

Abstract
Cell death plays an important role in the regulation of inflammation and may be the result of inflammation. The maintenance of tissue homeostasis necessitates both the recognition and removal of invading microbial pathogens as well as the clearance of dying cells. In the past few decades, emerging knowledge on cell death and inflammation has enriched our molecular understanding of the signaling pathways that mediate various programs of cell death and multiple types of inflammatory responses. This review provides an overview of the major types of cell death related to inflammation. Modification of cell death pathways is likely to be a logical therapeutic target for inflammatory diseases.
Keywords: Inflammation, Necroptosis, Apoptosis, Pyroptosis, Pyronecrosis, NETosis, Autophagy
Introduction
One of the most important factors in the development and homeostasis of organisms is the balance between cell survival and cell death. Early in 1960, apoptosis was considered the only standard programmed cell death form [1, 2], whereas necrosis was mostly considered an ‘accidental’ cell death that occurred in response to physical and chemical insults. Following the progression in cell death research, a tight link was demonstrated between molecularly defined cell death and inflammation. In host defense, programmed cell death can act in a protective manner; the death of infected cells may reduce microbial infections, separate uninfected neighboring cells, and alert the host through danger signals and inflammatory mediators. This review depicts intimate interconnections between cell death and inflammation and the pivotal protein in each special mechanistic module that executes the process of cell death and inflammation.
Review
Necrosis, necroptosis, and inflammation
Traditionally, necrosis is considered the primary form of cell death caused by inflammation. Necrosis was historically viewed as an accidental subroutine, largely resulting from very harsh physicochemical stimuli, including abrupt changes in temperature, osmotic pressure, or pH. Necrosis is morphologically identified by the swelling of organelles, increased cell volume, disruption of the plasma membrane, and loss of intracellular content. Necrosis is recognized as a cause of inflammation; the release of intracellular materials, which are termed as damage-associated molecular patterns (DAMPs), can trigger inflammatory reactions. DAMPs are the key to the pathogenesis of sterile inflammation, including gout, atherosclerosis, ischemia-reperfusion, and Alzheimer’s disease. For example, the DAMP molecule high-mobility group box 1 (HMGB1) can be released from necrotic cells and, in turn, stimulates neighboring cells via the receptor for advanced-glycation end-products (RAGE) to express proinflammatory cytokines, chemokines, and adhesion molecules, therefore inducing inflammation [3]. Recent studies using genetic approaches [4–7] and chemical inhibitors of necrosis [4, 8, 9] demonstrated the existence of multiple pathways of regulated necrosis.
Among the pathways of regulated necrosis, necroptosis is currently most frequently mentioned and investigated. Generally, necroptosis is defined as cell death mediated through a pathway that depends on the receptor-interacting protein kinase (RIP)1-RIP3 complex and that can be inhibited by Necrostatin-1 (Nec-1) [10] (Fig. 1). Necroptosis is induced by a class of death receptors that includes tumor necrosis factor receptor (TNFR)1, TNFR2, and Fas. Of these, the TNF-α/TNFR-induced pathway is the most widely studied. Binding of TNF-α to the extracellular portion of TNFR1 causes allosteric changes in the intracellular portion of TNFR1 followed by the release of the silencer of death domains (SODD) from the intracellular domain of TNFR1 [11]. TNFR1 and TNFR2 form complex I containing a death domain (e.g., TNF-α receptor-associated death domain (TRADD)), RIP1, Fas-associated death domain (FADD), and several E3 ubiquitin ligases, such as TNF-α receptor associated factor 2/5 (TRAF2/5) and inhibitor of apoptosis proteins (IAPs) cIAP1 and cIAP2 [12]. RIP1 is initially recruited to complex I and is polyubiquitinated by TRAF2/5, cIAP1, and cIAP2 [13, 14]. Because RIP1 exhibits a biphasic effect based on its ubiquitination state, complex I is situated at the crossroads of cell survival and death. Deubiquitination of RIP1 can inhibit the NF-κB pathway, which promotes cell death pathways. Whether TRADD is required for necroptosis potentially depends upon the type of stimulus. TNFR1 activation together with the absence of c-IAPs (IAP antagonist treatment), translation inhibition (cyclohexamide treatment), or RIP1 deubiquitination by the deubiquitinating enzyme (DUB) CYLD may promote the translocation of RIP1 to a secondary cytoplasmatic complex, Complex II [15–17]. Complex II is formed by the death domain containing protein FADD, caspase-8 and cFLIP. Complex II may activate either apoptotic or necroptotic downstream signaling pathways. Activation of caspase-8 drives complex II into a pro-apoptosis state by cleaving RIP1 and RIP3. However, when the apoptosis pathway is inhibited, a complex named the “necrosome” is formed (Fig. 1). The necrosome is primarily composed of RIP1 and RIP3 and distinctly enhances necroptosis [18].
Fig. 1.
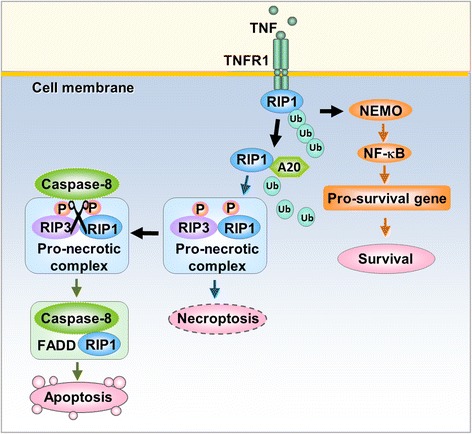
Model of TNF receptor signaling regulation of cell fate. Upon the binding of TNF to its receptor TNFR1, RIP1 is recruited to TNFR1 and is subsequently ubiquitinated. The polyubiquitinated RIP1, in turn, binds to NEMO, the regulatory subunit of NF-kB, to promote NF-κB activation, which leads to the induction of pro-survival genes to counter the death signals. Cell survival is a result of this pathway. The polyubiquitinated RIP1 can also migrate to the cytoplasm, where RIP1 is de-ubiquitinated by A20, the de-ubiquitylating enzyme. RIP1 and RIP3 can then form a pro-necrotic complex followed by phosphorylation on both kinases and induction of necroptosis. In circumstances in which caspase-8 is activated, RIP1 and RIP3 can be cleaved by caspase-8, and the pro-necrotic complex is blunted, which stimulates the cell to undergo apoptosis
The pseudokinase MLKL is a substrate of RIP3 required for necroptosis [4, 19]. Unlike its previous discovered function in regulating mitochondrial fission, MLKL recruitment and phosphorylation caused by RHIM-dependent oligomerization and intramolecular RIP3 autophosphorylation [20, 21] results in an activated state able to induce necroptosis [22]. Furthermore, several studies have deciphered a role for MLKL in necroptosis. MLKL oligomerization induced by RIP3 and plasma membrane localization is associated with its cytotoxicity [23–26]. MLKL binds to phosphatidylinositol phosphates (PIPs) [23, 25] and subsequently modifies sodium or calcium influx through ion channels, thereby increasing osmotic pressure and promoting plasma membrane rupture [24, 26, 27].
The mechanism by which the necrosome causes cell death remains unclear. Necroptosis shares some identical sub-cellular events with necrosis, such as oxidative burst, mitochondrial membrane hyperpolarization, lysosomal membrane permeabilization, and plasma membrane permeabilization. However, the mechanisms underlying those processes might be different [28]. Reactive oxygen species (ROS) potentially lead to cell death by directly oxidizing or triggering various downstream pathways in the mitochondria [29–31]. RIP3 accelerates mitochondrial ROS production and mitochondrial metabolism through the activation of a series of metabolism-related enzymes, including NADPH and JNK [32, 33]. Mitochondria also contribute to necrotic cell death through an ADP/ATP-related pathway in addition to ROS production. Adenine nucleotide translocase (ANT), an ADP/ATP carrier located in the inner mitochondrial membrane, is required for the synthesis of ATP in the mitochondria. RIP1-dependent inhibition of ANT is reportedly involved in the programmed necrosis induced by TNF-α and zVAD-fmk, whereas the later potentially blocks the ability of ANT to transport cytoplasmic ADP and thereby induces massive ATP depletion in mitochondria. The activity of ANT is potentially affected by interactions with VDAC and cyclophilin D (CYPD). Two other potential executional proteins are cPLA2 and lipoxygenase (LOXs). cPLA2 plays an important role in TNF-α-induced necrotic cell death in L929 cells and MEFs [34]. LOXs acts as a downstream effector of PLA2 and leads to the disruption of organelle and plasma membranes [35]. LOXs is reportedly involved in both apoptosis and necrosis induced by TNF-α, although the exact mechanism has yet to be defined [36, 37].
Necroptosis is able to trigger inflammation. This effect has been observed in a study using mice with deletion of FADD [38] or Casp8 [39] in intestinal epithelial cells (IECs) in which RIP3-dependent cell death caused intestinal inflammation. RIP3-mediated necroptosis may play a role in the pathogenesis of Crohn’s disease, as evidenced by the high RIP3 expression in Paneth cells of these patients [39]. Necroptosis has been found to stimulate the immune system to elicit inflammatory responses and has also been characterized in animal models of acute pancreatitis, ischemic injury, and neurodegeneration [9, 40–42]. RIP3−/− mice are protected from systemic inflammation caused by TNF stimulation and experimental sepsis induced by cecal ligation and puncture (CLP) [43, 44]. RIP1 and RIP3 also play crucial roles in the pathogenesis of Salmonella enterica serovar and S. typhimurium infection [45]. Necrotic macrophages have been observed in atherosclerosis lesions from both human patients and animals [46]. RIP3-dependent necroptosis is a key driver for inflammation in atherosclerosis; RIP3 deficiency alleviates macrophage necrosis in advanced atherosclerosis lesions in atherosclerosis-prone LDL-R−/− or ApoE−/− mice [47]. The contribution of RIP1-dependent necroptosis to multiple organ failure has also been observed in models of ischemia reperfusion (IR) and can be rescued by Nec-1 inhibitor [48–50]. In addition, necroptosis has been shown to contribute to neuronal damage in neonatal brain injury [51].
Taken together, necrosis and necroptosis are endogenous triggers of inflammation that influence host disease outcomes. Determining the relative contribution of necroptosis-dependent and -independent pathways in inflammation may lead to new and more specific therapeutic targets.
Apoptosis and inflammation
Apoptosis is one of the major types of cell death and has been well defined for many years. Two independent apoptotic signaling cascades, the extrinsic and intrinsic pathways, have been distinguished [52]. The extrinsic pathway is triggered by binding of Fas plasma membrane death receptor to Fas ligand (Fas-L) and other similar receptors, such as TNFR 1 and its relatives [53]. Fas-L combines with Fas to form a death complex. The Fas/Fas-L composite recruits death domain-containing protein (FADD) and pro-caspase-8, aggregating to become the death-inducing signaling complex (DISC). Consequently, the protein complex activates pro-caspase-8, which proceeds to trigger pro-caspase-3, the penultimate enzyme for the execution of the apoptotic process [54]. The intrinsic pathway also leads to apoptosis but under the control of mitochondrial pro-enzymes. When a cell is stimulated by either extracellular stimuli or intracellular signals, the outer mitochondrial membranes become permeable to internal cytochrome c, which is then released into the cytosol. Cytochrome c associates with the adaptor protein Apaf-1 to form the apoptosome, which triggers downstream caspase 9 [55]. Once activated, caspases-8, −9, and −10 process the executioner caspases-3 and −7. Mature caspases-3 and −7 cleave a large set of substrates, ultimately resulting in the characteristic morphological and biochemical hallmarks of apoptosis, such as phosphatidylserine exposure, nuclear condensation, membrane blebbing, and genomic DNA fragmentation.
Many factors and signaling pathways that are activated by inflammation are involved in the regulation of cell apoptosis. Absent in melanoma 2 (AIM2), a member of the pattern recognition receptors (PRRs) in the cytoplasm, has been found to activate caspase-3 in parallel with caspase-1 [56]. AIM2 can recognize DNA released by the cytosolic bacteria [57], whereas NLRP3, another member of the cytoplasmic PRRs, responds to the bacterial pore-forming toxin nigericin [58], both of which elicit apoptotic caspase activation [59, 60]. Apoptotic responses can be observed in wild type cells responding to AIM2 or NLRP3 stimuli [58]. AIM2 and NLRP3 inflammasome-dependent apoptosis requires caspase-8, which is recruited to the inflammasome through interaction between its DED domains and the PYD of apoptosis-associated speck-like protein containing a caspase activation and recruitment domains (CARD), an adaptor molecule of the inflammasome [57, 58, 61]. In contrast, BCL-2 can negatively regulate NLRP3 inflammasome activation by preventing the cytosolic release of mitochondrial DNA [62].
Apoptotic cells can expose “eat me” signals, which are either newly expressed molecules or existing molecules modified by oxidation, to initiate phagocytosis of the apoptotic cells [63]. The process of phagocytosis of apoptotic cells represents an anti-inflammatory mechanism. Phosphatidyl serine (PS) localized to the outer leaflet of the plasma membrane is the predominant “eat me” molecule upon apoptosis [63, 64]. Specific molecules such as milk fat globule epidermal growth factor 8 (MFG-E8) links PS to phagocyte avb3 integrin [63], whereas growth-arrest-specific 6 (GAS6) links PS to the receptor tyrosine kinase MER [63]. PS acts as a ligand for the T-cell immunoglobulin domain and mucin domain (TIM)-4 molecule on macrophages and dendritic cells (DC) [65], and TIM-4 helps promote the uptake of apoptotic cells [66]. Two other molecules, brain-specific angiogenesis inhibitor 1 (BAI1) and stabilin-2, have also been shown to mediate uptake of apoptotic cells via recognition of PS [67, 68].
Apoptotic cells are rarely detected under physiological conditions, but the presence of uncleared apoptotic cells has been linked to several different diseases, including infection and inflammation. PAMPs and DAMPs are detected by the tissue-resident cells in response to an acute infection or tissue injury. Next, leukocytes aggregate near to the site of inflammation; innate immune cells, such as neutrophils, are often the first cells to appear, whereas mononuclear cells and macrophages accumulate later [69]. This initial robust immune response is designed to destroy invading pathogens and enhance tissue repair [70, 71]. After eliminating the initial threat, leukocyte recruitment ceases, and the previously recruited cells are disposed. The main clearance route of leukocytes is local neutrophil apoptosis and subsequent phagocytosis [72, 73], although they can be cleared through transepithelial migration into the airway lumen in the context of lung inflammation [74] or via lymphatic vessels [75]. The phagocytosis of pathogens, such as Escherichia coli or Staphylococcus aureus, promotes neutrophil apoptosis following neutrophil recruitment, which is termed phagocytosis-induced cell death (PICD) [76]. This response is believed to be primarily protective for the host, and incidentally, pharmacological acceleration of neutrophil apoptosis is protective in pneumococcal meningitis by reducing the incidence of brain hemorrhage [77]. The failed clearance of apoptotic neutrophils can lead to a prolonged inflammatory response, and this phenomenon has been observed in disease, including chronic obstructive pulmonary disease (COPD) [78], pulmonary fibrosis [79] and cystic fibrosis [80]. The production of ROS by neutrophils involves this impaired phagocytosis process, in which ROS activate the GTPase RHOA in surrounding phagocytes and reduces apoptotic cell engulfment by neighboring cells [81–84]. Alveolar macrophages from patients with severe asthma and children with poorly controlled asthma are defective in clearing apoptotic cells [85, 86]. As the mainstay of treatment in asthma, corticosteroids not only induce eosinophil apoptosis [87] but also enhance monocyte-derived macrophage engulfment [88]. The mechanism underlying the enhanced clearance seems dependent on the binding of protein S to apoptotic cells and the upregulation of tyrosine-protein kinase MER on the surface of macrophages [89]. Recently, airway epithelial cells have been found to be capable of engulfing neighboring apoptotic cells, and deficiency of this engulfing function increases pro-inflammatory mediator production and exacerbates airway inflammation [90]. Apoptotic cells are well established to induce the synthesis of anti-inflammatory mediators such as TGF-β, prostaglandin E2, and platelet activating factor by macrophages [91, 92].
To summarize, contrary to traditional model, specific PRRs may activate apoptotic signaling pathways. More importantly, the clearance of apoptotic cells and neutrophil apoptosis in the host further affects inflammation. Therapeutic induction of neutrophil apoptosis at the inflammatory site may be a powerful pro-resolution intervention and could fulfill the clinical need to prevent the harmful consequences of inflammation.
Pyroptosis and inflammation
Pyroptosis is a form of cell death that depends on caspase-1 activation. Pyroptosis features rapid plasma-membrane rupture and release of proinflammatory intracellular content. Cell lysis during pyroptosis results from caspase-1-mediated processes [93–101]. Plasma membrane pores dependent on caspase-1 dissipate cellular ionic gradients, producing a net increased osmotic pressure, water influx, cell swelling, and eventual osmotic lysis, followed by release of inflammatory intracellular content [102]. Cell death due to pyroptosis results in a measurable cellular size increase and cleavage of chromosomal DNA [95, 97, 102–105].
The inflammasome, a caspase-1-containing complex that activates the proinflammatory cytokines IL-1β and IL-18 and results in proinflammatory cell death, is one of the drivers of pyroptosis. The inflammasome activates caspase-1 through a Nod-like receptor (NLRP1, 3, 6, 7, 12, NLRC4), AIM2, or Pyrin, all of which contain a CARD or pyrin domain (PYD) [106, 107]. Many inflammasomes recruit the ASC adaptor via homotypic interactions. Additional ASC molecules are incorporated via CARD-CARD and PYD-PYD interactions, until all ASC molecules are collected into a single focus. The recruitment of procaspase-1 into the ASC focus via CARD-CARD interactions results in its dimerization and proximity-induced autoproteolytic processing into the p10 and p20 subunits. This processed and catalytically active caspase-1 cleaves pro-IL-1β and pro-IL-18.
We recently reported that HMGB1 acting through RAGE on macrophages (or macrophage membrane) triggers dynamin-dependent endocytosis of HMGB1, which in turn initiates a cascade of cellular and molecular events. These events include cathepsin B activation and release from ruptured lysosomes, followed by pyroptosome formation and caspase-1 activation, which serves as a mechanism underlying the HMGB1-induced macrophage pyroptosis (Fig. 2) [108].
Fig. 2.
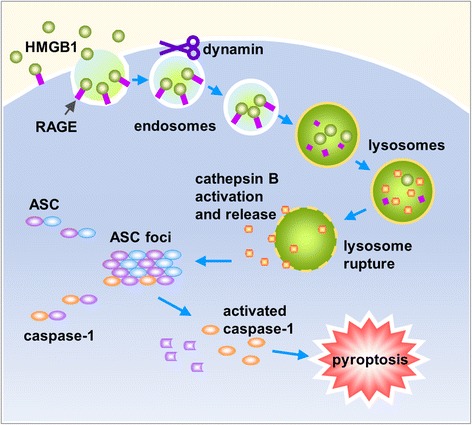
Model of macrophage endocytosis of HMGB1 induces pyroptosis. HMGB1 acting through RAGE on macrophages triggers dynamin-dependent endocytosis of HMGB1, which in turn initiates a cascade of cellular and molecular events. These include CatB activation and release from ruptured lysosomes followed by pyroptosome formation and caspase-1 activation, which promotes HMGB1-induced pyroptosis
A recent study demonstrated that after pyroptosis, ASC specks accumulate in the extracellular space, where they promote further maturation of IL-1β [109]. In addition, phagocytosis of ASC specks by macrophages induces lysosomal damage and nucleation of soluble ASC as well as activation of IL-1β in recipient cells [109]. These findings indicate that pyroptotic cell-released inflammasomes serve as danger signals promoting enhanced activation of macrophages.
Pyroptotic cells secrete the inflammatory cytokines IL-1β and IL-18 following caspase-1 activation. IL-1β is a potent endogenous pyrogen that stimulates fever, leukocyte tissue migration and expression of diverse cytokines and chemokines [110]. IL-18 induces IFNγ production and is important for the activation of T cells, macrophages and other cell types [111]. Cytokine secretion occurs through caspase-1-dependent pores in the plasma membrane. Pharmacological inhibition of cell lysis does not prevent caspase-1-dependent pore formation and cytokine secretion, suggesting that lysis is not required for the release of active IL-1β and IL-18 [102]. Thus, cytokine secretion and cell lysis are both downstream consequences of caspase-1-dependent pore formation. Notably, caspase-1 activation cannot trigger pyroptosis in all cell types; specifically, epithelial cells use caspase-1 activation to prevent cell death [112]. For example, caspase-1 activation stimulates lipid production and membrane repair in response to the pore-forming toxins aerolysin and α-toxin [112].
In addition to caspase-1, caspase-11 has also been found to be involved in pyroptosis [113–115]. A recent study revealed that caspase-11 participates in the process of non-canonical inflammasome activation downstream of a cytosolic ligand released from bacteria [116, 117].
Pyroptosis may protect against infection and induces pathological inflammation. However, exuberant or inappropriate caspase-1 activation and pyroptosis can be detrimental. During infection, caspase-1 activation helps to clear pathogens, such as Salmonella [118, 119], Francisella [120], Legionella [101, 121], Shigella [122], Anaplasma phagocytophilum [123], Burkholderia thailandensis [124], Burkholderia pseudomallei [125] and Listeria [126]. Mutations in NLR proteins can lead to improper caspase-1 activation and can cause hereditary autoinflammatory syndromes [127]. Moreover, caspase-1 is involved in the pathogenesis of several diseases characterized by inflammation and cell death, including myocardial infarction [128], cerebral ischaemia [129], neurodegenerative diseases [130], inflammatory bowel disease [131], and endotoxic shock [132].
As one of the most recently recognized types of cell death, pyroptosis exhibits a particular relationship with common pathogens, and clinic inflammatory disease for caspase-1 connects to both cell death and pro-inflammation directly. Pyroptosis and other caspase 1-dependent processes are therefore relevant to our understanding of the pathophysiology of inflammatory disease.
Pyronecrosis and inflammation
Pyronecrosis is another necrosis-like cell death process that is independent of caspase-1 and caspase-11 but is dependent on ASC and lysosomal protein cathepsin B. Pyroptosis results in the cellular secretion of the pro-inflammatory mediator HMGB1 [133]. Recent studies have demonstrated that pyronecrosis can be induced by several pathogens, including Neisseria gonorrhoeae [134], Toxoplasma gondii parasitophorous [135], Bacillus anthracis lethal toxin [136] and Staphylococcus aureus [137]. The mechanism underlying pyronecrosis remains unclear at present and requires further investigation.
NETosis and inflammation
NETosis is a special form of polymorphonuclear neutrophil (PMN) death that releases neutrophil extracellular traps (NETs) [138]. NETs are web-like structures released by neutrophils that are composed of decondensed chromatin in complex with different neutrophil proteins that can capture, neutralize, and kill microbes. These large extracellular structures provide a physical barrier to prevent microbial dissemination and increase the local concentration of antimicrobial effectors [139, 140]. There are two types of NETosis that can be distinguished by the occurrence time as early and late. The more frequently observed type is late NETosis, as NET release via cell death is a slow process (120–240 min) and is defined as suicidal NETosis. This form of suicide is an NADPH oxidase–dependent cellular death process requiring chromatin decondensation, followed by nuclear envelope disintegration and mixing of nucleic acids and granule proteins within a large intracellular vacuole [141]. However, it remains unclear how oxidants participate in the dismantling of the nuclear envelope and mixing of the NET components. Classically, suicidal NETosis occurs following stimulation by phorbol myristate acetate (PMA) through activation of protein kinase C and the Raf–mitogen-activated protein kinase (MEK)–extracellular signal-regulated kinase (ERK) pathway. NADPH assists in the translocation of neutrophil elastase from cytosolic granules into the nucleus, where it aids in chromatin breakdown via histone cleavage. Myeloperoxidase (MPO) is required for chromatin and nuclear envelope breakdown and granular mixing within the NET vacuole. One hundred twenty minutes after intracellular NET formation, the neutrophil outer membrane ruptures, and the mature NET is extruded.
The early form of NETosis occurs rapidly in response to a pathogen, e.g., after in vitro Staphylococcus aureus stimulation for 5–60 min. Early NETosis has also been termed vital NETosis in some studies [142]. In general, NETosis begins when the nucleus loses its characteristic lobulated architecture. Subsequently, nuclear membranes disassemble, and the chromatin decondenses into the cytoplasm while the plasma membrane remains intact. Finally, the plasma membrane bursts, leading to NET released [138]. This process is mainly dependent on ROS, such as superoxide generated by the NADPH oxidase Nox2. This mechanism spares the PMN outer membrane, thereby allowing the PMN to continue to function, even to the point of becoming anuclear. There are three major differences between suicidal NETosis and vital NETosis, including the nature of the inciting stimuli and the timing, the functional capacity of the PMNs during NET release, and the mechanisms employed to make and release NETs. In addition to PMN, NETosis has also been observed in eosinophils and mast cells [143]. Therefore, the more generalized term ‘ETosis’ maybe more accurate [144].
NETs can kill a number of pathogenic bacteria directly, beyond just capturing and immobilization [145–148]. Studies demonstrate that NETs can inactivate bacterial virulence factors, such as IpaB from S. flexneri [138]. NETs may also serve to opsonize certain fungi, such as A. fumigatus via long pentraxin 3 [149]. NETs generated from PMNs can inhibit the growth of Aspergillus [145] and kill C. albicanscan, even the opportunistic pathogen P. aeruginosa [150]. The gram-negative bacterium K. pneumoniae is not sufficient to induce NETosis in isolated neutrophils ex vivo but is a good inducer in a mouse lung infection model [151]. Human immunodeficiency virus (HIV)-1 has been shown to induce NETosis through a cell death pathway [152]. Feline leukemia virus (FeLV) was able to inhibit neutrophil activation by inhibiting the activation of PKC to reduce ROS production [153].
Numerous types of inflammation are associated with NETs and NETosis. NETs are observed in acute lung injury (ALI) models of both infection- or sterile- related by influenza virus [154, 155], bacteria or bacterial component LPS [156–158], fungi [148, 159, 160], and transfusion [161, 162]. Among them, human neutrophil antigen (HNA)-3a causes the most severe transfusion-related ALI and has been shown to promote NETosis in human neutrophils in vitro [161]. Extracellular neutrophil elastase release via NETosis may be an important cause of lung tissue damage and cystic fibrosis progression [163]. NETs have been shown to form scaffolds in circulation that promote thrombus formation by interacting with the endothelium, platelets, coagulation factors and red blood cells, which cause deep vein thrombosis. IL-8 and ROS release from endothelial cells can recruit and trigger neutrophils to form NETs, which subsequently promote damage to the endothelium through the binding of histones [164].
As a specific cell death type for neutrophils, NETosis help capture numerous pathogenic bacteria and virus. Further insight into the interaction between NETs and invaders would deepen the understanding of the inflammation process. Furthermore, NETotic products could be treated as prognostic biomarkers for inflammatory disorders, and whether the produces correlate with clinical outcome in a variety of diseases requires further translational investigation.
Autophagy and inflammation
Autophagy is a genetically regulated and evolutionarily conserved pathway for the degradation of subcellular components [165, 166]. Autophagy has previously been classified as a form of programmed cell death to describe a form of caspase-independent necrosis-like cell death associated with the accumulation of autophagosomes in cells [167]. This classification is now controversial, and the casual relationship between autophagy and cell death remains uncertain [168, 169].
Autophagy formation begins when an autophagic isolation membrane (also known as a phagophore) engulfs a portion of cytoplasm [170]. Beclin 1, the serine/threonine protein kinase ULK1, autophagy-related LC3 proteins, and γ-aminobutyric acid receptor-associated proteins are key regulators of phagophore formation [170]. A phagophore sequesters captured cytoplasmic cargo, and a double-membraned autophagosome is formed following elongation and closure. Autophagosome formation is largely controlled by mammalian target of rapamycin (mTOR). Inhibition of mTOR leads to the interaction between ULK1 and AMPK [171, 172], which in turn recruits the type III PI3 kinase VPS34 to promote the development of autophagosome [173, 174]. The degradation of the captured cargo begins when the double-membraned autophagosome matures into a single membrane-delimited autolysosome [175, 176]. Following this step, lysosomes can be recycled from autolysosomes, thereby permitting the cell to reuse a critical component required for further autophagy.
PRR signaling induced by PAMPs and DAMPs can activate autophagy. For instance, TLRs can cooperate with autophagy in response to PAMPs [177, 178], and NLRs interact with ATGs to localize autophagy [179, 180]. Inflammatory cytokines such as IL-1 family members [181, 182] and IFNγ [183–185] are also involved in the activation of autophagy, whereas TH2 cell-associated cytokines, IL-4 and IL-13, inhibit autophagy [184].
Multiple studies have confirmed the important role of autophagy during the infection process. Autophagy protects organism from infectious disease by degrading intracellular bacteria, viruses, and protozoan pathogens [186–188].
The role of autophagy in regulating inflammation has been demonstrated in Crohn’s disease and sepsis. Crohn’s disease is a type of chronic inflammation. Polymorphisms in the genes encoding the autophagy-related proteins Atg2a, Atg4a, Atg4d, death-associated protein, immunity-related GTPase family M protein (IRGM), and ULK-1 have been found to be associated with susceptibility to Crohn’s disease [189–191]. NOD2 mutations cause impairment in autophagosome induction and bacterial clearance [179]. Autophagy formation downstream of NOD2 activation controls IL-1β and IL-6 release [192, 193] and results in the tolerogenic presentation of commensal bacterial components on MHC class II complexes in dendrite cells [180]. Inhibition of autophagy in septic mice boosts inflammatory cytokine levels and increases mortality. This effect may due to the failure to clear damaged or dysfunctional mitochondria, which activate the NLRP3 inflammasome [194]. We have recently demonstrated that hemorrhagic shock (HS) acting through HMGB1/TLR4 signaling upregulates NOD2 expression in alveolar macrophages (AM) and subsequently sensitizes AM to the NOD2 ligand MDP, which leads to exacerbated inflammation in the lung. Moreover, upregulated NOD2 signaling induces autophagy in AM, which in turn negatively regulates lung inflammation via a mechanism that involves suppression of NOD2-RIP2 signaling and inflammasome activation. PMNs counteract this anti-inflammatory effect of autophagy via a NAD(P)H oxidase-derived ROS mechanism; therefore, PMNs enhance post-HS lung inflammation [195].
Although the relationship between autophagy and cell death remains uncertain, several members of the inflammation process are involved in autophagy. The function of autophagy in related inflammatory diseases requires further investigation. A better understanding of the relevance of the contribution of autophagy to inflammatory diseases has great clinical potential.
Conclusion and prospective
Emerging evidence has demonstrated the tight links between cell death and inflammation. A better appreciation of the cross-regulatory relationships between different forms of cell death and pathways will be crucial for understanding their roles in the inflammation process. It is important that we realize the therapeutic possibility of targeting programed cell death in patients. Our understanding of the molecular pathways of programed cell death will allow the development of reagents that control cell death, thereby serving as a novel strategy for interventions in inflammatory diseases. Some types of cell death that do not seem to be related to inflammation may also be considered in future studies in light of their possible interaction with inflammation; these approaches will help us better understand the entire inflammatory process network.
Acknowledgment
This work was supported by the USA National Institutes of Health Grant R01-HL-079669, USA National Institutes of Health Center Grant P50-GM-53789, and a USA VA Merit Award.
Abbreviations
- AIM
Absent in melanoma
- ALI
Acute lung injury
- AM
Alveolar macrophages
- ANT
Adenine nucleotide translocase
- BAI
Brain-specific angiogenesis inhibitor
- CARD
Caspase activation and recruitment domains
- CLP
Cecal ligation and puncture
- COPD
Chronic obstructive pulmonary disease
- CYPD
Cyclophilin D
- DAMPs
Damage-associated molecular patterns
- DC
Dendritic cells
- DISC
Death-inducing signaling complex
- ERK
Extracellular signal-regulated kinase
- FADD
Fas-associated death domain
- Fas-L
Fas ligand
- FeLV
Feline leukemia virus
- GAS
Growth-arrest-specific
- HIV
Human immunodeficiency virus
- HMGB1
High-mobility group box 1
- HNA
Human neutrophil antigen
- HS
Hemorrhagic shock
- IAPs
Inhibitor of apoptosis proteins
- IECs
Intestinal epithelial cells
- IR
Ischemia reperfusion
- IRGM
Immunity-related GTPase family M protein
- LOXs
Lipoxygenase
- MEK
Raf-mitogen-activated protein kinase
- MFG-E
Milk fat globule epidermal growth factor
- MPO
Myeloperoxidase
- mTOR
Mammalian target of rapamycin
- NETs
Neutrophil extracellular traps
- PICD
Phagocytosis-induced cell death
- PIPs
Phosphatidylinositol phosphates
- PMA
Phorbol myristate acetate
- PMN
Polymorphonuclear neutrophil
- PRRs
Pattern recognition receptors
- PS
Phosphatidyl serine
- PYD
Pyrin domain
- RAGE
Receptor for advanced-glycation end-products
- RIP
Receptor-interacting protein kinase
- ROS
Reactive oxygen species
- Nec-1
Necrostatin-1
- SODD
Silencer of death domains
- TIM
T-cell immunoglobulin domain and mucin domain
- TNFR
Tumor necrosis factor receptor
- TRADD
TNF-α receptor-associated death domain
- TRAF
TNF-α receptor associated factor
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YY collected the data and drafted the manuscript. GJ conceived and designed the study. PZ revised the manuscript. JF conceived and designed the study, reviewed and finalized the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yong Yang, Email: yyxyz19840408737@sina.com.
Gening Jiang, Email: jgn1121@163.com.
Peng Zhang, Email: zhangpeng1121@aliyun.com.
Jie Fan, Email: jif7@pitt.edu.
References
- 1.Suzanne M, Steller H. Shaping organisms with apoptosis. Cell Death Differ. 2013;20(5):669–75. doi: 10.1038/cdd.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–41. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 3.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 4.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 5.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 6.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19(10):2056–67. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Teng X, Degterev A, Jagtap P, Xing X, Choi S, Denu R, et al. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg Med Chem Lett. 2005;15(22):5039–44. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 9.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 10.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andera L. Signaling activated by the death receptors of the TNFR family. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2009;153(3):173–80. doi: 10.5507/bp.2009.029. [DOI] [PubMed] [Google Scholar]
- 12.Wertz IE, Dixit VM. Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth Factor Rev. 2008;19(3–4):313–24. doi: 10.1016/j.cytogfr.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 13.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A. 2008;105(33):11778–83. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283(36):24295–9. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Current Biology: CB. 2007;17(5):418–24. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–63. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30(6):689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 18.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138(2):229–32. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–7. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21(10):1511–21. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H, Song Y, et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014;21(11):1709–20. doi: 10.1038/cdd.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443–53. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193(4):1539–43. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110(33):E3109–18. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290–7. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105–21. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17(6):922–30. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 29.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22(15):3898–909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jezek P, Hlavata L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005;37(12):2478–503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278(38):36027–31. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 32.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–9. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 33.Wu YT, Tan HL, Huang Q, Sun XJ, Zhu X, Shen HM. zVAD-induced necroptosis in L929 cells depends on autocrine production of TNFalpha mediated by the PKC-MAPKs-AP-1 pathway. Cell Death Differ. 2011;18(1):26–37. doi: 10.1038/cdd.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayakawa M, Ishida N, Takeuchi K, Shibamoto S, Hori T, Oku N, et al. Arachidonic acid-selective cytosolic phospholipase A2 is crucial in the cytotoxic action of tumor necrosis factor. J Biol Chem. 1993;268(15):11290–5. [PubMed] [Google Scholar]
- 35.van Leyen K, Duvoisin RM, Engelhardt H, Wiedmann M. A function for lipoxygenase in programmed organelle degradation. Nature. 1998;395(6700):392–5. doi: 10.1038/26500. [DOI] [PubMed] [Google Scholar]
- 36.Maccarrone M, Melino G, Finazzi-Agro A. Lipoxygenases and their involvement in programmed cell death. Cell Death Differ. 2001;8(8):776–84. doi: 10.1038/sj.cdd.4400908. [DOI] [PubMed] [Google Scholar]
- 37.Festjens N, Kalai M, Smet J, Meeus A, Van Coster R, Saelens X, et al. Butylated hydroxyanisole is more than a reactive oxygen species scavenger. Cell Death Differ. 2006;13(1):166–9. doi: 10.1038/sj.cdd.4401746. [DOI] [PubMed] [Google Scholar]
- 38.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477(7364):330–4. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 39.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477(7364):335–9. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 41.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7(4):302–13. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Artal-Sanz M, Tavernarakis N. Proteolytic mechanisms in necrotic cell death and neurodegeneration. FEBS Lett. 2005;579(15):3287–96. doi: 10.1016/j.febslet.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 43.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35(6):908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 44.Linkermann A, Brasen JH, De Zen F, Weinlich R, Schwendener RA, Green DR, et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-alpha-induced shock. Mol Med. 2012;18:577–86. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13(10):954–62. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin J, Li H, Yang M, Ren J, Huang Z, Han F, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3(1):200–10. doi: 10.1016/j.celrep.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 48.Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81(8):751–61. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 49.Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107(4):270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 50.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88(7):1569–76. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chavez-Valdez R, Martin LJ, Northington FJ. Programmed Necrosis: A Prominent Mechanism of Cell Death following Neonatal Brain Injury. Neurol Res Int. 2012;2012:257563. doi: 10.1155/2012/257563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eum KH, Lee M. Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem. 2011;348(1–2):61–8. doi: 10.1007/s11010-010-0638-8. [DOI] [PubMed] [Google Scholar]
- 53.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–98. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258(6):479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 55.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55(3):178–94. doi: 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- 56.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323(5917):1057–60. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 57.Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, et al. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012;19(10):1709–21. doi: 10.1038/cdd.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20(9):1149–60. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdelaziz DH, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, et al. Asc-dependent and independent mechanisms contribute to restriction of legionella pneumophila infection in murine macrophages. Front Microbiol. 2011;2:18. doi: 10.3389/fmicb.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Puri AW, Broz P, Shen A, Monack DM, Bogyo M. Caspase-1 activity is required to bypass macrophage apoptosis upon Salmonella infection. Nat Chem Biol. 2012;8(9):745–7. doi: 10.1038/nchembio.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masumoto J, Dowds TA, Schaner P, Chen FF, Ogura Y, Li M, et al. ASC is an activating adaptor for NF-kappa B and caspase-8-dependent apoptosis. Biochem Biophys Res Commun. 2003;303(1):69–73. doi: 10.1016/s0006-291x(03)00309-7. [DOI] [PubMed] [Google Scholar]
- 62.Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20(10):1381–92. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7(12):964–74. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 64.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182(5):1545–56. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–9. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 66.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27(6):927–40. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450(7168):430–4. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 68.Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15(1):192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- 69.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21(2):325–32. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, et al. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci U S A. 2011;108(38):15990–5. doi: 10.1073/pnas.1110144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Farnworth SL, Henderson NC, Mackinnon AC, Atkinson KM, Wilkinson T, Dhaliwal K, et al. Galectin-3 reduces the severity of pneumococcal pneumonia by augmenting neutrophil function. Am J Pathol. 2008;172(2):395–405. doi: 10.2353/ajpath.2008.070870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83(3):865–75. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S5–11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 74.Persson CG, Uller L. Resolution of cell-mediated airways diseases. Respir Res. 2010;11:75. doi: 10.1186/1465-9921-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. 2011;117(4):1196–204. doi: 10.1182/blood-2009-11-254490. [DOI] [PubMed] [Google Scholar]
- 76.Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol. 1996;156(10):3986–92. [PubMed] [Google Scholar]
- 77.Koedel U, Frankenberg T, Kirschnek S, Obermaier B, Hacker H, Paul R, et al. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog. 2009;5(5):e1000461. doi: 10.1371/journal.ppat.1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81(4):289–96. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [DOI] [PubMed] [Google Scholar]
- 79.Morimoto K, Janssen WJ, Terada M. Defective efferocytosis by alveolar macrophages in IPF patients. Respir Med. 2012;106(12):1800–3. doi: 10.1016/j.rmed.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vandivier RW, Fadok VA, Ogden CA, Hoffmann PR, Brain JD, Accurso FJ, et al. Impaired clearance of apoptotic cells from cystic fibrosis airways. Chest. 2002;121(3 Suppl):89S. doi: 10.1378/chest.121.3_suppl.89s. [DOI] [PubMed] [Google Scholar]
- 81.McPhillips K, Janssen WJ, Ghosh M, Byrne A, Gardai S, Remigio L, et al. TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J Immunol. 2007;178(12):8117–26. doi: 10.4049/jimmunol.178.12.8117. [DOI] [PubMed] [Google Scholar]
- 82.Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem. 2006;281(13):8836–42. doi: 10.1074/jbc.M510972200. [DOI] [PubMed] [Google Scholar]
- 83.Moon C, Lee YJ, Park HJ, Chong YH, Kang JL. N-acetylcysteine inhibits RhoA and promotes apoptotic cell clearance during intense lung inflammation. Am J Respir Crit Care Med. 2010;181(4):374–87. doi: 10.1164/rccm.200907-1061OC. [DOI] [PubMed] [Google Scholar]
- 84.Cepkova M, Matthay MA. Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. J Intensive Care Med. 2006;21(3):119–43. doi: 10.1177/0885066606287045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fitzpatrick AM, Holguin F, Teague WG, Brown LA. Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. J Allergy Clin Immunol. 2008;121(6):1372–8. doi: 10.1016/j.jaci.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huynh ML, Malcolm KC, Kotaru C, Tilstra JA, Westcott JY, Fadok VA, et al. Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med. 2005;172(8):972–9. doi: 10.1164/rccm.200501-035OC. [DOI] [PubMed] [Google Scholar]
- 87.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96(25):14541–6. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1(6):496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- 89.Methot N, Huang J, Coulombe N, Vaillancourt JP, Rasper D, Tam J, et al. Differential efficacy of caspase inhibitors on apoptosis markers during sepsis in rats and implication for fractional inhibition requirements for therapeutics. J Exp Med. 2004;199(2):199–207. doi: 10.1084/jem.20031791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493(7433):547–51. doi: 10.1038/nature11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101(4):890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109(1):41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci U S A. 1999;96(5):2396–401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen Y, Smith MR, Thirumalai K, Zychlinsky A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. Embo J. 1996;15(15):3853–60. [PMC free article] [PubMed] [Google Scholar]
- 95.Bergsbaken T, Cookson BT. Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 2007;3(11):e161. doi: 10.1371/journal.ppat.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kelk P, Johansson A, Claesson R, Hanstrom L, Kalfas S. Caspase 1 involvement in human monocyte lysis induced by Actinobacillus actinomycetemcomitans leukotoxin. Infect Immun. 2003;71(8):4448–55. doi: 10.1128/IAI.71.8.4448-4455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sun GW, Lu J, Pervaiz S, Cao WP, Gan YH. Caspase-1 dependent macrophage death induced by Burkholderia pseudomallei. Cell Microbiol. 2005;7(10):1447–58. doi: 10.1111/j.1462-5822.2005.00569.x. [DOI] [PubMed] [Google Scholar]
- 98.Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A. 2008;105(11):4312–7. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thumbikat P, Dileepan T, Kannan MS, Maheswaran SK. Mechanisms underlying Mannheimia haemolytica leukotoxin-induced oncosis and apoptosis of bovine alveolar macrophages. Microb Pathog. 2005;38(4):161–72. doi: 10.1016/j.micpath.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 100.Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2006;2(3):e18. doi: 10.1371/journal.ppat.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med. 2006;203(4):1093–104. doi: 10.1084/jem.20051659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 103.Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38(1):31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- 104.Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci U S A. 1996;93(18):9833–8. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hilbi H, Chen Y, Thirumalai K, Zychlinsky A. The interleukin 1beta-converting enzyme, caspase 1, is activated during Shigella flexneri-induced apoptosis in human monocyte-derived macrophages. Infect Immun. 1997;65(12):5165–70. doi: 10.1128/iai.65.12.5165-5170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- 107.Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34(5):755–68. doi: 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21(8):1229–39. doi: 10.1038/cdd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation. Nat Immunol. 2014;15(8):727–37. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Delaleu N, Bickel M. Interleukin-1 beta and interleukin-18: regulation and activity in local inflammation. Periodontol 2000. 2004;35:42–52. doi: 10.1111/j.0906-6713.2004.003569.x. [DOI] [PubMed] [Google Scholar]
- 111.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001;19:423–74. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- 112.Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126(6):1135–45. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 113.Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem. 1996;271(34):20580–7. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- 114.Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92(4):501–9. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 115.Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol. 2000;149(3):613–22. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–3. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341(6151):1246–9. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 118.Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med. 2006;203(6):1407–12. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74(8):4922–6. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202(8):1043–9. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol. 2006;7(3):318–25. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 122.Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, et al. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12(5):581–90. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- 123.Pedra JH, Sutterwala FS, Sukumaran B, Ogura Y, Qian F, Montgomery RR, et al. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. J Immunol. 2007;179(7):4783–91. doi: 10.4049/jimmunol.179.7.4783. [DOI] [PubMed] [Google Scholar]
- 124.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339(6122):975–8. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS Pathog. 2011;7(12):e1002452. doi: 10.1371/journal.ppat.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, et al. Roles of caspase-1 in Listeria infection in mice. Int Immunol. 2004;16(2):335–43. doi: 10.1093/intimm/dxh041. [DOI] [PubMed] [Google Scholar]
- 127.Simon A, van der Meer JW. Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R86–98. doi: 10.1152/ajpregu.00504.2006. [DOI] [PubMed] [Google Scholar]
- 128.Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R, et al. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol. 2003;35(6):685–94. doi: 10.1016/s0022-2828(03)00113-5. [DOI] [PubMed] [Google Scholar]
- 129.Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J Cereb Blood Flow Metab. 1998;18(2):180–5. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- 130.Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399(6733):263–7. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 131.Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A. 2001;98(23):13249–54. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80(3):401–11. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 133.Willingham SB, Bergstralh DT, O'Connor W, Morrison AC, Taxman DJ, Duncan JA, et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2(3):147–59. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Duncan JA, Gao X, Huang MT, O'Connor BP, Thomas CE, Willingham SB, et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182(10):6460–9. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 2009;5(2):e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Averette KM, Pratt MR, Yang Y, Bassilian S, Whitelegge JP, Loo JA, et al. Anthrax lethal toxin induced lysosomal membrane permeabilization and cytosolic cathepsin release is Nlrp1b/Nalp1b-dependent. Plos One. 2009;4(11):e7913. doi: 10.1371/journal.pone.0007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol. 2012;92(5):1069–81. doi: 10.1189/jlb.0112014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 139.Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5(8):577–82. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 140.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30(11):513–21. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 141.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–41. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–94. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 143.Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011;18(4):581–8. doi: 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wartha F, Henriques-Normark B. ETosis: a novel cell death pathway. Sci Signal. 2008;1(21):e25. doi: 10.1126/stke.121pe25. [DOI] [PubMed] [Google Scholar]
- 145.Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114(13):2619–22. doi: 10.1182/blood-2009-05-221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185(12):7413–25. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 147.Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16(4):396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 148.Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006;8(4):668–76. doi: 10.1111/j.1462-5822.2005.00659.x. [DOI] [PubMed] [Google Scholar]
- 149.Jaillon S, Peri G, Delneste Y, Fremaux I, Doni A, Moalli F, et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med. 2007;204(4):793–804. doi: 10.1084/jem.20061301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mulcahy H, Charron-Mazenod L, Lewenza S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008;4(11):e1000213. doi: 10.1371/journal.ppat.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–91. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 2012;12(1):109–16. doi: 10.1016/j.chom.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 153.Wardini AB, Guimaraes-Costa AB, Nascimento MT, Nadaes NR, Danelli MG, Mazur C, et al. Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J Gen Virol. 2010;91(Pt 1):259–64. doi: 10.1099/vir.0.014613-0. [DOI] [PubMed] [Google Scholar]
- 154.Ng HH, Narasaraju T, Phoon MC, Sim MK, Seet JE, Chow VT. Doxycycline treatment attenuates acute lung injury in mice infected with virulent influenza H3N2 virus: involvement of matrix metalloproteinases. Exp Mol Pathol. 2012;92(3):287–95. doi: 10.1016/j.yexmp.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 155.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Barletta KE, Cagnina RE, Burdick MD, Linden J, Mehrad B. Adenosine A(2B) receptor deficiency promotes host defenses against gram-negative bacterial pneumonia. Am J Respir Crit Care Med. 2012;186(10):1044–50. doi: 10.1164/rccm.201204-0622OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol. 2011;187(4):1856–65. doi: 10.4049/jimmunol.1004201. [DOI] [PubMed] [Google Scholar]
- 158.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–62. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010;6(4):e1000873. doi: 10.1371/journal.ppat.1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hosogi S, Iwasaki Y, Yamada T, Komatani-Tamiya N, Hiramatsu A, Kohno Y, et al. Effect of inducible nitric oxide synthase on apoptosis in Candida-induced acute lung injury. Biomed Res. 2008;29(5):257–66. doi: 10.2220/biomedres.29.257. [DOI] [PubMed] [Google Scholar]
- 161.Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood. 2012;119(26):6335–43. doi: 10.1182/blood-2012-01-405183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661–71. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Roghanian A, Sallenave JM. Neutrophil elastase (NE) and NE inhibitors: canonical and noncanonical functions in lung chronic inflammatory diseases (cystic fibrosis and chronic obstructive pulmonary disease) J Aerosol Med Pulm Drug Deliv. 2008;21(1):125–44. doi: 10.1089/jamp.2007.0653. [DOI] [PubMed] [Google Scholar]
- 164.Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584(14):3193–7. doi: 10.1016/j.febslet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 165.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793(4):664–73. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 166.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90(4):1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 167.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6(12):1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 168.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–10. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7(5):457–65. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- 170.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 171.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22(2):132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 172.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. Plos One. 2010;5(11):e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010;38(2):265–79. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Simonsen A, Birkeland HC, Gillooly DJ, Mizushima N, Kuma A, Yoshimori T, et al. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117(Pt 18):4239–51. doi: 10.1242/jcs.01287. [DOI] [PubMed] [Google Scholar]
- 175.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–69. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 176.Furuta N, Fujita N, Noda T, Yoshimori T, Amano A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell. 2010;21(6):1001–10. doi: 10.1091/mbc.E09-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27(7):1110–21. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 180.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–7. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 181.Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–97. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–63. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 184.Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27(3):505–17. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 185.Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. 2010;12(12):1154–65. doi: 10.1038/ncb2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27(1):11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–30. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–96. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464(7289):713–20. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Henckaerts L, Cleynen I, Brinar M, John JM, Van Steen K, Rutgeerts P, et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn's disease. Inflamm Bowel Dis. 2011;17(6):1392–7. doi: 10.1002/ibd.21486. [DOI] [PubMed] [Google Scholar]
- 192.Ferwerda G, Kramer M, de Jong D, Piccini A, Joosten LA, Devesaginer I, et al. Engagement of NOD2 has a dual effect on proIL-1beta mRNA transcription and secretion of bioactive IL-1beta. Eur J Immunol. 2008;38(1):184–91. doi: 10.1002/eji.200737103. [DOI] [PubMed] [Google Scholar]
- 193.Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, et al. Crohn's disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut. 2011;60(9):1229–35. doi: 10.1136/gut.2010.228908. [DOI] [PubMed] [Google Scholar]
- 194.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wen Z, Fan L, Li Y, Zou Z, Scott MJ, Xiao G, et al. Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: role in posthemorrhagic shock acute lung inflammation. J Immunol. 2014;193(9):4623–33. doi: 10.4049/jimmunol.1400899. [DOI] [PMC free article] [PubMed] [Google Scholar]