

Abstract
Fungal fatty acid synthases Type I (FAS I) are up to 2.7 MDa large molecular machines composed of large multifunctional polypeptides. Half of the amino acids in fungal FAS I are involved in structural elements that are responsible for scaffolding the elaborate barrel-shaped architecture and turning fungal FAS I into highly efficient de novo producers of fatty acids. Rhodosporidium toruloides is an oleaginous fungal species and renowned for its robust conversion of carbohydrates into lipids to over 70% of its dry cell weight. Here, we use cryo-EM to determine a 7.8-Å reconstruction of its FAS I that reveals unexpected features; its novel form of splitting the multifunctional polypeptide chain into the two subunits α and β, and its duplicated ACP domains. We show that the specific distribution into α and β occurs by splitting at one of many possible sites that can be accepted by fungal FAS I. While, therefore, the specific distribution in α and β chains in R. toruloides FAS I is not correlated to increased protein activities, we also show that the duplication of ACP is an evolutionary late event and argue that duplication is beneficial for the lipid overproduction phenotype.
Keywords: mega-enzyme, multifunctional proteins, protein assembly, acyl carrier protein, biofuel
Introduction
Fatty acid synthases Type I (FAS I) are responsible for de novo fatty acid synthesis in eukaryotes and CMN group bacteria (Corynebacterium, Mycobacterium, and Nocardia). FAS I occur in MDa-sized assemblies of elaborate architecture in which fatty acids are synthesized in a machine-like fashion. FAS I are made-up of multifunctional polypeptide chains, and distinct from the “conventional” Type II FAS, which are present in most bacteria and mitochondria and provide the catalytic functions for fatty acid synthesis on separate proteins.1–3
FAS I can be classified in metazoan and microbial (fungal and bacterial). Metazoan FAS I occur as dimeric complexes of about 0.5 MDa.4 Current structural progress in the characterization of polyketide synthases Type I (PKS I) strongly suggests that the metazoan FAS I protein fold is widely distributed as an architectural blueprint in the PKS family.4–8 Microbial FAS I forms multi-oligomeric complexes of sizes between 1.9 MDa (bacterial FAS I) and 2.7 MDa (fungal). The best studied representative of microbial FAS I is the Saccharomyces cerevisiae FAS I. It has been extensively analyzed during several decades; for example, its characterization was largely responsible for awarding the Nobel prize to Lynen 50 years ago (1964, awarded jointly to Konrad Bloch and Feodor Lynen for their discoveries in cholesterol and fatty acid metabolism).9 S. cerevisiae FAS I (and Thermomyces lanuginosus FAS I) has also been recently analyzed by X-ray crystallography (Fig. 1),10–13 and the wealth of available functional data have been correlated to structural data, leading to a superior understanding of fungal Type I fatty acid synthesis.14 For an overview of the catalytic cycle of fatty acid synthesis as performed by fungal FAS I see Supporting Information Fig. S1.
Figure 1.
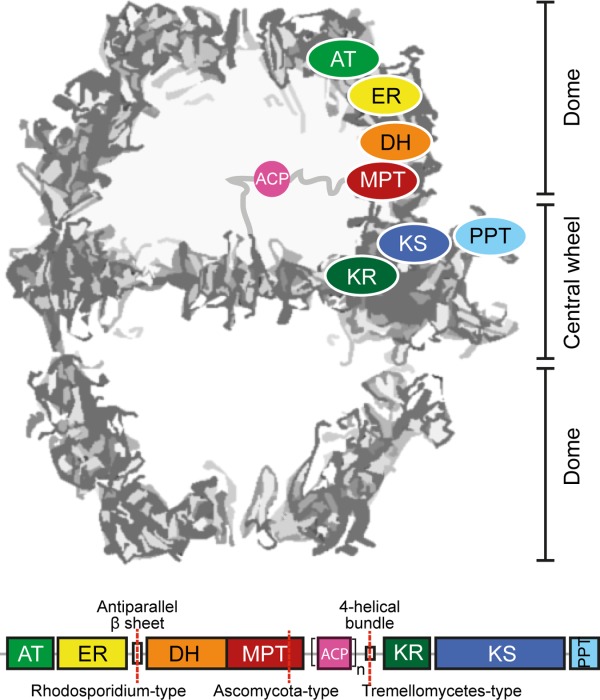
Architecture and domain organization of fungal FAS I. Fungal FAS I is an overall D3 symmetric barrel-shaped complex of homohexameric or heterodocameric (α6β6) oligomerization. For clarity of the structure representation, the protein is abstracted as a cross-section along the threefold axis. Fatty acid synthesis proceeds in two compartments, each of which is lined by three full sets of catalytic domains. The positioning of a set of domains is indicated for the upper compartment. The domains ketoacyl synthase (KS), ketoacyl reductase (KR), and phosphopantetheine transferase (PPT) comprise the central wheel part, while the other domains, acetyl-transferase (AT), enoyl reductase (ER), dehydratase (DH), and malonyl-palmitoyl-transferase (MPT) make up the dome-like structure. Fungal FAS I can be encoded by a single gene, as in the case of the group of Ustilaginomcetes, or split into two separate genes (FAS1 encoding the β-chain and FAS2 encoding the α-chain) as indicated. A mobile acyl carrier protein (ACP) as a single (n = 1) or duplicated domain (n = 2) spans the inner volume of the compartments, tethered by flexible linkers to the center of the wheel and the wall of the dome (abstracted by gray lines).
Acyl carrier protein (ACP)-mediated substrate shuttling is a key characteristic of fatty acid synthesis.3,15 ACP is a small globular protein that after post-translational modification by phosphopantetheine transferases (PPT) is able to bind substrates and intermediates via thioester formation.16 In Type I systems, ACP is part of the polypeptide chain and in fungal and bacterial FAS I located inside compartmented reaction chambers, where it delivers substrates and intermediates to the active sites of the different enzymatic domains17,18 (see Fig. 1). A peculiar structural feature that was observed in fungal FAS I is the positioning of the PPT (part of the polypeptide chain in fungal FAS I) at the perimeter of the central wheel.12 The spatial separation of PPT at the outside of the barrel from ACP in the inside has dramatic consequences for the assembly of the fungal FAS I: The post-translational activation of the ACP domain is not possible in the assembled barrel-like state, but requires an architecturally distinct preassembled state.12,14,19
We have started the biochemical and structural characterization of the Rhodosporidium toruloides FAS I because of our interest in the evolution and the assembly of FAS I. As a major event in the development of fungal FAS, single gene-encoding FAS split into two gene-encoding (FAS1 and FAS2) variants at different sites (see Fig. 1).11,14,20 In this light, R. toruloides FAS I is a topologically interesting fungal FAS I.21 It is composed of α- (FAS2 encoded) and β-chain (FAS1) with the splitting site within a β-sheet domain that connects the domains ER and the DH, representing a rare fungal FAS I variant. Further, it shows a duplicated ACP domain, which might correlate to the high oleagenic capacity of the organism. Here, we present the functional and cryo-EM structural characterization of R. toruloides FAS I. We discuss our data in light of the evolution of fungal FAS I toward the highly efficient molecular machines as they appear in the oleaginous strain R. toruloides, and we propose that the assembly of the up to 2.7 MDa large protein complexes is a robust sequential process, running through a defined sequence of events.
Results
Recombinant expression of R. toruloides FAS I
Fully assembled and functional wild-type R. toruloides FAS I was successfully expressed in Escherichia coli at a yield of more than 6 mg per liter culture. The FAS I complex was purified via a multi-step purification protocol [Fig. 2(A)]. Final size exclusion chromatography shows a monodisperse UV-absorption peak at an apparent molecular weight of about 2.7 MDa representing the α6β6 heterododecameric complex [Fig. 2(B)]. Tailing of the high oligomeric peak indicates minor impurities or protein degradation, as also observable in SDS-PAGE. In SDS-PAGE, a faint band was observed appearing with a higher apparent molecular weight than 460 kDa, which we speculated to occur from stable interactions of α- and β-chain to a higher oligomeric species that does not dissociate during sample preparation. However, variations in the strength of the denaturing conditions did not lead to varying band intensities [see Fig. 2(A)].
Figure 2.
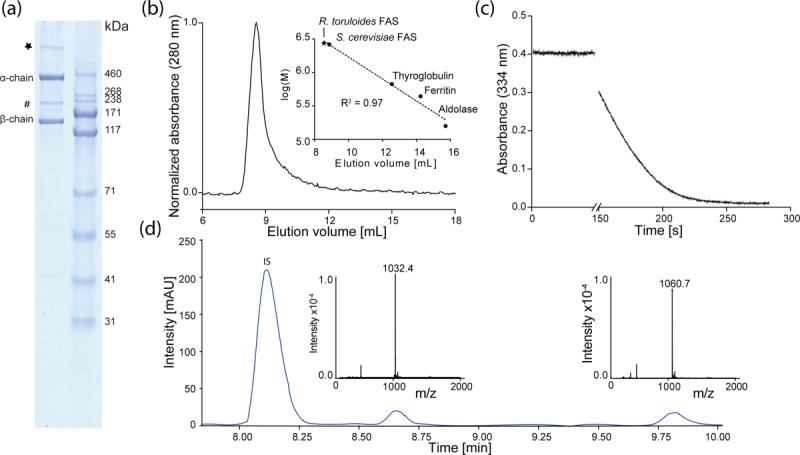
Purification and enzymatic properties of R. toruloides FAS I. (a) SDS-PAGE (NuPage Bis-Tris 4–12%, Life Technologies, USA) of purified recombinant FAS I with β-chain (138 kDa) and α-chain (321). A band at about 200 kDa (#) and a band at an apparent molecular weight clearly larger than 500 kDa (*) reflect main impurities. (b) Absorption profile from size exclusion chromatography of R. toruloides FAS I on a Superose 6 10/300 column (GE Healthcare). Linear regression of calibration with standard proteins and S. cerevisiae FAS I is shown in inset. (c) Absorption profile from activity assay of R. toruloides FAS I. Activity at 25°C was monitored by decrease in absorption at 334 nm from consumption of NADPH. (d) HPLC-MS analysis of the R. toruloides FAS I product spectrum. Main products are stearic (m/z = 1032.4) and arachidic acid (m/z = 1060.7); IS, internal standard (isoheptadecanoyl-CoA).
Enzymatic analysis
The enzymatic activity of R. toruloides FAS I was tested by monitoring NADPH consumption via absorption at 334 nm [Fig. 2(C)]. As an average of three independent protein preparations, we obtained a specific activity of 476 ± 159 mU/mg with respect to the consumption of two equivalents NADPH per reaction, reflecting one reaction cycle and the elongation of the fatty acid by one C2-unit. The individual protein batches showed specific activities of 618 ± 132 mU/mg, 506 ± 85 mU/mg and 304 ± 31 mU/mg, respectively. S. cerevisiae FAS I from native preparations yielded activities ranging from 343 mU/mg22 to 3500 mU/mg.23,24 Considering the particular architecture of the duplicated ACP as a means for increasing the capacity of substrate shuttling, R. toruloides FAS I activity is lower than expected. Negative stain EM, initially performed for monitoring protein quality, and cryo-EM show high protein homogeneity. It is tempting to speculate that low protein activities might reflect incomplete post-translational phosphopantetheinylation as a consequence of the heterogeneous environment during recombinant expressions. Alternatively, it might be resulted from insufficient cofactor (FMN) binding or the presence of protein tags. We also analyzed the product spectrum of R. toruloides FAS I by HPLC-MS, and detected C18- and C20-acyl CoA esters as the main products [Fig. 2(D)]. This is different to reported data, showing exclusively production of C16- and C18-fatty acids in vivo,25–27 and might reflect the difference between in vitro assay conditions and the in vivo environment; for example, different relative levels of malonyl- and acetyl-CoA concentrations28 or interfering macromolecules.29,30
Three-dimensional cryo-EM map
We generated a 3D map of R. toruloides FAS I from 3296 particles (Supporting Information Fig. S2). The resolution of the final map was measured by the Fourier shell correlation (FSC) method at the FSC 0.143 criterion as 7.8 Å.31 The map resolves all features of fungal FAS I that were seen before in X-ray11–13,18 and cryo-EM studies17 [Fig. 3(A)]. Docking of available structural information revealed that the map is more similar to published X-ray structures of T. lanuginosus and S. cerevisiae FAS I than to the cryo-EM structure of S. cerevisiae FAS I. In the S. cerevisiae FAS I cryo-EM structure,17 differences in domain positions were observed that lead to overall shorter (by ∼20 Å) but wider (by ∼18 Å) barrel-like structure. Most significant was a ∼9° rotated arrangement of the trimerization domains (TM) at the threefold axis.17 Our data revise the initial assumption that the X-ray structures of yeast FAS I might have been affected by crystal contacts, but suggest a breathing-like motion as real conformational variability of fungal FAS I. Unlike the S. cerevisiae FAS I cryo-EM map, the R. toruloides map showed no evidence of heterogeneity (Supporting Information Fig. S3), which characterizes the R. toruloides FAS I barrel as rigid which is in line with previous data.1,14 Density for the ACP domains was not visible in the map, indicating that they occupy varying positions inside the reaction chambers.
Figure 3.
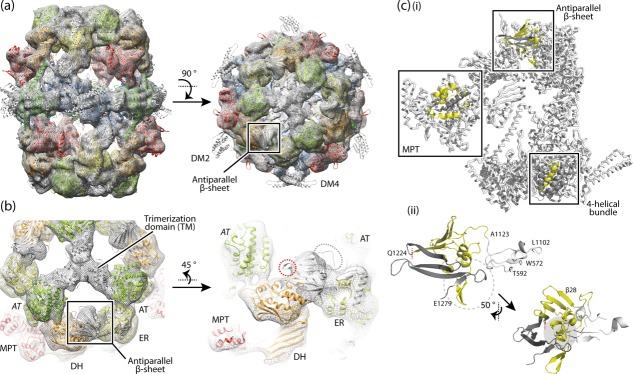
The structure of R. toruloides FAS I. (a) Cryo-EM map at 7.8 Å resolution with the X-ray structure of T. lanuginosus FAS I (pdb codes 2uva, 2uvb)11 fitted into the map as a rigid body. The protein is shown in side and top view. The polypeptides are colored according to the scheme above (see Fig. 1). Structural elements and the antiparallel β-sheet are highlighted. (b) 3D map and structural model as in (a) with zoom onto the antiparallel β-sheet domain. Domains are labeled. AT (italic letters) is provided from the neighboring polypeptide chain. The splitting site as occurring in R. toruloides FAS I is highlighted by a red circle; the loop connecting the β-sheet with the ER by a gray circle. (c, i) A single α-chain and a single β-chain are extracted from the fungal FAS I (T. lanuginosus FAS I; pdb codes 2uva, 2uvb)11 and splitting sites as occurring in fungal FAS I types Ascomycota (splitting site within MPT domain), Tremellomycetes (splitting site within 4-helical bundle) and Rhodosporidium (splitting site within antiparallel β-sheet) are indicated. In the highlighted structural elements, the β-chain part is shown in yellow and the α-chain part in dark gray. (ii) Antiparallel β-sheet composed of the β-chain and α-chain of R. toruloides FAS I; model and coloring as in (i); the left figure is roughly in the orientation of the side view in (a). The interface is comprised by formation of a curved β-sheet that interacts with α-helices at the concave face (highlighted by gray background), intertwined hairpin loops (highlighted by dashed circle), and the hairpin loop of α-chain interacting with a globular fold of the ER domain (colored in white). Residue numbers are given for defining borders (T. lanuginosus FAS I numbering).
By carrying the gene splitting site within the β-sheet domain encoding sequence, R. toruloides FAS I of the Sporidiobolales clade represents a specific case of two gene-encoding fungal FAS I variant. Docking of the T. lanuginosus FAS I X-ray structure into the EM map supports sequence analysis (Supporting Information Fig. S4) and does not reveal any unexpected features [Fig. 3(A,B)]. The loop connecting the antiparallel β-sheet with the ER and the loop carrying the splitting site protrude from the electron density [Fig. 3(B)]. These properties might be due to minor local structural differences between R. toruloides FAS I (map) and T. lanuginosus FAS I (model), or to local conformational variability, which cannot be traced at this resolution. Here it is worth to mention that also in the 3.1 Å X-ray structure the connecting linker as well as the first β-strand of the antiparallel β-sheet (β28) could not be traced in electron density (K1122-N1140, corresponding T. lanuginosus FAS I numbering).18
Discussion
We have recently started the structural and functional characterization of R. toruloides FAS I from recombinant expressions in E. coli because of our interest in the evolutionary development of fungal FAS I. R. toruloides is a high lipid producing fungal species, and initial characterizations speculated on R. toruloides encoding FAS I with improved fatty acid productivity.21
In R. toruloides FAS I, we were particularly interested in the novel form of splitting the multifunctional polypeptide into a β-chain which solely carries the domains AT and ER, and in an α-chain which includes all other domains. As expected, our data do not reveal any features emerging from the domain distribution that might be correlated with increased protein activity. Interestingly, the interaction of the α- and β-chain is different as compared to the other topical variants. In S. cerevisiae and T. lanuginosus FAS I, both representing the Ascomycota fungal FAS I variant, the α- and β-chain interact via termini that intertwine in forming a helical bundle as part of the MPT domain [Fig. 3(C)].11,18 From homology modeling with available structural information, it can be assumed that also FAS of the Tremellomycetes variant share the characteristic of chains interacting via intertwined termini. Here, the C-terminus of the β-chain and the N-terminus of the α-chain form a 4-helical bundle in the interface of the KR and the KS domain [Fig. 3(C)]. The interaction of chains in R. toruloides FAS I is conceptually different. Similar to the Ascomycota and the Tremellomycetes variants, the polypeptide chains interact within a domain (β-sheet domain, L1102-E1279, T. lanuginosus numbering). However, rather than showing intertwined termini, a large interface of 1725 Å2 (PDB ID: 2uva; T. lanuginosus)11 is formed [Fig. 3(C)]. Cryo-EM data are clearly at too low resolution to trace details of the interaction of the polypeptide chains, but the smooth incorporation of the T. lanuginosus FAS I structural model into R. toruloides FAS I cryo-EM density [see Fig. 3(A,B)] implies that there are no substantial differences in the fold of the ER-DH connecting β-sheet domain, in line with alignments and secondary structure predictions (see Supporting Information Fig. S4).
Gene splitting in two gene-encoded variants is an evolutionary late event,14,32 and the topological variants currently identified from available sequence information are relevant for understanding the assembly of fungal FAS I. Recently, the idea of the evolutionary conservation of assembly pathways has been established.33 For fungal FAS I, evolutionary pressure for the conservation of the assembly pathway arises from the structural particularity of the spatially separated domains ACP and PPT in the mature barrel-like structure. As they are hindered in forming productive interactions in the mature structure, the phosphopantetheinylation of ACP by PPT has to occur in a preassembled state.12,19 From the common motif of the interactions of chains within domains, we speculate that in the process of assembly, the two gene-encoding variants form pseudo-single chains so that all variants align into a single pathway in an early step. Subsequently, these initial assemblies arrange into the preassembled states competent for post-translational modification of ACP. From the structural appearance of the interaction of chains in two gene-encoded fungal FAS I, it is tempting to speculate that the assembly pathway of fungal FAS I is a robust sequential process, that putatively accepts many splitting sites, as long as the interactions of the polypeptide chains is strong enough, either by intertwining termini, as observed in Ascomycota and the Tremellomycetes variants, or by forming large interfaces as observed for the Rhodosporidium variant.
Another interesting feature of R. toruloides FAS I is the duplication of the ACP domain. As evident from the structural characterization of R. toruloides FAS I (see Supporting Information Fig. S3) and reported for S. cerevisiae FAS I before,17,18 the extensive scaffolding in fungal FAS I leads to conformationally restricted proteins in which essentially only the substrate shuttling ACP is mobile owing to conformationally variable linkers. The ACP domains are 77% sequence identical and show particular high conservation in regions relevant for docking (Supporting Information Fig. S6). From sequence conservation, a specialization among the two ACP domains for any of the steps relevant for fatty acid synthesis is unlikely. Similar to what was shown for multienzyme proteins in polyunsaturated fatty acid (PUFA) production that carry tandem ACP domains, the purpose of ACP duplication in R. toruloides FAS I seems to lie in the increase in its apparent ACP concentration and, consequently, the increase in the capacity for shuttling substrates and intermediates.34,35 Additional benefit arises from crowding of the reaction chamber with a second ACP domain and a connecting linker.36 Our cryo-EM data did not allow assignment of ACP positions, reflecting the positional variability of the ACP as an inherent feature of FAS I systems.1,17 An interesting aspect is the occurrence of the duplicated ACP across the phylogenetic clades that define the fungal FAS I variants (Supporting Information Fig. S5). This gives evidence that the duplication of ACP is an evolutionary recent development that occurred after the evolutionary event of the splitting of the single gene-encoded fungal FAS I in the currently known two gene-encoded variants.
Conclusion
FAS I are among the most complex proteins analyzed in eukaryotes.1,14 They are made up of multifunctional polypeptides that assemble to up to 2.7 MDa large complexes of elaborate architecture. Fungal FAS I are highly evolved. Their development comprises hallmarks as follows: (i) The extension of individual monofunctional proteins with scaffolding domains.32 (ii) Fusion events of the monofunctional proteins to a stable single gene-encoded multifunctional FAS I protein.32 This FAS I ancestor protein is distributed in Corynebacteria, Mycobacteria, and Nocardia. (iii) Further scaffolding to a single gene-encoded fungal FAS I.20,37 (iv) Gene splitting into two gene-encoded variants. Many splitting sites may be tolerated as long as they do not interfere in FAS I assembly. (v) ACP duplication in certain species, as R. toruloides FAS I, for increasing the substrate shuttling capacity.
Material and Methods
Plasmid construction
Total RNA was prepared as described previously.21 cDNA synthesis was performed with PrimeScript High Fidelity RT-PCR Kit (Takara, Dalian, China). FAS coding genes were amplified with primer pairs (FAS1-5-NdeI, GGCATTCCATATGGCAAGCTGGAGCCACCCGCAGTTCGAAAAGGGTGCAATGAACGGCCGAGCGACGCG, FAS1-3-EcoRI, GGAATTCTCAGAGCCCGCCGAAGACG, FAS2-5-HindIII, GCCCAAGCTTATGGTCGCGGCGCAGGACTTG and FAS2-3-NotI, CCGCATTGCGGCCGCCTTCTGGGCGATGACGACGGC). The β-chain encoding fragment inserted into pET22b(+) vector (Novagen, Darmstadt, Germany) yielded pET22b-RtFAS1, while the α-chain fragment inserted into pET24b(+) vector (Novagen, Darmstadt, Germany) gave pET24b-RtFAS2-WT. Plasmids were coding for a β-chain carrying an N terminal Strep-II-tag (MASWSHPQFEKGA-), and the α-chain modified with an N terminal T7-tag (MASMTGGQQMGRDPNSSSVDKL-) and a C terminal His6-tag (-AAALEHHHHHH).
Recombinant expression and purification of Rhodosporidium FAS
E. coli strain BL21 (DE3; Novagen, Darmstadt, Germany) was co-transformed with plasmids pET22b-RtFAS1 and pET24b-RtFAS2-WT via the heat shock method and selected on LB agar supplemented with an appropriate amount of antibiotics (100 µg/mL ampicillin and 50 µg/mL kanamycin). Expression and purification of the recombinant Rhodosporidium FAS I was described before.38 Briefly, single colonies were used to inoculate 5 mL of a LB preculture, which finally was used to inoculate 1.6 L TB media (with 100 µg/mL ampicillin and 50 µg/mL kanamycin). After growth to OD600 of 0.6–0.8, the expression was induced by adding IPTG to the medium to a final concentration of 0.5 mM. The culture was incubated at 200 rpm and 16°C for 30 h. Cells were harvested by centrifugation at 8000 rpm (6200g) for 10 min. The cell pellet (about 12 g) was resuspended in 36 mL lysis buffer (100 mM KPi pH 7.0, 5 mM EDTA, 5 mM β-mercaptoethanol, 1 mM phenylmethylsulfonylfluoride, 3 mM MgCl2, 0.5 mg/mL lysozyme (Genview, Beijing, China), protease inhibitor tablets (Roche, Basel, Switzerland) and homogenized by ultrasonication for 20 min. The lysate was cleared by centrifugation at 14,000 rpm (18,000g) for 30 min. Saturated (NH4)2SO4 solution was added to the supernatant (25%, v/v), and the mixture was stirred for 30 min. Precipitated protein was pelleted at 14,000 rpm (18,000g) for 30 min. A second protein pellet was obtained in a similar protocol, adding additional saturated (NH4)2SO4 solution to 33% (v/v). Pellets were resuspended in 16 mL buffer A (20 mM HEPES pH 8.0, 100 mM KCl, 5 mM EDTA, 5 mM β-mercaptoethanol), and loaded onto a 10–40% (w/v) sucrose density gradient (in buffer A). The centrifugation was performed in a SW32 rotor at 27,000 rpm (about 90,000g) and 4°C for 15 h. FAS I containing fractions were collected and ultrafiltrated. Subsequently, anion exchange chromatography (DEAE-sepharose) was performed. Proteins were eluted with KCl by gradients of buffer A and B (20 mM HEPES pH 8.0, 800 mM KCl, 5 mM EDTA, 5 mM β-mercaptoethanol). FAS I containing fractions were concentrated and a second run of sucrose density centrifugation was performed for further purification. More than 10 mg protein was obtained.
Size exclusion chromatography
Size exclusion chromatography was performed on a Superose 6 10/300 GL column (GE Healthcare). After equilibration of the column with buffer (100 mM sodium phosphate pH 7.2, 200 mM NaCl, 1 mM EDTA), 500 µL of protein in buffer A (1.4 mg/mL) was loaded onto the column. 1 mL fractions were collected with a flow rate of 0.3 mL/min, and absorption at 280 nm was recorded. During all steps, the protein was always kept at 4°C. The calibration of the chromatographic system was done with a high molecular weight gel filtration calibration kit (GE Healthcare).
Enzymatic activity assay
Enzyme activity was determined by recording the NADPH-consumption via the decrease in absorption at 334 nm in an UV/vis-spectrometer (Lambda 25; Perkin Elmer). The assay was performed with 25 µg protein in a buffered solution (200 mM potassium phosphate pH 7.3, 87.5 µM DTT, 250 mM NADPH, 417 mM acetyl-CoA, 500 mM malonyl-CoA) with a total volume of 120 µL at room temperature. The activity analysis comprises only the linear range of the absorption curve after malonyl-CoA-addition. One unit of FAS I activity is defined as the turnover of 1 µmol malonyl-CoA or 2 µmol NADPH per minute, respectively.
Enzymatic product spectrum
To analyze the product spectrum of R. toruloides FAS I, the enzymatic reaction was performed in buffered solution (200 mM potassium phosphate pH = 7.3, 87.5 µM DTT, 2.25 mM NADPH, 0.20 mM acetyl-CoA, 1.00 mM malonyl-CoA) with a total volume of 100 µL and with 20 µg of protein. The reaction was kept at room temperature for 18 h and stopped by freezing the samples in liquid nitrogen. Work-up was done by acetone precipitation of the protein and vacuum evaporation of the solvent. The residue was redissolved in 50 µL water and analyzed by HPLC-MS.
Cryo-EM data collection
A 1.3-mg/mL FAS I sample of 3 µL was applied to glow discharged Quantifoil R2/2 holey carbon grids (Quantifoil Micro Tools, Jena, Germany). The samples were vitrified using an FEI Vitrobot plunge-freezer. Data was collected on an FEI Tecnai Polara operating at 300 kV, using a back-thinned FEI Falcon II direct electron detector. The Falcon II camera was calibrated at the desired nominal magnification of 78,000×. The calibrated magnification on the 14 µm pixel camera was 106,000×, resulting in a 1.32 Å pixel size at the specimen. The camera system was set up to record 18 frames/s.39,40 Videos were collected for 1.5 s with a total of 24 frames with a calibrated dose of 3.5e−/Å2 per frame, at defocus values of 1.5–3 µm.
Image processing
The 24 frames of each video were aligned using the whole-image motion correction method described in Li et al.41 Particle picking was carried out using the manual procedure of EMAN Boxer,42 and the contrast transfer function of every image was determined using CTFFIND343 in the RELION workflow.44 The data set was refined with the gold standard refinement procedure of RELION,45 using the cryo-EM map of S. cerevisiae FAS I17 low-pass filtered to 60 Å as a starting model, using 20 frames (from frame 2 to frame 21). A postprocessing procedure implemented in RELION44 was applied to the final maps for appropriate masking, B-factor sharpening and resolution validation.46 The final map of 3296 particles has a resolution of 7.8 Å by the FSC 0.143 criterion31 after applying this postprocessing procedure. The local resolution of the map was estimated with the Resmap software (available at http://resmap.sourceforge.net).47 The map was displayed in USCF Chimera,48 and the structure of T. lanuginosus FAS I (pdb codes 2uva, 2uvb)11 was fitted into the map as a rigid body without further refinement.
Acknowledgments
We are grateful to Werner Kühlbrandt for his continuous support of our research focus on the structural characterization of FAS mega-enzymes.
Glossary
- ACP
acyl carrier protein
- AT
acetyl transferase
- DH
dehydratase
- ER
enoyl reductase
- FAS I
fatty acid synthase Type I
- KR
ketoacyl reductase
- KS
ketoacyl synthase
- MPT
malonyl-palmitoyl transferase
- PPT
phosphopantetheine transferase.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- Maier T, Leibundgut M, Boehringer D, Ban N. Structure and function of eukaryotic fatty acid synthases. Q Rev Biophys. 2010;43:373–422. doi: 10.1017/S0033583510000156. [DOI] [PubMed] [Google Scholar]
- Gago G, Diacovich L, Arabolaza A, Tsai SC, Gramajo H. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol Rev. 2011;35:475–497. doi: 10.1111/j.1574-6976.2010.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer E, Hofmann J. Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol Mol Biol Rev. 2004;68:501–517. doi: 10.1128/MMBR.68.3.501-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay AT. The structures of type I polyketide synthases. Nat Prod Rep. 2012;29:1050–1073. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, Narayan AR, Håkansson K, Sherman DH, Smith JL, Skiniotis G. Structure of a modular polyketide synthase. Nature. 2014;510:512–517. doi: 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AL, Matsui T, Weiss TM, Khosla C. Architectures of whole-module and bimodular proteins from the 6-deoxyerythronolide B synthase. J Mol Biol. 2014;426:2229–2245. doi: 10.1016/j.jmb.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittner A, Grininger M. Modular polyketide synthases (PKSs): a new model fits all? Chembiochem. 2014;15:2489–2493. doi: 10.1002/cbic.201402432. [DOI] [PubMed] [Google Scholar]
- Lynen F. From "activated acetic acid" to terpenes and fatty acids. Umsch Wiss Tech. 1965;65:321–326. [Google Scholar]
- Jenni S, Leibundgut M, Maier T, Ban N. Architecture of a fungal fatty acid synthase at 5 Å resolution. Science. 2006;311:1263–1267. doi: 10.1126/science.1123251. [DOI] [PubMed] [Google Scholar]
- Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Structure of fungal fatty acid synthase and implications for iterative substrate shuttling. Science. 2007;316:254–261. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- Lomakin IB, Xiong Y, Steitz TA. The crystal structure of yeast fatty acid synthase, a cellular machine with eight active sites working together. Cell. 2007;129:319–332. doi: 10.1016/j.cell.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Johansson P, Wiltschi B, Kumari P, Kessler B, Vonrhein C, Vonck J, Oesterhelt D, Grininger M. Inhibition of the fungal fatty acid synthase type I multienzyme complex. Proc Natl Acad Sci U S A. 2008;105:12803–12808. doi: 10.1073/pnas.0805827105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grininger M. Perspectives on the evolution, assembly and conformational dynamics of fatty acid synthase type I (FAS I) systems. Curr Opin Struct Biol. 2014;25:49–56. doi: 10.1016/j.sbi.2013.12.004. [DOI] [PubMed] [Google Scholar]
- White SW, Zheng J, Zhang Y-M, Rock CO. The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- Beld J, Sonnenschein EC, Vickery CR, Noel JP, Burkart MD. The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Nat Prod Rep. 2014;31:61–108. doi: 10.1039/c3np70054b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson P, Mills DJ, Wouts R, Grininger M, Vonck J, Kuhlbrandt W. Direct structural insight into the substrate-shuttling mechanism of yeast fatty acid synthase by electron cryomicroscopy. Proc Natl Acad Sci USA. 2010;107:9164–9169. doi: 10.1073/pnas.0913547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibundgut M, Jenni S, Frick C, Ban N. Structural basis for substrate delivery by acyl carrier protein in the yeast fatty acid synthase. Science. 2007;316:288–290. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- Johansson P, Mulinacci B, Koestler C, Vollrath R, Oesterhelt D, Grininger M. Multimeric options for the auto-activation of the Saccharomyces cerevisiae FAS type I megasynthase. Structure. 2009;17:1063–1074. doi: 10.1016/j.str.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Boehringer D, Ban N, Leibundgut M. 7.5-Å cryo-em structure of the mycobacterial fatty acid synthase. J Mol Biol. 2013;425:841–849. doi: 10.1016/j.jmb.2012.12.021. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhang S, Liu H, Shen H, Lin X, Yang F, Zhou YJ, Jin G, Ye M, Zou H, Zhao ZK. A multi-omic map of the lipid-producing yeast Rhodosporidium toruloides. Nat Commun. 2012;3:1112. doi: 10.1038/ncomms2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki N, Funabashi H, Shimazawa R, Furukawa J, Kawaguchi A, Okuda S, Iwasaki S. Effect of side-chain structure on inhibition of yeast fatty-acid synthase by cerulenin analogs. Eur J Biochem. 1993;211:111–115. doi: 10.1111/j.1432-1033.1993.tb19876.x. [DOI] [PubMed] [Google Scholar]
- Fichtlscherer F, Wellein C, Mittag M, Schweizer E. A novel function of yeast fatty acid synthase. Subunit alpha is capable of self-pantetheinylation. Eur J Biochem/FEBS. 2000;267:2666–2671. doi: 10.1046/j.1432-1327.2000.01282.x. [DOI] [PubMed] [Google Scholar]
- Wieland F, Renner L, Verfürth C, Lynen F. Studies on the multi-enzyme complex of yeast fatty-acid synthetase. Eur J Biochem. 1979;94:189–197. doi: 10.1111/j.1432-1033.1979.tb12885.x. [DOI] [PubMed] [Google Scholar]
- Davoli P, Mierau V, Weber RWS. Carotenoids and fatty acids in red yeasts Sporobolomyces roseus and Rhodotorula glutinis. Appl Biochem Microbiol. 2004;40:392–397. [PubMed] [Google Scholar]
- Easterling ER, French WT, Hernandez R, Licha M. The effect of glycerol as a sole and secondary substrate on the growth and fatty acid composition of Rhodotorula glutinis. Bioresource Technol. 2009;100:356–361. doi: 10.1016/j.biortech.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhao X, Wang W, Du W, Liu D. Microbial conversion of biodiesel byproduct glycerol to triacylglycerols by oleaginous yeast Rhodosporidium toruloides and the individual effect of some impurities on lipid production. Biochem Eng J. 2012;65:30–36. [Google Scholar]
- Sumper M, Oesterhelt D, Riepertinger C, Lynen F. Synthesis of various carboxylic acids by the multienzyme complex of fatty acid synthesis from yeast, and clarification of their structure. Eur J Biochem/FEBS. 1969;10:377–387. doi: 10.1111/j.1432-1033.1969.tb00701.x. [DOI] [PubMed] [Google Scholar]
- Peterson DO, Bloch K. Mycobacterium smegmatis fatty acid synthetase. Long chain transacylase chain length specificity. J Biol Chem. 1977;252:5735–5739. [PubMed] [Google Scholar]
- Zimhony O, Vilcheze C, Jacobs WR., Jr Characterization of Mycobacterium smegmatis expressing the Mycobacterium tuberculosis fatty acid synthase I (fas1) gene. J Bacteriol. 2004;186:4051–4055. doi: 10.1128/JB.186.13.4051-4055.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Bukhari HST, Jakob RP, Maier T. Evolutionary origins of the multienzyme architecture of giant fungal fatty acid synthase. Structure. 2014;22:1775–1785. doi: 10.1016/j.str.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Marsh JA, Hernández H, Hall Z, Ahnert SE, Perica T, Robinson CV, Teichmann SA. Protein complexes are under evolutionary selection to assemble via ordered pathways. Cell. 2013;153:461–470. doi: 10.1016/j.cell.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zirkle R, Metz JG, Braun L, Richter L, Van Lanen SG, Shen B. The role of tandem acyl carrier protein domains in polyunsaturated fatty acid biosynthesis. J Am Chem Soc. 2008;130:6336–6337. doi: 10.1021/ja801911t. [DOI] [PubMed] [Google Scholar]
- Trujillo U, Vázquez-Rosa E, Oyola-Robles D, Stagg LJ, Vassallo DA, Vega IE, Arold ST, Baerga-Ortiz A. Solution structure of the tandem acyl carrier protein domains from a polyunsaturated fatty acid synthase reveals beads-on-a-string configuration. PLoS One. 2013;8:e57859. doi: 10.1371/journal.pone.0057859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmi C, Grininger M, Gipson P, Faraldo-Gomez J. Mechanism of substrate shuttling by the acyl-carrier protein within the fatty acid mega-synthase. J Am Chem Soc. 2010;132:12357–12364. doi: 10.1021/ja103354w. [DOI] [PubMed] [Google Scholar]
- Ciccarelli L, Connell SR, Enderle M, Mills DJ, Vonck J, Grininger M. Structure and conformational variability of the Mycobacterium tuberculosis fatty acid synthase multienzyme complex. Structure. 2013;21:1251–1257. doi: 10.1016/j.str.2013.04.023. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhang S, Lin X, Liu W, Zhao ZK. Expression, purification and characterization of a novel fatty acid synthase from Rhodosporidium toruloides. Chin J Biotech. 2014;30:1414–1423. [PubMed] [Google Scholar]
- Allegretti M, Mills DJ, McMullan G, Kühlbrandt W, Vonck J. Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. eLife. 2014;3:e01963. doi: 10.7554/eLife.01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X-c, Fernandez IS, McMullan G, Scheres SHW. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife. 2013;2:e00461. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat Methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SHW, Henderson R. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information