of Medicine, University of Florida, PO Box 100226, Gainesville, FL 32610-0226, USA
J. R. Forder, Department of Radiology, JHMHC, College of Medicine, University of Florida, PO Box 100374, Gainesville, FL 32610-0374, USA
J. J. Shuster, Department of Health Outcomes and Policy, JHMHC, College of Medicine, University of Florida, PO Box 100177, Gainesville, FL 32610-0177, USA
D. A. Wagner, Metabolic Solutions, Inc., 460 Amherst Street, Nashua, NH 03063, USA
P. W. Stacpoole, Email: pws@ufl.edu, Department of Medicine, JHMHC, College of Medicine, University of Florida, PO Box 100226, Gainesville, FL 32610-0226, USA. Department of Biochemistry and Molecular Biology, JHMHC, College of Medicine, University of Florida, PO Box 100245, Gainesville, FL 32610-0245, USA
References
- 1.Yung WK, Albright RE, Olson J, Fredericks R, Fink K, Prados MD, Brada M, Spence A, Hohl RJ, Shapiro W, Glantz M, Greenberg H, Selker RG, Vick NA, Rampling R, Friedman H, Phillips P, Bruner J, Yue N, Osoba D, Zaknoen S, Levin VA. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83(5):588–593. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villà S, Weber DC, Moretones C, Mañes A, Combescure C, Jové J, Puyalto P, Cuadras P, Bruna J, Verger E, Balañà C, Graus F. Validation of the new Graded Prognostic Assessment scale for brain metastases: a multicenter prospective study. Radiat Oncol. 2011;6:23. doi: 10.1186/1748-717X-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furman W. Everolimus for Treating Pediatric Patients with Recurrent or Refractory Tumors. 2005 Available from http://clinicaltrials.gov/ct2/show/NCT00187174?term=recurrent+tumors+AND+brain&rank=12.
- 4.Caffo M, Barresi V, Caruso G, Cutugno M, La Fata G, Venza M, Alafaci C, Tomasello F. Innovative therapeutic strategies in the treatment of brain metastases. Int J Mol Sci. 2013;14(1):2135–2174. doi: 10.3390/ijms14012135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65(24):3981–3999. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72(2):560–567. doi: 10.1158/0008-5472.CAN-11-1215. [DOI] [PubMed] [Google Scholar]
- 8.Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 2013;3:38. doi: 10.3389/fonc.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38(11):1124–1144. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- 10.Park JM, Recht LD, Josan S, Merchant M, Jang T, Yen YF, Hurd RE, Spielman DM, Mayer D. Metabolic response of glioma to dichloroacetate measured in vivo by hyperpolarized (13)C magnetic resonance spectroscopic imaging. Neuro Oncol. 2013;15(4):433–441. doi: 10.1093/neuonc/nos319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stacpoole PW. The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine–both or neither? Environ Health Perspect. 2011;119(2):155–158. doi: 10.1289/ehp.1002554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shroads AL, Langaee T, Coats BS, Kurtz TL, Bullock JR, Weithorn D, Gong Y, Wagner DA, Ostrov DA, Johnson JA, Stacpoole PW. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J Clin Pharmacol. 2012;52(6):837–849. doi: 10.1177/0091270011405664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berendzen K, Theriaque DW, Shuster J, Stacpoole PW. Therapeutic potential of dichloroacetate for pyruvate dehydrogenase complex deficiency. Mitochondrion. 2006;6(3):126–135. doi: 10.1016/j.mito.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Simpson NE, Han Z, Berendzen KM, Sweeney CA, Oca-Cossio JA, Constantinidis I, Stacpoole PW. Magnetic resonance spectroscopic investigation of mitochondrial fuel metabolism and energetics in cultured human fibroblasts: effects of pyruvate dehydrogenase complex deficiency and dichloroacetate. Mol Genet Metab. 2006;89(1–2):97–105. doi: 10.1016/j.ymgme.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Glushakova LG, Judge S, Cruz A, Pourang D, Mathews CE, Stacpoole PW. Increased superoxide accumulation in pyruvate dehydrogenase complex deficient fibroblasts. Mol Genet Metab. 2011;104(3):255–260. doi: 10.1016/j.ymgme.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 17.Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS, Michelakis ED. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2013;32(13):1638–1650. doi: 10.1038/onc.2012.198. [DOI] [PubMed] [Google Scholar]
- 18.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31):31–34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 19.Zhou ZH, McCarthy DB, O’Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA. 2001;98(26):14802–14807. doi: 10.1073/pnas.011597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smolle M, Prior AE, Brown AE, Cooper A, Byron O, Lindsay JG. A new level of architectural complexity in the human pyruvate dehydrogenase complex. J Biol Chem. 2006;281(28):19772–19780. doi: 10.1074/jbc.M601140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J Pharmacol Exp Ther. 2008;324(3):1163–1171. doi: 10.1124/jpet.107.134593.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchell G, Lambert M, Tanguay R. Hypertyrosinemia. In: Schriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. McGraw-Hill, Inc., Health Professions Division; New York: 1995. pp. 1077–106. [Google Scholar]
- 23.Karnofsky D, editor. The clinical evaluation of chemotherapeutic agents in cancer. Columbia University Press; New York: 1949. [Google Scholar]
- 24.Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, Carbone PP. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5(6):649–655. [PubMed] [Google Scholar]
- 25.Nabors LB, Supko JG, Rosenfeld M, Chamberlain M, Phuphanich S, Batchelor T, Desideri S, Ye X, Wright J, Gujar S, Grossman SA. New Approaches to Brain Tumor Therapy (NABTT) CNS Consortium (2011) Phase I trial of sorafenib in patients with recurrent or progressive malignant glioma. Neuro Oncol. 13(12):1324–1330. doi: 10.1093/neuonc/nor145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grossman SA, Ye X, Peereboom D, Rosenfeld MR, Mikkelsen T, Supko JG, Desideri S Adult Brain Tumor Consortium. Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro Oncol. 2012;14(4):511–517. doi: 10.1093/neuonc/nor230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117(5):1519–1531. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- 28.Kreisl TN, Kim L, Moore K, Duic P, Kotliarova S, Walling J, Musib L, Thornton D, Albert PS, Fine HA. A phase I trial of enzastaurin in patients with recurrent gliomas. Clin Cancer Res. 2009;15(10):3617–3623. doi: 10.1158/1078-0432.CCR-08-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubicek GJ, Werner-Wasik M, Machtay M, Mallon G, Myers T, Ramirez M, Andrews D, Curran WJ, Jr, Dicker AP. Phase I trial using proteasome inhibitor bortezomib and concurrent temozolomide and radiotherapy for central nervous system malignancies. Int J Radiat Oncol Biol Phys. 2009;74(2):433–439. doi: 10.1016/j.ijrobp.2008.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan Z, Henderson GN, James MO, Stacpoole PW. Determination of dichloroacetate and its metabolites in human plasma by gas chromatography–mass spectrometry. J Chromatogr B Biomed Sci Appl. 1997;703(1–2):75–84. doi: 10.1016/s0378-4347(97)00404-0. [DOI] [PubMed] [Google Scholar]
- 31.Henderson GN, Whalen PO, Darr RA, Curry SH, Derendorf H, Baumgartner TG, Stacpoole PW. Development of an oral drug formulation for dichloroacetate and thiamine. Drug Dev Indust Pharm. 1994;20(15):2425–2437. [Google Scholar]
- 32.Langaee T, Ronaghi M. Genetic variation analyses by pyrosequencing. Mutat Res. 2005;673:96–102. doi: 10.1016/j.mrfmmm.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 33.Stephens M, Smith NK, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stacpoole PW, Wagner DW. Rapid breath test for in vivo determination of human pyruvate dehydrogenase complex activity. UMDF Annual Meeting; Bethesda, MD. June 12–16.2012. [Google Scholar]
- 35.Wagner DA, Schatz R, Coston R, Curington C, Bolt D, Toskes PP. A new 13C breath test to detect vitamin B12 deficiency: a prevalent and poorly diagnosed health problem. J Breath Res. 2011;5(4):046001. doi: 10.1088/1752-7155/5/4/046001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003;43(7):683–691. [PubMed] [Google Scholar]
- 37.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO European Organization for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group . Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 38.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 39.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB, Tsien C, Mikkelsen T, Wong ET, Chamberlain MC, Stupp R, Lamborn KR, Vogelbaum MA, van den Bent MJ, Chang SM. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28(11):1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 40.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329(Pt 1):191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66(3):324–330. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- 42.Abdelmalak M, Lew A, Ramezani R, Shroads AL, Coats BS, Langaee T, Shankar MN, Neiberger RE, Subramony SH, Stacpoole PW. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab. 2013;109:139–143. doi: 10.1016/j.ymgme.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121(5):e1223–e1228. doi: 10.1542/peds.2007-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010;127(11):2510–2519. doi: 10.1002/ijc.25499. [DOI] [PubMed] [Google Scholar]
- 45.Bersin RM, Stacpoole PW. Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am Heart J. 1997;134(5 Pt 1):841–855. doi: 10.1016/s0002-8703(97)80007-5. [DOI] [PubMed] [Google Scholar]
- 46.Piao L, Marsboom G, Archer SL. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J Mol Med (Berl) 2010;88(10):1011–1020. doi: 10.1007/s00109-010-0679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stacpoole PW. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell. 2012;11(3):371–377. doi: 10.1111/j.1474-9726.2012.00805.x. [DOI] [PubMed] [Google Scholar]
- 48.Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, Liu SH, Cheng AL. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108(1):72–81. doi: 10.1038/bjc.2012.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujiwara S, Kawano Y, Yuki H, Okuno Y, Nosaka K, Mitsuya H, Hata H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer. 2013;108(1):170–178. doi: 10.1038/bjc.2012.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N. Dichloroacetate improves immune dysfunction cuased by tumor-secreted lactic acid and increases anti-tumor immunoreactivity. Intr J Cancer. 2013 doi: 10.1002/ijc.28114. [DOI] [PubMed] [Google Scholar]
- 51.Sanchez WY, McGee SL, Connor T, Mottram B, Wilkinson A, Whitehead JP, Vuckovic S, Catley L. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer. 2013;108(8):1624–1633. doi: 10.1038/bjc.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu CA, Chao Y, Shiah SG, Lin WW. Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim Biophys Acta. 2013;1833(5):1147–1156. doi: 10.1016/j.bbamcr.2013.01.025. [DOI] [PubMed] [Google Scholar]