

Abstract
Amylase-binding protein A (AbpA) of a number of oral streptococci is essential for the colonization of the dental pellicle. We have determined the solution structure of residues 24–195 of AbpA of Streptococcus gordonii and show a well-defined core of five helices in the region of 45–115 and 135–145. 13Cα/β chemical shift and heteronuclear 15N-{1H} NOE data are consistent with this fold and that the remainder of the protein is unstructured. The structure will inform future molecular experiments in defining the mechanism of human salivary α-amylase binding and biofilm formation by streptococci.
Keywords: amylase-binding protein, AbpA, α-amylase, NMR, protein structure
Introduction
Human salivary α-amylase is the most abundant enzyme in the oral cavity of humans where it plays a key role in the hydrolysis of the α1,4 glycosidic bonds in dietary starch.1 α-Amylase, however, accumulates in the dental pellicle formed on the tooth surface where it acts as a site for colonization of a group of oral streptococci, including Streptococcus gordonii, S. mitis, S. parasanguis, S. cristatus and S. salivarius.2–4 Furthermore, these bacteria act as attachment points for nonamylase-binding bacteria and thus the development of the dental pellicle. The colonization of the dental pellicle by the attachment of these bacteria to α-amylase does not prevent the enzyme from hydrolysing starch.5,6 Thus, the continued activity of α-amylase supplies fermentable sugars to the oral microflora in the developing pellicle and the resultant production of lactic acid, which is an important causative agent of tooth decay.
Of these bacteria, S. gordonii is a primary colonizer of the tooth, and present in substantial quantities in the early dental pellicle. This initial attachment of S. gordonii to α-amylase is critical to the development of the pellicle in providing a colonization site for nonamylase-binding bacteria and therefore its disruption may be significant for controlling the development of pellicle and dental disease. To attach to α-amylase on the tooth surface, S. gordonii produces amylase-binding proteins, of which two have been well characterized: low-molecular weight protein (AbpA) and high-molecular weight protein (AbpB).7–9 Of these two proteins, AbpA (20 kDa) has been identified as the major amylase-binding protein of S. gordonii.10–13 AbpA-deficient mutants showed not only that the protein is required for adhesion and biofilm development, but important for growth of S. gordonii in human saliva.12 Currently, how AbpA binds α-amylase is not understood. Sequence alignments and truncation experiments suggest several conserved regions may be involved in α-amylase binding,13,14 but in the absence of structural data it is difficult to draw conclusions. Here we present the NMR solution structure of AbpA (24–195) from S. gordonii and show that the protein fold has a helical core with disordered N- and C-terminal regions. The structure will guide further structure–function experiments.
Results and Discussion
Amylase-binding protein A (AbpA) comprises 42 alanine residues (24.4% of the total amino acids) and often repeated along the sequence. Therefore sequence specific resonance assignments were manually assigned using several three dimensional NMR experiments: HNCACB, HN(CA)CO, H(CCO)NH, (H)CC(CO)NH, 15N-edited NOESY, 13C-edited (aliphatic) NOESY, and 13C-edited (aromatic) NOESY. 98.8% backbone amide signals were unambiguously assigned. Missing assignments include the first N-terminus residue (Ala24) and the histidine residue (His87) located in the loop connecting the helices α3 and α4. The near-complete backbone (98.7%, HN, N, C, Cα, Hα) and side chain (80%) assignments were useful to generate the automated structure determination using UNIO-ATNOS/CANDID15,16 and CYANA3.0.17 Table1 shows the structure calculation statistics of 20 conformers representing the NMR structure of AbpA.
Table 1.
Structure Calculation Statistics of the 20 NMR Conformers of the S. gordonii Amylase-Binding Protein A (AbpA)
| Quantity | Valuea |
|---|---|
| NOE upper distance limits | 1774 |
| Intraresidual | 402 |
| Short range | 642 |
| Medium range | 536 |
| Long range | 194 |
| Talos dihedral angle constraints (φ, ψ) | 126 |
| Residual target function value (Å2) | 2.04 ± 0.36 |
| Residual NOE violations | |
| Number ≥0.1 Å | 16 ± 4 |
| Maximum (Å) | 0.17 ± 0.11 |
| Residual dihedral angle violations | |
| Number ≥2.5° | 1 ± 1 |
| Maximum (°) | 3.3 ± 1.3 |
| AMBER energies (kcal/mol) | |
| Total | −6859 ± 131 |
| Van der Waals | −234 ± 32 |
| Electrostatic | −8056 ± 138 |
| RMSD from mean coordinatesb (Å) | |
| Backbone (47–117,134–142) | 0.74 ± 0.23 |
| All heavy atoms (47–117,134–142) | 1.25 ± 0.32 |
| Ramachandran plot statisticsc (24–195) | |
| Most favoured and additional allowed regions (%) | 93.9 |
| Generously allowed regions (%) | 3.8 |
| Disallowed regions (%) | 2.4 |
Except for the top six entries, average values, and standard deviations for the 20 energy-minimized conformers are given. The top five entries represent the output from the seventh cycle of UNIO-ATNOS/CANDID and CYANA 3.0.
The numbers in parentheses indicate the residues for which the RMSD was calculated.
As determined by PROCHECK.27 Residues in the disallowed regions were observed either in the loop or unstructured regions of the structure.
The final solution structure of AbpA showed apparent disorder in the N- and C-terminal regions. However, the region spanning residues 45–115 and 135–145 shows a well-ordered protein core [Fig. 1(A)]. The core consists of five helices. The first two, α1 (Ala47–His57) and α2 (Asp58-Ala67), are almost continuous; however, there is a bend between His57 and Asp58 that makes these two helices distinct. Pro69 breaks the main chain following α2 and is at the N-terminal end prior to α3 (Ala70–Ala80). After α3, a well-defined loop reverses the direction of the mainchain to form the long helix α4 (Asn87–Gln115). Following this helix is a poorly structured loop region and the final short helix, α5 (Thr134–Tyr142). Conserved hydrophobic residues [Fig. 1(C)] appear to form the hydrophobic core between α1-4 which is likely to be important for protein stability. These residues include Leu54, Ile61, Val71, Ala74, Leu78, Val81, Ala102, and Val111. Multiple sequence alignment suggests several polar residues are conserved [Fig. 1(C)], including His57, His86, Arg107 and Arg141. His57, Arg107, and Arg141 are located in close proximity; however, Arg107 is on the opposite side of the protein relative to the other two residues [Fig. 1(B)]. Interestingly, Lys109, together with His57 and Arg141 would form a positive patch [Fig. 1(B)], but Lys109 is not conserved. Extending from this positively charged cluster are four tyrosines, Tyr53, Tyr110, Tyr118, and Tyr129 [Fig. 1(B)]. None of these Tyr residues are strictly conserved; however, in all homologues at least three of these Tyr are present [Fig. 1(B)].
Figure 1.
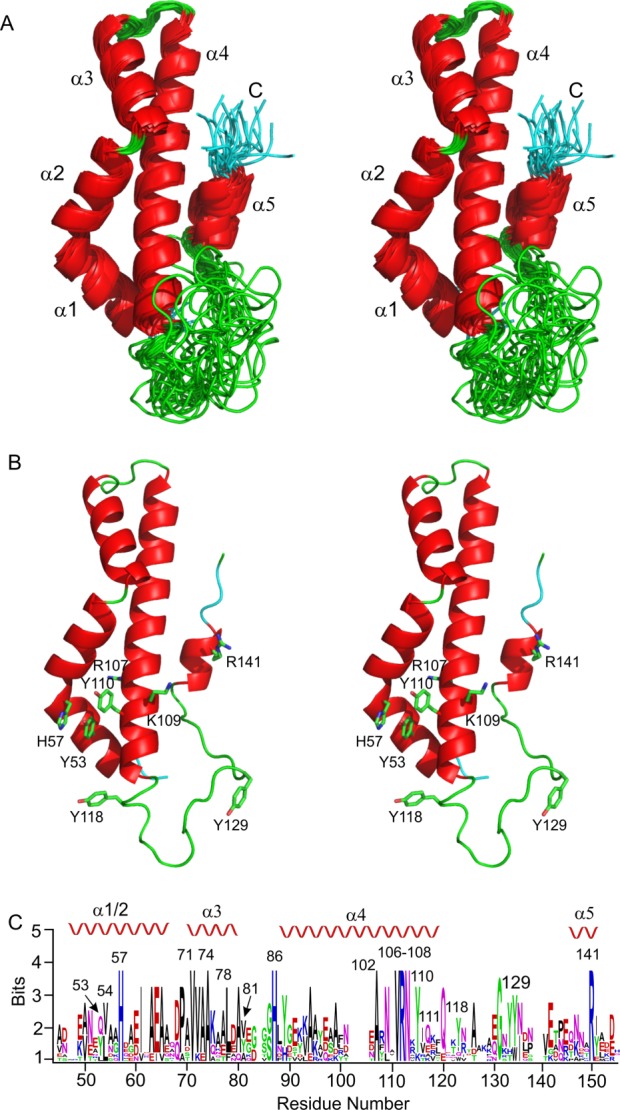
Structure of S. gordonii amylase-binding protein AbpA (pdb id 2MXX). A. Stereoview of a superposition of the best 20 conformers for the region 45–145 showing the well-defined five helices. The flow of the mainchain starts in the back of the plane. B. Stereoview of the best conformer (model 5) oriented as in (A). A cluster of Tyr and basic residues are annotated. C. Weblogo29 showing the conserved residues within the region 45–145 of AbpA. Residues according to the sequence of S. gordonii AbpA are marked and the position of the secondary structure is indicated above the logo. Nine homologues were used in the alignment: S. gordonii, S. cristatus, S. salivarius, S. parasanguinis, S. mitis, S. vestibularis, S. australis, S. infantis, and S. oralis. While not all Abps from these species have been shown to bind α-amylase, the strong sequence alignment suggests that these other Abps are likely to bind α-amylase.
Inspection of the 13Cα and 13Cβ chemical shift data is consistent with the description of the helical fold of the region 45–145 [Fig. 2(A)] and shows there is very little propensity for regular secondary structure either to the N- or C-terminal side of this region. Indeed the analysis of a heteronuclear 15N-{1H} NOE experiment shows outside of residues 45–115 and 135–145 there is considerable protein flexibility suggesting disordered structure in these regions [Fig. 2(B)]. Whether these regions adopt a structure within the cell-surface complex with AbpB and α-amylase remains to be determined.13
Figure 2.
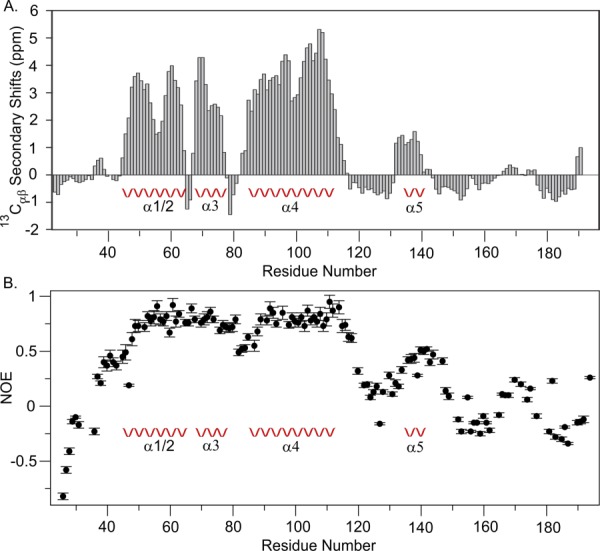
A. Secondary chemical shifts for 13Cα and 13Cβ of S. gordonii AbpA. Segments with >1 ppm are consistent with the presence of α-helix structure. 13Cαβ values were computed using the formula as described previously.28 B. Plot of 15N-{1H} NOE data at 700 MHz of S. gordonii AbpA. Well defined structure typically has an NOE value of ∼0.8 which is observed for helices α1–4. The N-terminal region 24–45 and the C-terminal 145–195 show NOE values less than 0.5 consistent with disordered structure. The loop between 115 and 135 also shows NOE values of less than 0.5 and helix α5 shows values ∼0.5, consistent with the observed disordered structure of the loop and structural heterogeneity of α5.
A DALI18 search using the core of AbpA (residues Ala47 to Tyr142) showed greatest similarity (Z-score 6.1) to a subdomain (residues 2229–2310) of the carboxyl transferase domain of acetyl CoA carboxylase (ACC2).19 The two regions show very little sequence similarity (∼9% identity), but the helices α1 to α4 are similarly positioned in both proteins. The fifth helix significantly differs in its orientation. This region in ACC2 forms an antiparallel helical extension and is a part of a dimerization surface, otherwise there does not appear to be a functional overlap with AbpA.
In an attempt to localize the functional regions of AbpA, N- and C-terminal truncations were expressed, tested for protein fold by circular dichroism (CD) and α-amylase binding by surface plasmon resonance.14 C-terminal truncation of the last 30 residues had little effect on the ability to bind salivary α-amylase, suggesting these residues are not important. The CD spectra showed an increase in helical content for this truncate,14 consistent with the removal of disordered structure within the C-terminal region (Fig. 2). N-terminal truncation of the first 30 residues weakened binding by 104, suggesting functionally important residues are within this region. CD spectra suggested little change in the structure compared to full-length protein (24–195). However, the structure presented here indicates that this truncation would remove the first two turns of helix α1, and while the fold may have been retained, the extent of any destabilization remains to be resolved. Nevertheless, this truncation would perturb the region of conserved positively charged residues and the aromatic cluster that have been identified [Fig. 1(B)], where the latter has been speculated to interact with aromatic clusters that have been identified in α-amylase.20
In summary, we have shown that the amylase-binding protein AbpA of S. gordonii has a well-defined fold of five helices within the regions of 45–115 and 135–145. Chemical shift and 15N-{1H} NOE data show that the remaining parts of the protein are unstructured. Analysis of the core shows a cluster of positively charged and aromatic residues which may be functionally important. This structure will advise future mutagenesis and binding studies in order to elucidate the mechanism of binding to human salivary α-amylase.
Materials and Methods
Protein expression and purification
The abpA gene spanning residues 24–195 from S. gordonii G9B was cloned into a pET29b vector as described previously.14 AbpA with a His6-tag (referred to as AbpA) was expressed in Escherichia coli BL21 (DE3) and cells were grown to OD600 of 0.6 at 37°C, and then transferred to 16°C and induced for 16 h. Cells were harvested, pelleted, and stored at −20°C. Cells were lysed using an Avestin EmulsiFlex C3 cell crusher and centrifuged at 4°C, 13,000g for 40 min to remove insoluble cell debris. AbpA was purified from the soluble fraction by affinity chromatography over His60 Ni Superflow resin (Clonetech, Takara), which was equilibrated with 20 mM Tris–HCl, 500 mM NaCl, and 5 mM imidazole at pH 8. AbpA was eluted with 400 mM imidazole in 20 mM Tris–HCl, 500 mM NaCl at pH 8.0. The eluted fusion protein was further purified by size exclusion chromatography using a HiLoad™ 16/60 Superdex™ 75 prep grade column (GE Healthcare), where it elutes as a single monomeric peak in 20 mM Tris–HCl, 150 mM NaCl at pH 7.4. Collected fractions were pooled, buffer exchanged to 50 mM sodium phosphate, 100 mM NaCl at pH 5.8 for recording NMR experiments. The purified AbpA was further characterized by mass spectrometry. All NMR samples were uniformly 15N or 13C, 15N labelled according to the protocol of Cai et al.21 The AbpA structure was solved as fusion with the His6 tag.
NMR spectroscopy
NMR samples of 325 µM of S. gordonii AbpA (residues 24–195, with an additional Met at the N-terminus and a C-terminal His6-tag) were dissolved in 50 mM phosphate buffer at pH 5.8 containing 100 mM NaCl and 7% D2O. Spectra were recorded at 25°C on Bruker Avance IIIHD 700 MHz and Avance II 800 MHz spectrometers equipped with CryoProbes: 3D HNCACB, HN(CA)CO, H(CCO)NH, (H)CC(CO)NH, 15N-edited NOESY, 13C-edited (aliphatic) NOESY, and 13C-edited (aromatic) NOESY. All NOESY data were collected at 800 MHz with mixing times of 150 ms. To assess protein flexibility a 2D 15N-{1H} NOE spectrum was acquired at 700 MHz with 4 s of 1H saturation and an additional 5 s of relaxation. NMR data were processed using either Topspin3.2 or NMRPipe,22 chemical shifts were assigned using CARA,23 and the analysis of the 2D 15N-{1H} NOE in relax.24
Structure calculation
NMR structure calculation was performed using UNIO interface (UNIO'10 Version 2.0.2). The chemical shift assignments, TALOS+ constraints,25 and three NOESY spectra were used as the input of UNIO-ATNOS/CANDID15,16 and CYANA 3.017 Forty conformers with lowest target function after cycle7 were energy minimized with OPALp26 and canonical hydrogen bonds in the region of secondary structures of the protein were identified by MOLMOL.27 The structure calculation were then repeated and refined in the presence of hydrogen bond constraints. An ensemble of 20 NMR conformers was selected using the validation criteria as described previously28 and analyzed by MOLMOL. Structure images were generated using PyMol (http://www.pymol.org).
Data deposition
NOE distance constraints and chemical shifts were deposited in the BioMagResBank (http://www.bmrb.wisc.edu) with BMRB code 25435. The atomic coordinates of 20 NMR conformers were deposited in the Protein Data Bank (http://www.pdb.org) with PDB code 2MXX.
References
- Aguirre A, Levine MJ, Cohen RE, Tabak LA. Immunochemical quantitation of alpha-amylase and secretory IgA in parotid saliva from people of various ages. Arch Oral Biol. 1987;32:297–301. doi: 10.1016/0003-9969(87)90024-0. [DOI] [PubMed] [Google Scholar]
- Douglas CW. Bacterial–protein interactions in the oral cavity. Adv Dental Res. 1994;8:254–262. doi: 10.1177/08959374940080021901. [DOI] [PubMed] [Google Scholar]
- Kilian M, Nyvad B. Ability to bind salivary alpha-amylase discriminates certain viridans group streptococcal species. J Clin Microbiol. 1990;28:2576–2577. doi: 10.1128/jcm.28.11.2576-2577.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannapieco FA. Saliva–bacterium interactions in oral microbial ecology. Crit Rev Oral Biol Med. 1994;5:203–248. doi: 10.1177/10454411940050030201. [DOI] [PubMed] [Google Scholar]
- Douglas CW, Heath J, Gwynn JP. Enzymic activity of salivary amylase when bound to the surface of oral streptococci. FEMS Microbiol Lett. 1992;71:193–197. doi: 10.1016/0378-1097(92)90511-l. [DOI] [PubMed] [Google Scholar]
- Scannapieco FA, Bhandary K, Ramasubbu N, Levine MJ. Structural relationship between the enzymatic and streptococcal binding sites of human salivary alpha-amylase. Biochem Biophys Res Commun. 1990;173:1109–1115. doi: 10.1016/s0006-291x(05)80900-3. [DOI] [PubMed] [Google Scholar]
- Brown AE, Rogers JD, Haase EM, Zelasko PM, Scannapieco FA. Prevalence of the amylase-binding protein A gene (abpA) in oral streptococci. J Clin Microbiol. 1999;37:4081–4085. doi: 10.1128/jcm.37.12.4081-4085.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwynn JP, Douglas CW. Comparison of amylase-binding proteins in oral streptococci. FEMS Microbiol Lett. 1994;124:373–379. doi: 10.1111/j.1574-6968.1994.tb07311.x. [DOI] [PubMed] [Google Scholar]
- Scannapieco FA, Haraszthy GG, Cho MI, Levine MJ. Characterization of an amylase-binding component of Streptococcus gordonii G9B. Infect Immun. 1992;60:4726–4733. doi: 10.1128/iai.60.11.4726-4733.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri B, Rojek J, Vickerman MM, Tanzer JM, Scannapieco FA. Interaction of salivary alpha-amylase and amylase-binding-protein A (AbpA) of Streptococcus gordonii with glucosyltransferase of Sgordonii and Streptococcus mutans. BMC Microbiol. 2007;7:60. doi: 10.1186/1471-2180-7-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JD, Haase EM, Brown AE, Douglas CW, Gwynn JP, Scannapieco FA. Identification and analysis of a gene (abpA) encoding a major amylase-binding protein in Streptococcus gordonii. Microbiology. 1998;144:1223–1233. doi: 10.1099/00221287-144-5-1223. [DOI] [PubMed] [Google Scholar]
- Rogers JD, Palmer RJ, Jr, Kolenbrander PE, Scannapieco FA. Role of Streptococcus gordonii amylase-binding protein A in adhesion to hydroxyapatite, starch metabolism, and biofilm formation. Infect Immun. 2001;69:7046–7056. doi: 10.1128/IAI.69.11.7046-7056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitkova AE, Haase EM, Scannapieco FA. Taking the starch out of oral biofilm formation: molecular basis and functional significance of salivary alpha-amylase binding to oral streptococci. Appl Environ Microbiol. 2013;79:416–423. doi: 10.1128/AEM.02581-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P, Ragunath C, Vyas V, Shanmugam M, Ramasubbu N. Probing the interaction of human salivary alpha-amylase and amylase binding protein A(AbpA) of Streptococcus gordonii. Mol Biol. 2013;2:6. [Google Scholar]
- Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madauss KP, Burkhart WA, Consler TG, Cowan DJ, Gottschalk WK, Miller AB, Short SA, Tran TB, Williams SP. The human ACC2 CT-domain C-terminus is required for full functionality and has a novel twist. Acta Crystallogr D. 2009;65:449–461. doi: 10.1107/S0907444909008014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunath C, Manuel SG, Venkataraman V, Sait HB, Kasinathan C, Ramasubbu N. Probing the role of aromatic residues at the secondary saccharide-binding sites of human salivary alpha-amylase in substrate hydrolysis and bacterial binding. J Mol Biol. 2008;384:1232–1248. doi: 10.1016/j.jmb.2008.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Huang Y, Sakaguchi K, Clore GM, Gronenborn AM, Craigie R. An efficient and cost-effective isotope labeling protocol for proteins expressed in Escherichia coli. J Biomol NMR. 1998;11:97–102. doi: 10.1023/a:1008222131470. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Keller RLJ. 2004. ) The computer aided resonance assignment tutorial, Cantina, Switzerland.
- d'Auvergne EJ, Gooley PR. Optimisation of NMR dynamic models II. A new methodology for the dual optimisation of the model-free parameters and the Brownian rotational diffusion tensor. J Biomol NMR. 2008;40:121–133. doi: 10.1007/s10858-007-9213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luginbuhl P, Guntert P, Billeter M, Wuthrich K. The new program OPAL for molecular dynamics simulations and energy refinements of biological macromolecules. J Biomol NMR. 1996;8:136–146. doi: 10.1007/BF00211160. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. , 29–32. [DOI] [PubMed] [Google Scholar]
- Serrano P, Pedrini B, Mohanty B, Geralt M, Herrmann T, Wuthrich K. The J-UNIO protocol for automated protein structure determination by NMR in solution. J Biomol NMR. 2012;53:341–354. doi: 10.1007/s10858-012-9645-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]