

Abstract

The newly discovered light-dependent transcription factor CarH uses adenosylcobalamin as a light sensor to regulate expression of protective genes in bacteria upon exposure to sunlight. This use of adenosylcobalamin is a clever adaptation of a classic enzyme cofactor, taking advantage of its photolabile Co–C bond. However, it is also puzzling in that photolysis of adenosylcobalamin generates the 5′-deoxyadenosyl radical that could damage DNA. Here, using liquid chromatography and spectroscopic techniques, we demonstrate that CarH suppresses release of the 5′-deoxyadenosyl radical and instead effects conversion to a nonreactive 4′,5′-anhydroadenosine. In this manner, CarH safeguards use of adenosylcobalamin in light-dependent gene regulation.
Adenosylcobalamin (AdoCbl, coenzyme B12) is best known as a cofactor for enzymes that catalyze carbon skeleton rearrangements or elimination reactions through radical-based chemistry.1 Recently, however, AdoCbl was shown to be used as a light sensor by the light-dependent transcription factor CarH.2 Light exposure of AdoCbl leads to cleavage of its covalent Co–C bond, and this cleavage is harnessed by CarH to activate transcription. In particular, in the dark, intact AdoCbl mediates formation of a CarH tetramer that binds to DNA and represses transcription, whereas light exposure leads to tetramer disassembly, dissociation from DNA, and transcription activation.2 A primary function of CarH-type transcription factors is to activate responses that mitigate light-induced damage. In Myxococcus xanthus,2−4Thermus thermophilus,5 and other bacteria, CarH activates expression of carotenoid biosynthetic genes upon light exposure, resulting in production of carotenoids, which then protect the cell from photooxidative damage by quenching light-induced reactive oxygen species. In this context, the use of AdoCbl as a light sensor appears to be paradoxical: light-induced cleavage of the Co–C bond leads to formation of a 5′-deoxyadenosyl radical, which itself could rapidly generate reactive oxygen species or react with and damage DNA, the type of damage that this mode of light sensing is meant to prevent. Given that release of the reactive 5′-deoxyadenosyl radical adjacent to DNA could be detrimental to the cell, we hypothesized that CarH might possess a mechanism to prevent the release of this radical. Therefore, we sought to investigate the photochemistry of CarH-bound AdoCbl by probing the state of the cobalamin after photolysis and the chemical nature of the product derived from the AdoCbl 5′-deoxyadenosyl group, hereafter termed the CarH photolysis product.
The photochemistry of free AdoCbl has been studied extensively.6−11 Briefly, exposure of AdoCbl to <550 nm light leads to homolytic cleavage of the Co–C bond,6−8 generating five-coordinate cob(II)alamin [cob(II)] and a 5′-deoxyadenosyl radical (Figure S1 of the Supporting Information). Under aerobic conditions, the 5′-deoxyadenosyl radical rapidly reacts with molecular oxygen to first form 5′-peroxyadenosine, which then decomposes to adenosine 5′-aldehyde with adenosine and adenine as minor products.11 Cob(II) also is oxidized, either by 5′-peroxyadenosine or by molecular oxygen, followed by ligation of water to form aquacob(III)alamin (OH2Cbl).11 Under anaerobic conditions, the major products are cob(II) and 5′,8-cycloadenosine, generated by cyclization of the 5′-deoxyadenosyl radical.10,12
To probe the state of CarH-bound cobalamin after photolysis under both aerobic and anaerobic conditions, CarH was exposed to light and characterized by ultraviolet–visible (UV–vis) and electron paramagnetic resonance (EPR) spectroscopy. Photolysis of CarH-bound AdoCbl under aerobic conditions results in the formation of six-coordinate cob(III)alamin [cob(III)] with a UV–vis spectrum similar to that of OH2Cbl (Figure 1a). This species is EPR-silent. The UV–vis spectrum after anaerobic photolysis of CarH-bound AdoCbl resembles that of cob(II), including a peak at 312 nm and a broad feature around 480 nm (Figure 1b).13 Exposure of this species to molecular oxygen resulted in quantitative conversion to cob(III) (Figure 1a), suggesting that the species formed upon anaerobic photolysis is chemically competent to form the same cob(III) product observed under aerobic conditions. Additionally, the EPR spectrum after anaerobic photolysis of CarH-bound AdoCbl closely resembles that of free cob(II), with a distinct axial signal, hyperfine splitting arising from the 59Co nucleus (I = 7/2), and superhyperfine splitting arising from an axial 14N ligand (I = 1) (Figure 1c).14 Thus, these data indicate that cob(II) is the product of CarH photolysis under anaerobic conditions and that the presence of molecular oxygen results in oxidation to cob(III).
Figure 1.
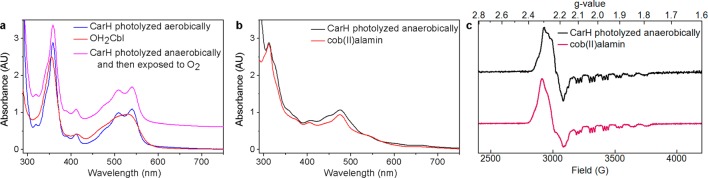
Fate of CarH cobalamin after photolysis. (a) The UV–vis spectrum of aerobically photolyzed CarH-bound AdoCbl (blue) has characteristic features of six-coordinate cob(III). The spectrum of OH2Cbl is shown as a comparison (red). In addition, the spectrum of anaerobically photolyzed CarH after exposure to molecular oxygen (pink, spectrum shifted by 0.6 AU) is identical to that of aerobically photolyzed CarH. (b) The UV–vis spectrum of anaerobically photolyzed CarH-bound AdoCbl (black) has characteristic features of cob(II). The spectrum of free cob(II) is shown as a comparison (red). (c) X-Band EPR spectrum of anaerobically photolyzed CarH (black), recorded at 77 K. The spectrum has features characteristic of cob(II). The spectrum of pure cob(II), generated from anaerobic photolysis of AdoCbl (red), is shown as a comparison.
To characterize the fate of the 5′-deoxyadenosyl group after photolysis, liquid chromatography–mass spectrometry (LC–MS) experiments were performed on light-exposed CarH. Because cobalamin remains associated with CarH after photolysis, the products derived from the 5′-deoxyadenosyl group are easily separated from cobalamin-bound CarH using a 10 kDa cutoff filter (Figure 2, red trace). Notably, under aerobic conditions, only a single product with a molecular mass of 249 Da (m/z 250) is detected. This result is in contrast to the observation of multiple products for aerobic photolysis of the free cofactor, all of which have molecular masses of >249 Da9,11 (Figure S1). As a control, we reproduced the literature reports for free AdoCbl, identifying all of the same products: adenosine 5′-aldehyde (m/z 266 for the free aldehyde and m/z 284 for the aldehyde hydrate) and 5′-peroxyadenosine (m/z 284) as the main nucleoside species (Figure 2, black trace, and Figure S2 of the Supporting Information) as well as a smaller amount of adenosine (m/z 268). With free AdoCbl, species corresponding to cob(III) (the water ligand likely dissociated during ionization) and uncleaved AdoCbl are also observed (Figure S2).
Figure 2.
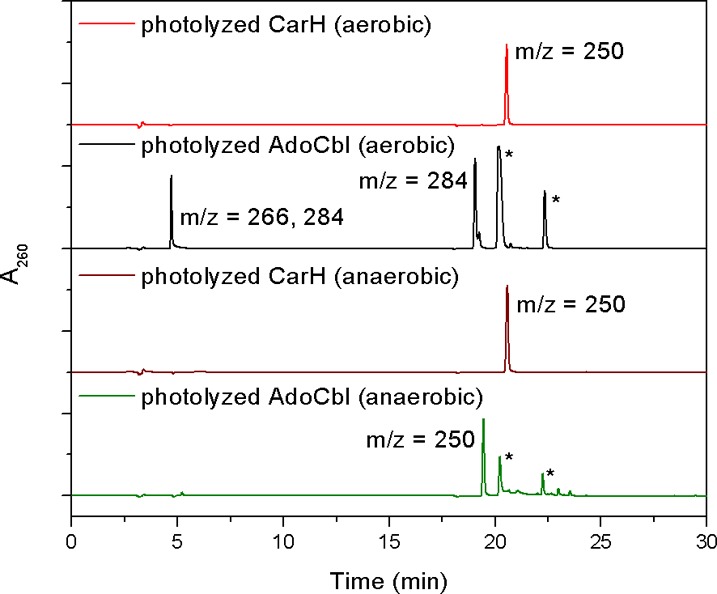
Fate of the CarH 5′-deoxyadenosyl group upon photolysis. Shown are A260 traces from LC of different light-exposed CarH and free AdoCbl samples. CarH photolyzed under aerobic (red) or anaerobic (brown) conditions yields only a single nucleoside product with a retention time and a molecular mass different from those of nucleoside products derived from free AdoCbl photolyzed under aerobic (black) or anaerobic (green) conditions. Mass-to-charge ratios of peaks, as determined by an in-line mass spectrometer, are indicated. Peaks denoted with an asterisk are cobalamin species, as indicated by additional absorbance at 350 and 520 nm.
Photolysis of AdoCbl-bound CarH was also conducted under anaerobic conditions. Again, only a single product with a molecular mass of 249 Da (m/z 250) was observed, and it had the same retention time as the aerobic CarH photolysis product (Figure 2, brown trace), indicating that the fate of the CarH 5′-deoxyadenosyl group is independent of the presence of oxygen.
The molecular mass of the product of both aerobic and anaerobic photolysis of CarH corresponds to that of 5′,8-cycloadenosine, which is the product of anaerobic photolysis of free AdoCbl (Figure S1). To determine if the CarH photolysis product is 5′,8-cycloadenosine or a different compound with the same molecular mass, 5′,8-cycloadenosine was generated as a standard by photolysis of AdoCbl under anaerobic conditions. Notably, 5′,8-cycloadenosine and the CarH photolysis product have different retention times during liquid chromatography (Figure 2, brown and green traces) and furthermore exhibit different fragmentation patterns during mass spectrometry. The mass spectrum of the CarH photolysis product reveals a major species with an ion at m/z 250 as well as a fragment with an ion at m/z 136, corresponding to adenine (Figure S3 of the Supporting Information). This pattern indicates that the CarH photolysis product undergoes fragmentation during ionization because of cleavage of the N-glycosidic bond. In contrast, the mass spectrum of 5′,8-cycloadenosine does not feature an adenine fragment (Figure S3).
To further compare the CarH photolysis product and 5′,8-cycloadenosine, susceptibility to acid treatment was examined. For this purpose, 8% (v/v) trifluoroacetic acid (TFA) was added to the samples after photolysis, and the LC–MS experiments were repeated. As expected, 5′,8-cycloadenosine was unaffected by TFA (Figure S4 of the Supporting Information). Addition of 8% (v/v) TFA to the CarH photolysis product, however, resulted in the disappearance of the nucleoside peak and the appearance of a broad peak with an ion at m/z 136, again corresponding to adenine (Figure S4). Addition of 70% (v/v) methanol did not alter the CarH photolysis product, suggesting that degradation was specific to the addition of acid (Figure S4 of the Supporting Information). These results indicate that acid treatment of the CarH photolysis product results in cleavage of the N-glycosidic bond to yield adenine and an undefined ribose derivative, and that the CarH photolysis product is chemically distinct from 5′,8-cycloadenosine and any other known compounds generated by photolysis of free AdoCbl.
To unambiguously determine the chemical structure of the CarH photolysis product, 0.22 μmol of the product was purified from 25 mg of protein and then characterized by a series of NMR experiments (see the Supporting Information). The chemical shifts and correlations obtained from one-dimensional 1H and two-dimensional (2D) 1H–1H DQCOSY, 1H–13C HSQC, and 1H–13C gHMBC spectra (Figure 3 and Figures S5–S8 of the Supporting Information) provided unambiguous support for the assignment of the CarH photolysis product as 4′,5′-anhydroadenosine.
Figure 3.

1H and 13C chemical shifts (1H in bold) as well as 2D peak correlations (Figures S6–S8) identify the photolysis product as 4′,5′-anhydroadenosine. Predicted shifts obtained from ACD/NMR Predictor (Advanced Chemistry Development, Inc.) are shown in parentheses. Predictions of chemical shifts of hydroxyl and amine protons are unreliable and were not obtained.
Notably, 4′,5′-anhydroadenosine had not previously been observed as a photolysis product of AdoCbl. Instead, 4′,5′-anhydroadenosine has been observed as a product of base-catalyzed degradation of 5′-deoxyadenosylcobaloxime,15 an AdoCbl analogue in which the corrin ring is substituted with cobaloxime, and as the inactivation product of the AdoCbl-dependent enzymes ethanolamine ammonia-lyase and diol dehydratase upon reaction with N2O (refs (16) and (17)) or substrate analogues,18 the relevance and mechanism of which are unclear.19 More importantly, 4′,5′-anhydroadenosine has been observed after thermolysis of AdoCbl or adenosylcobinamide (AdoCbl lacking the dimethylbenzimidazole nucleotide tail) in viscous solvents, attributed to β-H elimination from the 5′-deoxyadenosyl radical.20,21 Although organometallic β-H elimination reactions are commonly concerted and involve migration of a hydride (H–) to the metal, radical-based β-H eliminations have been observed after thermolysis of alkylcobalamins and other organometallic complexes.20−27 These radical-based eliminations occur in two steps, with initial homolytic cleavage of the metal–carbon bond followed by hydrogen atom migration in the caged radical pair,21,27 as demonstrated by radical trapping21 and kinetic experiments.25−27
The mechanism of 4′,5′-anhydroadenosine formation after AdoCbl thermolysis could illuminate the mechanism of its formation after photolysis of CarH. For free AdoCbl, 4′,5′-anhydroadenosine is observed only upon thermolysis in glycerol, accounting for 4–5% of the total product derived from the 5′-deoxyadenosyl group.20 Glycerol, a very viscous solvent, creates a strong solvent cage that slows escape of the 5′-deoxyadenosyl radical from the caged [Cob(II)·5′-deoxyadenosyl radical] pair and allows it to undergo cob(II)-mediated β-H elimination of the C4′ hydrogen to form 4′,5′-anhydroadenosine20 and the cob(III)alamin hydride [which can also be described as protonated cob(I)alamin].29,30 Although the existence of the related cobaloxime hydride has recently been called into question31 and although cob(III)alamin hydride is not well-characterized, studies of its reactivity appear to support its existence,29,30 and other cobalt(III) hydride compounds have been reported (see, for example, ref (32)).
By analogy, CarH might exert a strong cage effect after photolysis, favoring formation of 4′,5′-anhydroadenosine and cob(III)alamin hydride. Protein-induced cage effects have been observed in the cobalamin binding module of methionine synthase,33 which is related to CarH by sequence but binds methylcobalamin instead of AdoCbl. Similarly, the AdoCbl-dependent enzyme glutamate mutase exerts a cage effect upon AdoCbl photolysis, which could prevent loss of the 5′-deoxyadenosyl radical in the absence of substrate.34,35 However, in these enzyme examples, the cage effect is thought to enhance radical pair recombination and not β-H elimination. Thus, although precedent exists for protein cage effects, the outcome of the effect appears to be different for CarH, perhaps in line with its modified function as a light sensor.
The literature precedent described above, along with the data presented here, allows us to propose the mechanism outlined in path 1 of Scheme 1: homolytic Co–C bond cleavage induced by light exposure is followed by β-H elimination from C4′ to form 4′,5′-anhydroadenosine and cob(III)alamin hydride bound to CarH. Cob(III)alamin hydride is thought to decompose to cob(II) and molecular hydrogen,21,25,26 which would explain our observation of cob(II) as the final product under anaerobic conditions. For direct decomposition, two cob(III)alamin hydride molecules bound to CarH would need to come into direct contact, which seems unlikely but could be facilitated by the proximity of the CarH monomers either before, during, or after light-induced tetramer disassembly.2 Alternatively, decomposition to cob(II) could occur via a cob(I)alamin intermediate that is not detected on the time scale of the spectroscopy experiments. Under aerobic conditions, both cob(I) or cob(II) should oxidize forming cob(III), as observed spectroscopically, or the cob(III)alamin hydride could directly react with oxygen to generate cob(III).
Scheme 1. Proposed CarH Photolysis Mechanism.

See the text for details. C4′ and C5′ of the 5′-deoxyadenosyl group are labeled. Cobalamin is shown as a rhombus with the Co oxidation state indicated. The lower axial nitrogen ligand is denoted as N. The adenine base is abbreviated as Ad.
Although all of our data are consistent with path 1 of Scheme 1, we note that photolysis might also proceed through heterolytic cleavage of the Co–C bond, forming the 5′-deoxyadenosyl anion and cob(III), followed by β-hydride elimination to form 4′,5′-anhydroadenosine and cob(III)alamin hydride (path 2 in Scheme 1). Notably, both paths share the same final products. We currently favor the homolytic pathway because of its precedent in AdoCbl photochemistry, but distinction between these mechanisms will require time-resolved spectroscopic experiments to directly probe the cobalamin intermediates and more detailed mechanistic studies.
The discovery that the transcription factor CarH uses AdoCbl to achieve light-dependent gene regulation was at the same time fascinating and confusing. Although this function represents an innovative use of the light-sensitive Co–C bond of AdoCbl in biology, it also predicted that reactive 5′-deoxyadenosyl radicals would be generated very close to DNA upon light exposure. Here, we demonstrate that CarH safeguards the use of AdoCbl as a light sensor by altering the products of the photolysis reaction such that 5′-deoxyadenosyl radicals are not released. Whereas AdoCbl is usually used as an enzyme cofactor, with the 5′-deoxyadenosyl radical as the working species and photolysis as an unwanted side reaction, the roles are reversed in the use of AdoCbl as a light sensor by CarH: here, photolysis is desired, and generation of the 5′-deoxyadenosyl radical is an unwanted side reaction. These results extend our understanding of the functional repurposing of AdoCbl by CarH, showing a classic cofactor in a new light.
Acknowledgments
We thank Dr. S. Padmanabhan (IQFR Madrid, Madrid, Spain) and Dr. Montserrat Elías-Arnanz (University of Murcia, Murcia, Spain) for generously donating the CarH expression vector and helpful discussions and Dr. Koli Taghizadeh, Yifeng Wei, Vinita Lukose, Dr. Garrett Whitworth, and Dr. Joris de Schutter [all at Massachusetts Institute of Technology (MIT)] for technical assistance and advice. NMR, EPR, and mass spectra were recorded at the MIT Department of Chemistry Instrumentation Facility. LC–MS was performed at the MIT Center for Environmental Health Sciences facilities, which are supported by National Institute of Environmental Health Sciences Grant P30-ES002109.
Supporting Information Available
HPLC traces and NMR spectra of the photolysis product. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00416.
This work was supported by National Institutes of Health Grant GM069857 (to C.L.D.). C.L.D. is a Howard Hughes Medical Institute Investigator.
The authors declare no competing financial interest.
Supplementary Material
References
- Frey P. A. (2010) in Comprehensive Natural Products II. Chemistry and Biology (Mander L., and Liu H.-W., Eds.) pp 501–546, Elsevier, Oxford, U.K. [Google Scholar]
- Ortiz-Guerrero J. M.; Polanco M. C.; Murillo F. J.; Padmanabhan S.; Elias-Arnanz M. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 7565–7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Marin M. C.; Padmanabhan S.; Polanco M. C.; Murillo F. J.; Elias-Arnanz M. (2008) Mol. Microbiol. 67, 804–819. [DOI] [PubMed] [Google Scholar]
- Elias-Arnanz M.; Padmanabhan S.; Murillo F. J. (2011) Curr. Opin. Microbiol. 14, 128–135. [DOI] [PubMed] [Google Scholar]
- Takano H.; Kondo M.; Usui N.; Usui T.; Ohzeki H.; Yamazaki R.; Washioka M.; Nakamura A.; Hoshino T.; Hakamata W.; Beppu T.; Ueda K. (2011) J. Bacteriol. 193, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J. F.; Netzel T. L. (1979) J. Am. Chem. Soc. 101, 4000–4002. [Google Scholar]
- Chen E. F.; Chance M. R. (1990) J. Biol. Chem. 265, 12987–12994. [PubMed] [Google Scholar]
- Walker L. A.; Shiang J. J.; Anderson N. A.; Pullen S. H.; Sension R. J. (1998) J. Am. Chem. Soc. 120, 7286–7292. [Google Scholar]
- Hogenkamp H. P. C.; Ladd J. N.; Barker H. A. (1962) J. Biol. Chem. 237, 1950–1952. [PubMed] [Google Scholar]
- Hogenkamp H. P. C. (1963) J. Biol. Chem. 238, 477–480. [Google Scholar]
- Schwartz P. A.; Frey P. A. (2007) Biochemistry 46, 7284–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law P. Y.; Wood J. M. (1973) Biochim. Biophys. Acta 331, 451–454. [DOI] [PubMed] [Google Scholar]
- Stich T. A.; Buan N. R.; Brunold T. C. (2004) J. Am. Chem. Soc. 126, 9735–9749. [DOI] [PubMed] [Google Scholar]
- Huhta M. S.; Chen H. P.; Hemann C.; Hille C. R.; Marsh E. N. G. (2001) Biochem. J. 355, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauzer G. N.; Sibert J. W. (1970) J. Am. Chem. Soc. 92, 1022–1030. [DOI] [PubMed] [Google Scholar]
- Schrauzer G. N.; Stadlbauer E. A. (1975) Bioinorg. Chem. 4, 185–198. [DOI] [PubMed] [Google Scholar]
- Katz R. N.; Vickrey T. M.; Schrauzer G. N. (1976) Angew. Chem., Int. Ed. 15, 542–543. [DOI] [PubMed] [Google Scholar]
- Krouwer J. S.; Schultz R. M.; Babior B. M. (1978) J. Biol. Chem. 253, 1041–1047. [PubMed] [Google Scholar]
- Sandala G. M.; Smith D. M.; Radom L. (2005) J. Am. Chem. Soc. 127, 8856–8864. [DOI] [PubMed] [Google Scholar]
- Garr C. D.; Finke R. G. (1993) Inorg. Chem. 32, 4414–4421. [Google Scholar]
- Garr C. D.; Finke R. G. (1992) J. Am. Chem. Soc. 114, 10440–10445. [Google Scholar]
- Jacobsen E. N.; Bergman R. G. (1985) J. Am. Chem. Soc. 107, 2023–2032. [Google Scholar]
- Sweany R. L.; Halpern J. (1977) J. Am. Chem. Soc. 99, 8335–8337. [Google Scholar]
- Baldwin D. A.; Betterton E. A.; Chemaly S. M.; Pratt J. M. (1985) J. Chem. Soc., Dalton Trans. 1613–1618. [Google Scholar]
- Kim S. H.; Chen H. L.; Feilchenfeld N.; Halpern J. (1988) J. Am. Chem. Soc. 110, 3120–3126. [Google Scholar]
- Ng F. T. T.; Rempel G. L.; Mancuso C.; Halpern J. (1990) Organometallics 9, 2762–2772. [Google Scholar]
- Schrauzer G. N.; Holland R. J. (1971) J. Am. Chem. Soc. 93, 4060–4062. [DOI] [PubMed] [Google Scholar]
- Grate J. H.; Schrauzer G. N. (1979) J. Am. Chem. Soc. 101, 4601–4611. [Google Scholar]
- Lacy D. C.; Roberts G. M.; Peters J. C. (2015) J. Am. Chem. Soc. 137, 4860–4864. [DOI] [PubMed] [Google Scholar]
- Marinescu S. C.; Winkler J. R.; Gray H. B. (2012) Proc. Natl. Acad. Sci. U.S.A. 109, 15127–15131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett J. T.; Drennan C. L.; Amaratunga M.; Scholten J. D.; Ludwig M. L.; Matthews R. G. (1996) Bioorg. Med. Chem. 4, 1237–1246. [DOI] [PubMed] [Google Scholar]
- Sension R. J.; Cole A. G.; Harris A. D.; Fox C. C.; Woodbury N. W.; Lin S.; Marsh E. N. G. (2004) J. Am. Chem. Soc. 126, 1598–1599. [DOI] [PubMed] [Google Scholar]
- Sension R. J.; Harris D. A.; Stickrath A.; Cole A. G.; Fox C. C.; Marsh E. N. G. (2005) J. Phys. Chem. B 109, 18146–18152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.