

Abstract
The existence of N-formylated sugars in the O-antigens of Gram-negative bacteria has been known since the middle 1980s, but only recently have the biosynthetic pathways for their production been reported. In these pathways, glucose-1-phosphate is first activated by attachment to a dTMP moiety. This step is followed by a dehydration reaction and an amination. The last step in these pathways is catalyzed by N-formyltransferases that utilize N10-formyltetrahydrofolate as the carbon source. Here we describe the three-dimensional structure of one of these N-formyltransferases, namely VioF from Providencia alcalifaciens O30. Specifically, this enzyme catalyzes the conversion of dTDP-4-amino-4,6-dideoxyglucose (dTDP-Qui4N) to dTDP-4,6-dideoxy-4-formamido-d-glucose (dTDP-Qui4NFo). For this analysis, the structure of VioF was solved to 1.9 Å resolution in both its apoform and in complex with tetrahydrofolate and dTDP-Qui4N. The crystals used in the investigation belonged to the space group R32 and demonstrated reticular merohedral twinning. The overall catalytic core of the VioF subunit is characterized by a six stranded mixed β-sheet flanked on one side by three α-helices and on the other side by mostly random coil. This N-terminal domain is followed by an α-helix and a β-hairpin that form the subunit:subunit interface. The active site of the enzyme is shallow and solvent-exposed. Notably, the pyranosyl moiety of dTDP-Qui4N is positioned into the active site by only one hydrogen bond provided by Lys 77. Comparison of the VioF model to that of a previously determined N-formyltransferase suggests that substrate specificity is determined by interactions between the protein and the pyrophosphoryl group of the dTDP-sugar substrate.
Keywords: N-formyltransferase; lipopolysaccharide; 4,6-dideoxy-4-formamido-d-glucose; O-antigen; protein structure; bacterial sugar biosynthesis; formylated sugars; Providencia alcalifaciens; VioF
Introduction
Gram-negative bacteria belonging to the genus Providencia can cause acute infections of the human urinary and gastrointestinal tracts, especially in immunocompromised individuals.1 These opportunistic pathogens, often found in polluted waters or sewage, have been isolated from a wide range of organisms including chickens, dogs, and cows. Although human infections by these bacteria are relatively rare and typically nosocomial, there have been recent reports linking Providencia exposure to human pericarditis,2 meningitis,3 endocarditis,4 and food poisoning outbreaks.5 Importantly, many Providencia species are becoming resistant to various types of antibiotics including the tetracyclines, the penicillins, and the cephalosporins.6
To date, the genus Providencia consists of eight species, which differ significantly in the carbohydrate compositions of their O-antigens.1 The O-antigens are the outermost components of the bacterial lipopolysaccharides that are highly variable from species to species. Some contain quite unusual sugars such as l-quinovose, l-colitose, d-galacturonamide, and 4,6-dideoxy-4-formamido-d-glucose (Qui4NFo), among others.1,7 These carbohydrates are synthesized in vivo as nucleotide-linked sugars before being transferred to the growing O-antigens chains. Importantly, the O-antigens have been implicated in bacterial virulence by providing protection against the host immune response.8
The genes encoding the enzymes required for the biosynthesis of these unique sugars are typically found within clusters. Recently the DNA sequence of the O-antigen gene cluster from Providencia alcalifaciens O30 was reported, and the genes encoding enzymes required for dTDP-Qui4NFo biosynthesis were identified (Scheme 1).9 In the first step of the pathway, glucose-1-phosphate is attached to a dTMP moiety to yield dTDP-glucose. The next two steps involve a dehydration and an amination to form dTDP-4-amino-4,6-dideoxyglucose (dTDP-Qui4N). From the study of Liu et al.,9 Orf1 was demonstrated to encode a protein, referred to as VioF, which catalyzes the last step in the dTDP-Qui4NFo biosynthetic pathway, namely the N-formylation of dTDP-Qui4N to yield dTDP-Qui4NFo (Scheme 1). In this elegant investigation, it was further established that VioF requires N10-formyltetrahydrofolate (N10-formyl-THF) for activity (Scheme 2).
Scheme 1.
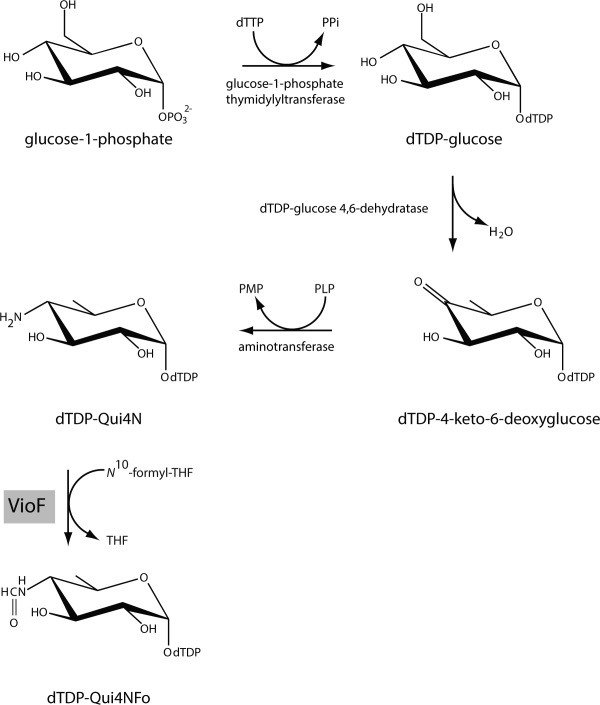
Predicted pathway for the production of dTDP-Qui4NFo.
Scheme 2.
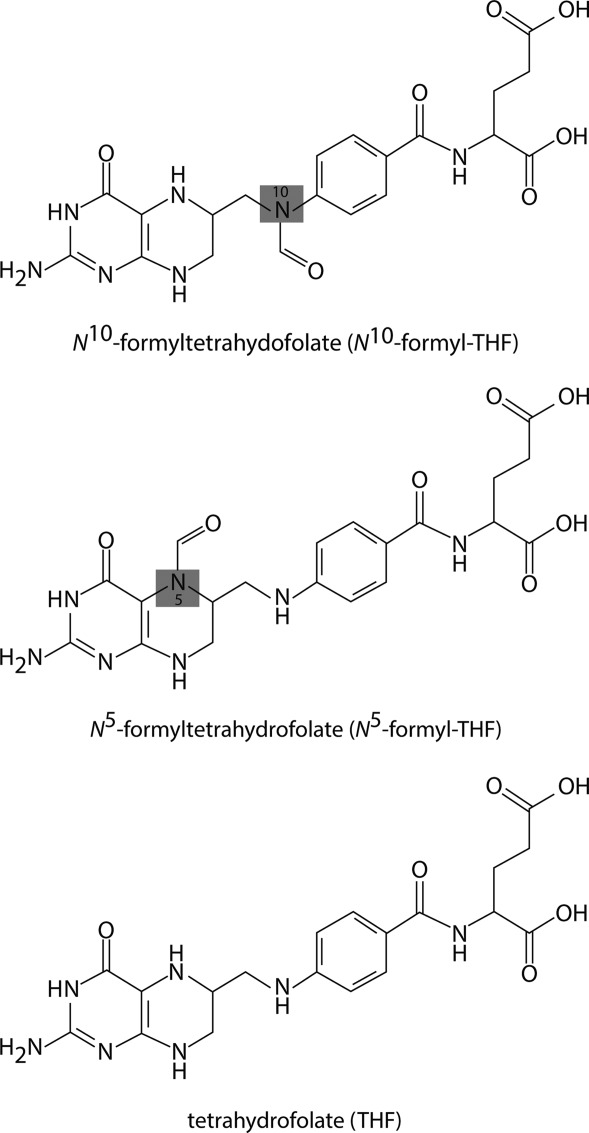
Structure of tetrahydrofolate and related derivatives.
Within the last several years, our laboratory has reported on the structures of three N-formyltransferases that function on nucleotide-linked sugars.10–12 One of them, WbtJ from Francisella tularensis (the causative agent of rabbit fever), catalyzes the same reaction as VioF (Scheme 1).11 Whereas it was possible to determine the structure of WbtJ in the presence of a dTDP-sugar ligand, it was never possible to solve its structure with bound dTDP-sugar and cofactor. Another N-formyltransferase recently investigated in our laboratory was WlaRD from Campylobacter jejuni. In contrast to that observed for VioF and WbtJ, WlaRD catalyzes an N-formylation reaction at the C-3′ rather than the C-4′ amino group to yield dTDP-3,6-dideoxy-3-formamido-d-glucose or dTDP-Qui3NFo (Scheme 3). Seven different X-ray structures of WlaRD, in complexes with various dTDP-linked sugars and tetrahydrofolate cofactors, were determined to resolutions of 1.9 Å or better.
Scheme 3.
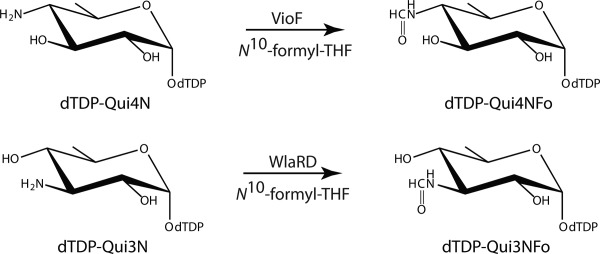
Reactions catalyzed by VioF and WlaRD.
Curious as to the manner in which the C-3′ versus C-4′ amino N-formyltransferases accommodate their respective substrates and cofactors into the active site pockets, we initiated an X-ray crystallographic analysis of VioF with the goal of capturing a “ternary complex” of the enzyme. Here we report the high-resolution structures of both the apoform of VioF and the enzyme with bound tetrahydrofolate (Scheme 2) and dTDP-Qui4N. Comparisons of the molecular architectures of VioF, WbtJ, and WlaRD provide new details with respect to the substrate specificities of these fascinating enzymes.
Results and Discussion
Overall structure of VioF
The crystals utilized in this investigation contained a dimer in the asymmetric unit and diffracted to a nominal resolution of 1.9 Å. They grew as six-sided stellate rods suggesting the possibility of twinning (Fig. 4). Their diffraction patterns appeared normal, however, and indicated that they belonged to a hexagonal/trigonal/rhombohedral space group. Integration of the X-ray data sets under any of these types of space groups showed centrosymmetric diffraction intensities with pseudo obverse/reverse systematic absences. Given this, it became clear that the crystals were highly twinned, thereby making this structural analysis especially problematic as described in the Materials and Methods. Once this was discovered, the overlapping lattices became obvious as can be seen in the sample diffraction pattern presented in Figure 5.
Figure 1.
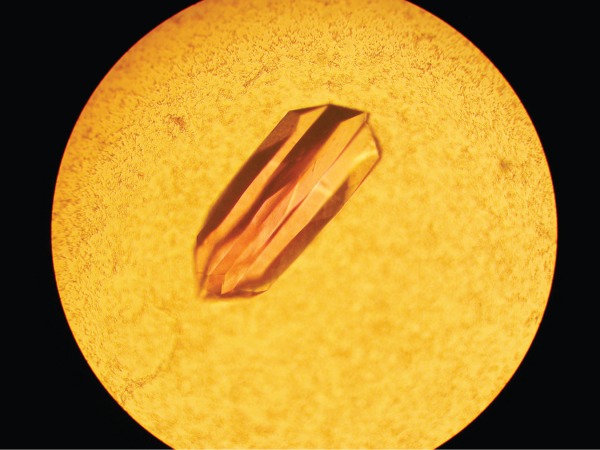
Crystal of VioF. All of the VioF crystals grew as six-sided stellate rods. The crystals demonstrated reticular merohedral twinning.
Figure 2.

Diffraction from a VioF crystal. Shown in (a) is a diffraction pattern collected from a VioF crystal. At first glance, it appears like that from a typical single crystal. Upon X-ray data processing it became clear that the crystal was twinned. Shown in (b) are the predicted overlays for the diffraction patterns with domains 1 and 2 circled in red and yellow, respectively. A RLATT representation of the Bragg reflections harvested for indexing is displayed in (c). This view illustrates the manner in which the two lattices intersect.
Regardless of the technical difficulties, the structure of the ternary complex of VioF with tetrahydrofolate (THF) and dTDP-Qui4N was ultimately solved and refined to an Roverall of 18.9% and an Rfree of 24.4%. Shown in Figure 6(a) is a ribbon representation of the dimer. It has overall dimensions of ∼70 Å x 73 Å x 102 Å and a total buried surface area of ∼3100 Å2. The subunit:subunit interface is formed primarily by an extended stretch of polypeptide chain (Leu 199 to Ser 204), an α-helix (Leu 205 to Ala 214), and a β-hairpin motif (Ala 223 to Ser 240). This interface is similar to that observed for WbtJ.11 The N-terminal domain harbors the active site and is dominated by a six-stranded mixed β-sheet flanked on one side by three α-helices and on the other side by mostly random coil. In light of the fact that the α-carbons for the two subunits of the dimer superimpose with a root-mean-square deviation of 0.21 Å, the following discussion refers only to the first monomer in the X-ray coordinate file.
Figure 3.
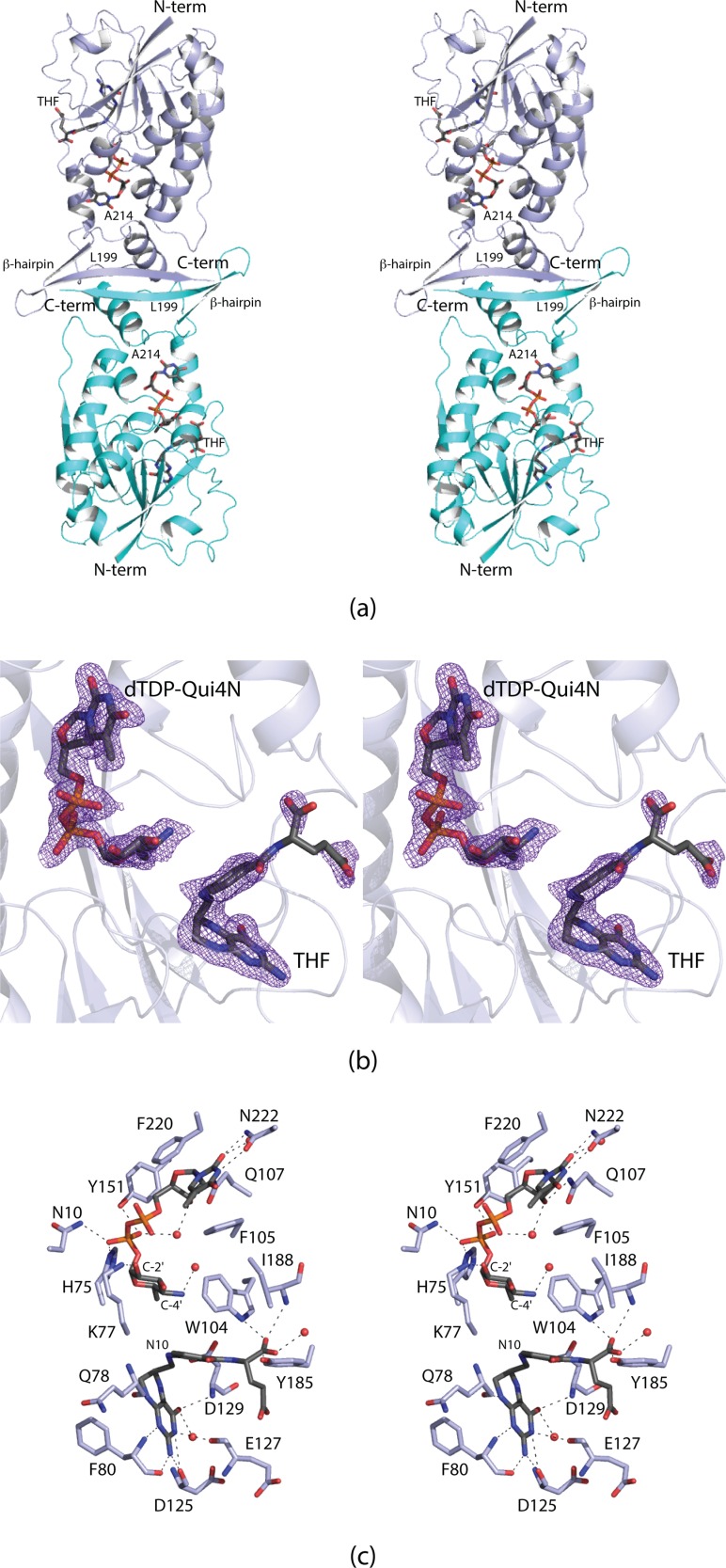
Structure of VioF. Shown in (a) is a ribbon representation of the VioF dimer with subunits 1 and 2 displayed in light blue and cyan, respectively. The ligands are drawn in stick representations. The electron density corresponding to the bound ligands in subunit 1 is shown in (b). The electron density map was calculated with coefficients of the form Fo − Fc, where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. The map was contoured at 3σ. The electron density for the glutamate portion of the cofactor is weak most likely due to the fact that there are few interactions between it and the protein. A close-up view of the active site is provided in (c). Distances within 3.2 Å between the protein and ligand atoms are indicated by the dashed lines. Ordered water molecules are depicted as red spheres. All figures were prepared with the software package PyMOL.13
For preparation of the ternary complex described here, crystals of the apoenzyme were soaked in solutions containing 5 mM dTDP-Qui4N and 5 mM N10-formyl-THF, with the goal of trapping the products, dTDP-Qui4NFo and THF, in the active site. Shown in Figure 6(b) are the electron densities corresponding to the ligands, which unambiguously reveal the presence of dTDP-Qui4N and THF. Given that N10-formyl-THF is not particularly stable, the soaking conditions utilized most likely promoted the loss of the formyl group in a noncatalytic fashion. Regardless, the structure represents the first model of a ternary complex for any sugar N-formyltransferase that functions on C-4′ amino sugars.
The active site of VioF, as displayed in Figure 6(c), is quite shallow and close to the surface of the molecule. The side chains of Phe 105 and Phe 220 form parallel stacking interactions with the thymine ring of dTDP-Qui4N, whereas the side chain of Asn 222 provides hydrogen-bonding interactions. The pyrophosphoryl moiety of dTDP-Qui4N is anchored into the active site by the side chains of Asn 10, His 75, and Tyr 151. The side chain of Lys 77 mediates the only specific interaction occurring between the pyranosyl moiety of the dTDP-sugar and the protein. The bicyclic ring of THF is anchored into the active site by hydrogen-bonding interactions with protein backbone atoms (Gln 78, Phe 80, Asp 125, Glu 127, and Asp 129). The distance between N-10 of THF and the C-4′ amino group of the sugar is 4.8 Å. In WlaRD, as discussed below, this distance is shorter at 4.0 Å.
In order to assess the changes in conformation that occur upon substrate/cofactor binding, the three-dimensional structure of the apoenzyme was also determined to 1.89 Å resolution (Roverall of 18.4%, Rfree of 23.8%). The α-carbons for the models of the apoenzyme and the ternary complex superimpose with a root-mean-square deviation of 0.30 Å thereby indicating no major structural perturbations. In the apoenzyme structure, water molecules simply line the cavity that is occupied by the dTDP-sugar and the cofactor in the ternary complex. The only notable difference between the two models occurs in the loop defined by Gly 218 to Asn 222 (Fig. 7). The aromatic side chain of Phe 220, which forms a stacking interaction with the thymine ring of the dTDP-sugar, swings out of the binding pocket in the apoenzyme structure.
Figure 4.
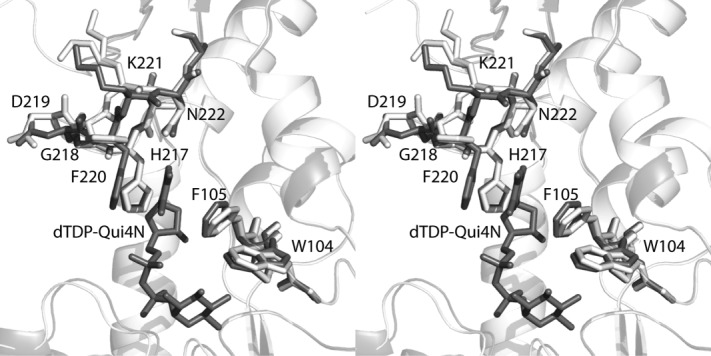
Comparison of the VioF apoenzyme and the ternary complex structures. A close-up view of the active sites is shown with the models of the apoenzyme and the ternary complex highlighted in white and gray, respectively.
Comparison of VioF to WbtJ
As discussed in the Introduction, the first reported X-ray model for an N-formyltransferase that functions on C-4′ amino sugars was that of WbtJ.11 Unfortunately, all attempts to trap a folate analog in the active site of WbtJ were unsuccessful, and thus the molecular architecture of the ternary complex was never defined.11 Given that the amino acid sequence identities and similarities between VioF and WbtJ are 48% and 65%, respectively, it is not surprising that the α-carbons for the two proteins superimpose with a root-mean-square deviation of 0.90 Å. As such, the VioF/dTDP-sugar/THF complex structure described here provides information regarding the manner in which a folate-derived cofactor might bind in the active site of WbtJ as well.
A superposition of the WbtJ and VioF active sites is shown in Figure 8(a). As can be seen, all of the residues responsible for anchoring the THF cofactor into the VioF active site are conserved in WbtJ. The only minor change is the substitution of Phe 220 in VioF with a tyrosine residue in WbtJ. A structural alignment of VioF and WbtJ with other N-formyltransferases such as Escherichia coli ArnA,14 E. coli l-methionyl-tRNA N-formyltransferase,15 and E. coli glycinamide ribonucleotide transformylase16 reveals three conserved residues, Asn 92, His 94, and Asp 129 (VioF numbering) that are strictly conserved. The positions of these residues in VioF and WbtJ are shown in Figure 8(a,b). These are thought to play key roles in catalysis with the conserved histidine functioning as an active site base. In WbtJ, the distance between the C-4′ amino group and the imidazole side chain of His 92 is ∼7.9 Å. Due to this rather long distance, it was originally suggested that the dTDP-sugar adopted a non-productive conformation in the crystalline enzyme due to the lack of a bound folate derivative.11 This conclusion, in retrospect, was wrong. As is obvious from Figure 8(a,b), the dTDP-sugar, when bound to VioF in a ternary complex, adopts a similar curved conformation as that observed in WbtJ. In VioF, the C-4′ amino group and the catalytic histidine are still too far apart at 7.1 Å. Clearly in a productive VioF/dTDP-Qui4N/N10-formyl-THF complex, the distance between the C-4′ amino group and the catalytic histidine must be considerably shorter as discussed below. The reported catalytic efficiencies of VioF (1.2 × 103 M−1 s−1) and WbtJ (1.7 × 104 M−1 s−1) differ by approximately an order of magnitude.9,11
Figure 5.
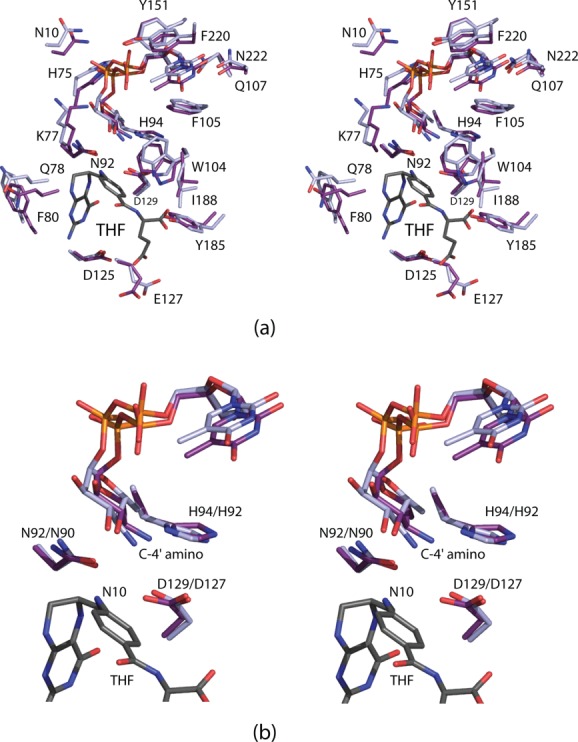
Comparison of the active sites for VioF and WbtJ. The THF cofactor, as observed binding in the VioF active site, is colored in gray (a). Those residues belonging to VioF are presented in violet whereas those belonging to WbtJ are displayed in light blue. A close-up view of the region surrounding the conserved catalytic triad is presented in (b) with the same coloring scheme as described in (a). The first and second numbers in the amino acid labels correspond to VioF and WbtJ, respectively.
Comparison of VioF to WlaRD
WlaRD from C. jejuni catalyzes an N-formylation at the C-3′ amino group (Scheme 3), and has a catalytic efficiency similar to that of WbtJ (1.1 × 104 M−1 s−1).10 A superposition of the ribbon representations for the VioF and WlaRD subunits is presented in Figure 9(a). The two models correspond with a root-mean-square deviation of 1.6 Å for 190 structurally equivalent α-carbons. The core catalytic domains, which consist of ∼220 amino acid residues, adopt similar molecular architectures. Not unexpectedly, the amino acid sequence identities and similarities between the VioF and WlaRD catalytic cores are ∼24% and 43%, respectively. The structures of the two enzymes diverge, however, around Asn 222 (VioF numbering) whereby the longer polypeptide chain of WlaRD folds into a four-stranded antiparallel β-sheet. As a consequence, the dimeric quaternary structures of the two enzymes are decidedly different.
Figure 6.
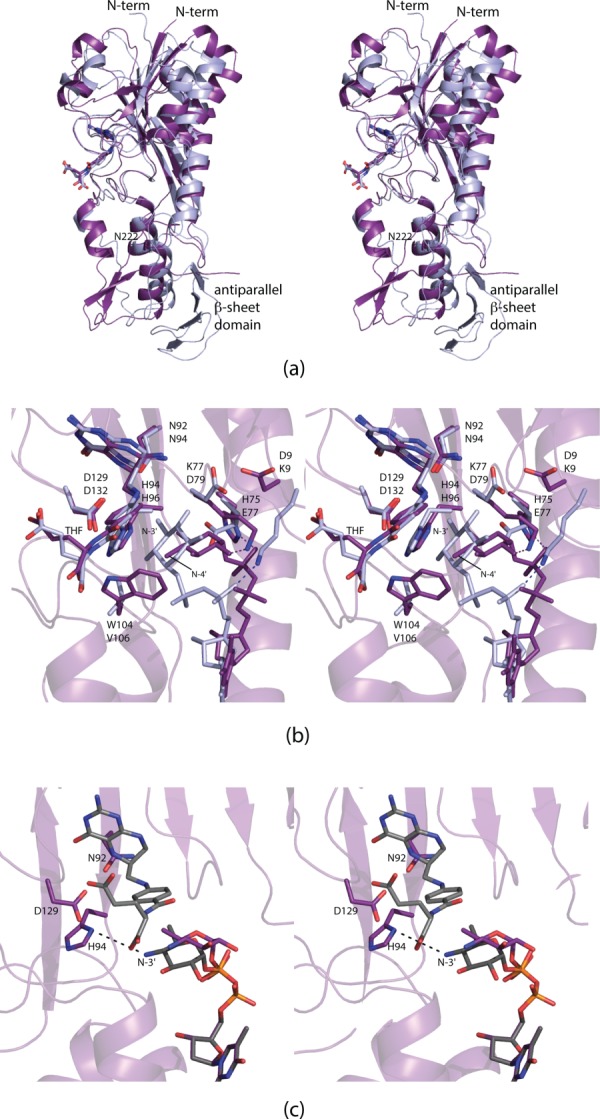
Comparison of VioF with WlaRD. A superposition of the ribbon representations for the VioF (violet) and the WlaRD (light blue) subunits is presented in (a). A close-up view of their active sites is shown in (b). Note the close correspondence in the positions of the THF cofactors and the catalytic triads. It is the pyranosyl moieties of the dTDP-sugar substrates that assume markedly different locations within the active site regions. Simple rotations about the β-phosphoryl group of the dTDP-Qui4N ligand can position the C-4' amino group of the sugar to within 4.5 Å of His 94, the presumed catalytic base. This is shown in (c) where the observed conformation of the substrate pyranosyl group is highlighted in purple bonds and the “model” is displayed in gray bonds.
A superposition of the VioF and WlaRD active sites is shown in Figure 9(b). The positions of the conserved catalytic triads (Asn 92, His 94, and Asp 129) as well as the THF cofactors are, within experimental error, identical. It is the orientations of the dTDP-sugars that differ considerably between VioF and WlaRD. In both VioF and WbtJ, which function on C-4′ amino groups, there is a conserved tryptophan (Trp 104) that occludes the active site in such a manner that the dTDP-sugar substrate could never adopt the orientation observed in WlaRD [Fig. 9(b)]. There are two additional and significant changes in the active site compositions of these two enzymes. In VioF, the pyranosyl group of the dTDP-sugar is anchored into the active site by Lys 77 [Figs. 6(c) and 9(b)]. This residue, in WlaRD, is replaced with Asp 79. Likewise, in WlaRD, the side chain of Lys 9 forms an electrostatic interaction with a phosphoryl oxygen of the dTDP-sugar. The corresponding residue in VioF is Asp 9. These three differences likely play a key role in determining substrate specificities by forcing the pyrophosphoryl groups of the substrates to adopt different dihedral angles, thereby leading to different orientations of the pyranosyl groups in the VioF and WlaRD active sites.
In the WlaRD/THF/dTDP-Qui3N abortive complex, the C-3′ amino group lies within 4.3 Å from the catalytic histidine (His 96). As discussed above, in VioF, the C-4′ amino group is located at 7.1 Å from His 94, which is too far for the imidazole side chain to function as a catalytic base. The active site of VioF is quite open, however, with little interactions occurring between the Qui4N moiety and the protein. Simple rotations about the β-phosphoryl group of the dTDP-sugar can position the C-4′ amino group to within ∼4.5 Å of His 94 as indicated in Figure 9(c).
Conclusions
Until recently there had been a paucity of structural information on those enzymes responsible for the formylation of nucleotide-linked sugars, although the existence of such carbohydrates was documented back in 1985.17 Indeed, the biological role of sugar N-formylation is still not well understood. It is intriguing, however, that a search of the amino acid sequence databases reveals that homologs of VioF, WbtJ, and WlaRD are also found in bacteria such as the plant pathogens Pantoea ananatis and Pseudomonas syringae. Strikingly, P. ananatis, though primarily a plant pathogen, has been shown to infect humans, and has also been found as a contaminant of aviation jet fuel tanks.18
Before our investigations on WlaRD, WbtJ, and now VioF, only the structure of one sugar N-formyltransferase had been reported, namely ArnA from E. coli.14 ArnA is a bifunctional enzyme with the N-terminal domain catalyzing the N-formylation of a C-4′ amino group and the C-terminal domain responsible for an oxidative decarboxylation reaction.19 Although informative, the structure of ArnA was never solved in the presence of a nucleotide-linked sugar.19,20 The X-ray analysis of VioF presented here provides a new three-dimensional scaffold to more fully understand the substrate specificities of the C-4′ versus the C-3′ amino sugar N-formyltransferases. In both types of enzymes, there are few interactions between the amino acid side chains of the proteins and the pyranosyl groups of the substrates. Rather it is the residues that line the pyrophosphoryl binding pockets that apparently play a role in substrate specificity. It will be of interest to compare the structures of additional enzymes involved in the production of N-formylated sugars. For example, 4,6-dideoxy-4-formamido-d-mannopyranosyl residues have been observed on the O-antigens of Brucella abortus and Brucella melitensis,21 and 2-formamido-2-deoxy-d-galacturonic acid has been reported in Pseudomonas aeruginosa O4.17 Investigations on the N-formyltransferases involved in the biosynthesis of these novel sugars are presently underway.
Materials and Methods
Cloning of the vioF gene
A synthetic gene encoding for VioF from P. alcalifaciens serogroup O30 was synthesized by DNA2.0 using E. coli optimized codons and was employed as a template for subsequent cloning experiments. New constructs were prepared via PCR using Platinum Pfx DNA polymerase (Invitrogen) and primers that incorporated NdeI and XhoI restriction sites. The PCR product was digested with NdeI and XhoI and ligated into pETDuet1, which had been previously modified to incorporate a C-terminal polyhistidine tag with the sequence SAHHHHHH after the XhoI restriction site (unpublished), resulting in LESAHHHHHH being added to the C-terminus of the protein. Cloning at the N-terminus was initiated at the second methionine in the deposited DNA sequence since homologous enzyme sequences all start at this position. Thus residue 1 in the structure reported here corresponds to residue 9 in the UniProt database (entry M9P0Q2).
Protein expression and purification
The pETDuet1-vioF plasmid was utilized to transform Rosetta2(DE3) E. coli cells (Novagen). The cultures were grown in lysogeny broth supplemented with ampicillin and chloramphenicol at 37°C with shaking until an optical density of 0.8 was reached at 600 nm. The flasks were cooled in an ice bath, and the cells were induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and allowed to express protein at 16°C for 24 h.
The cells were harvested by centrifugation and disrupted by sonication on ice. The lysate was cleared by centrifugation, and VioF was purified with Ni-NTA resin (Qiagen) according to the manufacturer's instructions. The protein was dialyzed against 10 mM Tris-HCl (pH 8.0) and 200 mM NaCl and concentrated to 20 mg/mL based on an extinction coefficient of 0.97 (mg/mL)−1 cm−1.
Crystallization and structural analysis of the apoenzyme
Crystallization conditions were surveyed by the hanging drop method of vapor diffusion using a laboratory-based sparse matrix screen at both room temperature and 4°C. X-ray diffraction quality crystals of the apoenzyme were subsequently prepared from precipitant solutions containing 8 to 10% poly(ethylene glycol) 8000, 1M tetramethylammonium chloride, and 100 mM Hepes (pH 7.5) at 4°C. As noted in the Discussion and Results, the crystals appeared as six-sided stellate rods (Fig. 4).
For X-ray data collection, the crystals were transferred to a cryo-protectant solution containing 15% poly(ethylene glycol) 8000, 1.3M tetramethylammonium chloride, 200 mM NaCl, 10% ethylene glycol, and 100 mM Hepes (pH 7.5). X-ray data were collected at 100 K with a Bruker AXS Platinum 135 CCD detector controlled by the Proteum software suite (Bruker AXS Inc.). The X-ray source was Cu-Kα radiation from a Rigaku RU200 X-ray generator equipped with Montel optics and operated at 50 kV and 90 mA.
Analysis with XPREP (Bruker AXS (2014) PROTEUM2, Version 2014, Bruker AXS Inc., Madison, WI) indicated that the crystals were obverse/reverse twins.22 The mean intensity and mean intensity-to-sigma ratios were both significantly lower for the reflection conditions −h + k + l = 3n and h − k + l = 3n in comparison with all other reflections. In addition, only every third reflection was present in the reciprocal space plot for layers l = 3n and in all other layers one third of the reflections were missing. The absence of these reflections had a noticeable effect on the intensity distribution with the data exhibiting a mean |E2 – 1| value much higher (1.101) than expected for a noncentrosymmetric space group. The obverse/reverse test in XPREP indicated that the fractional contribution of the second domain was 0.48. The space group was thus determined to be R32 with a twofold axis parallel to the threefold [001] as the twin law relating the two domains. Reflection arrays for the two domains were separated using the program RLATT (Bruker AXS (2014). PROTEUM2, Version 2014, Bruker AXS Inc., Madison, WI) and indexed to produce the corresponding rhombohedral cells. The data were integrated with SAINT (Bruker AXS (2014). PROTEUM2, Version 2014, Bruker AXS Inc., Madison, WI) using the orientation information for both domains. The raw intensities were placed on a common scale and detwinned using TWINABS (Bruker AXS (2014). PROTEUM2, Version 2014, Bruker AXS Inc., Madison, WI). The unit cell dimensions were a = b = 132.0 Å and c = 161.4 Å. There was one dimer per asymmetric unit. Relevant X-ray data collection statistics are listed in Table1.
Table 1.
X-ray Data Collection Statistics
| dTDP-Qui4N/THF complex | Apoenzyme | |
|---|---|---|
| Resolution limits (Å) | 50.0–1.90 (2.0–1.90) | 50.0–1.89 (1.94-1.89)a |
| No. independent reflections | 42,792 (6144) | 43,314 (3060) |
| Completeness (%) | 99.9 (99.1) | 99.8 (96.9) |
| Redundancy | 5.3 (4.5) | 6.8 (4.7) |
| Avg I/Avg σ(I) | 14.3 (2.4) | 17.0 (3.4) |
| Rsym (%)b | 5.3 (23.7) | 6.7 (37.9) |
Statistics for the highest resolution bin.
Rsym = 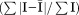 × 100.
× 100.
The structure of the apoenzyme was solved via molecular replacement with the software package PHASER23 and using as a search model the coordinates for WbtJ.11 Iterative rounds of model-building with COOT24,25 and refinement with REFMAC26 reduced the Rwork and Rfree to 18.1% and 23.8%, respectively, from 50 to 1.89 Å resolution. Model refinement statistics are listed in Table2.
Table 2.
Refinement Statistics
| dTDP-Qui4N/THF complex | Apoenzyme | |
|---|---|---|
| Resolution limits (Å) | 50–1.90 | 50–1.89 |
| aR-factor (overall)%/no. reflections | 18.9/42,766 | 18.4/43,313 |
| R-factor (working)%/no. reflections | 18.7/40,605 | 18.1/41,131 |
| R-factor (free)%/no. reflections | 24.4/2161 | 23.8/2182 |
| No. protein atoms | 3959 | 3971 |
| No. heteroatoms | 587 | 531 |
| Average B values | ||
| Protein atoms (Å2) | 21.8 | 22.0 |
| Ligand (Å2) | 31.3 | N/A |
| Solvent (Å2) | 26.8 | 29.5 |
| Weighted RMS deviations from ideality | ||
| Bond lengths (Å) | 0.012 | 0.011 |
| Bond angles (°) | 2.12 | 2.08 |
| Planar groups (Å) | 0.010 | 0.009 |
| Ramachandran regions (%)b | ||
| Most favored | 92.6 | 91.4 |
| Additionally allowed | 7.4 | 8.6 |
| Generously allowed | 0 | 0 |
R-factor = (Σ|Fo − Fc|/Σ|Fo|) × 100 where Fo is the observed structure-factor amplitude and Fc. is the calculated structure-factor amplitude.
Distribution of Ramachandran angles according to PROCHECK.27
Structural analysis of VioF ternary complex
Crystals of the apoenzyme were transferred to solutions containing 12% poly(ethylene glycol) 8000, 1M tetramethylammonium chloride, 200 mM NaCl, 5 mM dTDP-Qui4N, 5 mM N10-formyl-THF, and 100 mM Hepes (pH 7.5) for 2 days. The crystals were then transferred to a cryo-protectant solution composed of 15% poly(ethylene glycol) 8000, 1.3M tetramethylammonium chloride, 200 mM NaCl, 5 mM dTDP-Qui4N, 5 mM N10-formyl-THF, 10% ethylene glycol, and 100 mM Hepes (pH 7.5). Data were collected and processed as for the apoenzyme structure (relevant X-ray data collection statistics are listed in Table1.). The structure was solved via Difference Fourier techniques using the apoenzyme structure as a starting model. Iterative rounds of model-building with COOT24,25 and refinement with REFMAC26 reduced the Rwork and Rfree to 18.7% and 24.4%, respectively, from 50 to 1.90 Å resolution. Model refinement statistics are listed in Table2. The dTDP-Qui4N and N10-formyl-THF ligands were prepared as previously described.11,28
Acknowledgments
The authors thank both reviewers for their insightful comments. X-ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, N. J. (accession no. 4YFV and 4YFY).
Glossary
- dTDP
thymidine diphosphate
- dTDP-Qui4N
dTDP-4-amino-4,6-dideoxy-D-glucose
- dTDP-Qui4NFo
dTDP-4,6-dideoxy-4-formamido-d-glucose
- dTMP
thymidine monophosphate
- Hepes
N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
- Ni-NTA
Ni nitrilotriacetic acid
- PCR
polymerase chain reaction
- THF
tetrahydrofolate
- Tris
tris-(hydroxymethyl)aminomethane.
References
- Ovchinnikova OG, Rozalski A, Liu B, Knirel YA. O-antigens of bacteria of the genus Providencia: structure, serology, genetics, and biosynthesis. Biochemistry. 2013;78:798–817. doi: 10.1134/S0006297913070110. [DOI] [PubMed] [Google Scholar]
- Simon C, Dieli M, Brucato A, Pedrotti P, Brambilla P, Curri SF, Senni M, Pericotti S, Suter F, Ferrazzi P. Images in cardiovascular medicine. Bacterial pericarditis due to Providencia stuartii: an atypical case of relapsing pericarditis. Circulation. 2010;122:e401–e403. doi: 10.1161/CIRCULATIONAHA.110.943118. [DOI] [PubMed] [Google Scholar]
- Sipahi OR, Bardak-Ozcem S, Ozgiray E, Aydemir S, Yurtseven T, Yamazhan T, Tasbakan M, Ulusoy S. Meningitis due to Providencia stuartii. J Clin Microbiol. 2010;48:4667–4668. doi: 10.1128/JCM.01349-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krake PR, Tandon N. Infective endocarditis due to Providenca stuartii. South Med J. 2004;97:1022–1023. doi: 10.1097/01.smj.0000141308.19657.ba. [DOI] [PubMed] [Google Scholar]
- Murata T, Iida T, Shiomi Y, Tagomori K, Akeda Y, Yanagihara I, Mushiake S, Ishiguro F, Honda T. A large outbreak of foodborne infection attributed to Providencia alcalifaciens. J Infect Dis. 2001;184:1050–1055. doi: 10.1086/323458. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang J, Hao H, Qiu L, Liu H, Chen S, Dang R, Yang Z. Pathogenic Providencia alcalifaciens strain that causes fatal hemorrhagic pneumonia in piglets. Curr Microbiol. 2014;68:278–284. doi: 10.1007/s00284-013-0470-y. [DOI] [PubMed] [Google Scholar]
- Kocharova NA, Ovchinnikova OG, Torzewska A, Shashkov AS, Knirel YA, Rozalski A. The structure of the O-polysaccharide from the lipopolysaccharide of Providencia alcalifaciens O30. Carbohydr Res. 2006;341:786–790. doi: 10.1016/j.carres.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Knirel YA, Valvano MA. Bacterial Lipopolysaccharides. New York: Springer-Verlag; 2011. [Google Scholar]
- Liu B, Chen M, Perepelov AV, Liu J, Ovchinnikova OG, Zhou D, Feng L, Rozalski A, Knirel YA, Wang L. Genetic analysis of the O-antigen of Providencia alcalifaciens O30 and biochemical characterization of a formyltransferase involved in the synthesis of a Qui4N derivative. Glycobiology. 2012;22:1236–1244. doi: 10.1093/glycob/cws089. [DOI] [PubMed] [Google Scholar]
- Thoden JB, Goneau MF, Gilbert M, Holden HM. Structure of a sugar N-formyltransferase from Campylobacter jejuni. Biochemistry. 2013;52:6114–6126. doi: 10.1021/bi4009006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer AL, Thoden JB, Holden HM. Three-dimensional structure of a sugar N-formyltransferase from Francisella tularensis. Protein Sci. 2014;23:273–283. doi: 10.1002/pro.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford CR, Thoden JB, Holden HM. New role for the ankyrin repeat revealed by a study of the N-formyltransferase from Providencia alcalifaciens. Biochemistry. 2015;54:631–638. doi: 10.1021/bi501539a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- Williams GJ, Breazeale SD, Raetz CR, Naismith JH. Structure and function of both domains of ArnA, a dual function decarboxylase and a formyltransferase, involved in 4-amino-4-deoxy-l-arabinose biosynthesis. J Biol Chem. 2005;280:23000–23008. doi: 10.1074/jbc.M501534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E, Blanquet S, Mechulam Y. Structure of crystalline Escherichia coli methionyl-tRNA(f)Met formyltransferase: comparison with glycinamide ribonucleotide formyltransferase. EMBO J. 1996;15:4749–4758. [PMC free article] [PubMed] [Google Scholar]
- Almassy RJ, Janson CA, Kan CC, Hostomska Z. Structures of apo and complexed Escherichia coli glycinamide ribonucleotide transformylase. Proc Natl Acad Sci USA. 1992;89:6114–6118. doi: 10.1073/pnas.89.13.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knirel YA, Vinogradov EV, Shashkov AS, Dmitriev BA, Kochetkov NK, Stanislavsky ES, Mashilova GM. Somatic antigens of Pseudomonas aeruginosa. The structure of the O-specific polysaccharide chains of lipopolysaccharides of P. aeruginosa serogroup O4 (Lanyi) and related serotype O6 (Habs) and immunotype 1 (Fisher) Eur J Biochem. 1985;150:541–550. doi: 10.1111/j.1432-1033.1985.tb09055.x. [DOI] [PubMed] [Google Scholar]
- Coutinho TA, Venter SN. Pantoea ananatis: an unconventional plant pathogen. Mol Plant Pathol. 2009;10:325–335. doi: 10.1111/j.1364-3703.2009.00542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breazeale SD, Ribeiro AA, McClerren AL, Raetz CR. A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4-amino-4-deoxy-l-arabinose. Identification and function of UDP-4-deoxy-4-formamido-l-arabinose. J Biol Chem. 2005;280:14154–14167. doi: 10.1074/jbc.M414265200. [DOI] [PubMed] [Google Scholar]
- Gatzeva-Topalova PZ, May AP, Sousa MC. Crystal structure and mechanism of the Escherichia coli ArnA (PmrI) transformylase domain. An enzyme for lipid A modification with 4-amino-4-deoxy-l-arabinose and polymyxin resistance. Biochemistry. 2005;44:5328–5338. doi: 10.1021/bi047384g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundle DR, Gerken M, Peters T. Synthesis of antigenic determinants of the Brucella A antigen, utilizing methyl 4-azido-4,6-dideoxy-alpha-d-mannopyranoside efficiently derived from d-mannose. Carbohydr Res. 1988;174:239–251. doi: 10.1016/0008-6215(88)85094-8. [DOI] [PubMed] [Google Scholar]
- Parsons S. Introduction to twinning. Acta Crystallogr D. 2003;59:1995–2003. doi: 10.1107/s0907444903017657. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- Breazeale SD, Ribeiro AA, Raetz CR. Oxidative decarboxylation of UDP-glucuronic acid in extracts of polymyxin-resistant Escherichia coli. Origin of lipid a species modified with 4-amino-4-deoxy-l-arabinose. J Biol Chem. 2002;277:2886–2896. doi: 10.1074/jbc.M109377200. [DOI] [PubMed] [Google Scholar]