

Abstract
Aims
Understanding drug–drug interactions (DDI) is a critical part of the drug development process as polypharmacy has become commonplace in many therapeutic areas including the cancer patient population. The objectives of this study were to investigate cytochrome P450 (CYP)-mediated DDI profiles available for therapies used in the oncology setting and evaluate how models based on in vitro–in vivo extrapolation performed in predicting CYP-mediated DDI risk.
Methods
A dataset of 125 oncology therapies was collated using drug label and approval history information, incorporating in vitro and clinical PK data. The predictive accuracy of the basic and net effect mechanistic static models was assessed using this oncology drug dataset, for both victim and perpetrator potential of CYP3A-mediated DDI.
Results
The incidence of CYP3A-mediated interaction potential was 47%, 22% and 11% for substrates, inhibitors and inducers, respectively. The basic models for precipitants gave conservative predictions with no false negatives, whilst the mechanistic static models provided reasonable quantitative predictions (2.3–3-fold error). Further analysis revealed that incorporating DDI at the level of the intestine was in most cases over-predicting interaction magnitude due to overestimates of the rate and extent of oral absorption of the precipitant. Quantifying victim DDI potential was also demonstrated using fmCYP3A estimates from ketoconazole clinical DDI studies to predict the magnitude of interaction on co-administration with the CYP3A inducer, rifampicin (1.6–3.3 fold error).
Conclusions
This work illustrates the utility and limitations of current DDI risk assessment approaches applied to a range of contemporary anti-cancer agents, and discusses the implications for therapeutic combination strategies.
Keywords: anti-cancer therapy, CYP3A, CYP induction, CYP inhibition, drug interactions, pharmacokinetics
What is Already Known about this Subject
Recent years have seen the validation and enhancement of in vitro–in vivo extrapolation models for drug–drug interaction (DDI) risk assessment based on data from human in vitro test systems. Previous studies have established the utility of these models using large datasets of prototypic interacting drug pairs, historical data on older drugs or blinded proprietary data.
What this Study Adds
The focus of this work was on anti-cancer drugs including many novel molecularly targeted agents, some with recent approvals, such as ceritinib, ibrutinib, trametinib, dabrafenib and crizotinib, to gain further understanding of how these approaches perform for therapies that are reflective of contemporary drug development.
Introduction
Understanding drug–drug interactions (DDI) is a critical part of the drug development process since a clinically relevant change in exposure of a co-administered drug can lead to loss of efficacy or, conversely, an adverse drug reaction (ADR), depending on the therapeutic window of the victim drug. The latter becomes especially important with anti-cancer medications since they are typically administered at or close to the maximum tolerated dose 1. In the general population, 20–30% of all ADRs have been attributed to DDI 2, whilst DDI are estimated to be the cause of death in approximately 4% of cancer patients 3.
The study of DDI potential is initiated early in drug discovery with preclinical assessment and characterization using appropriate in vitro tools of human systems, which depending on the outcomes of a thorough risk assessment may require formal clinical studies in support of labelling requirements and prescribing information. Within that continuum, advances in modelling and simulation approaches have enabled a more quantitative perspective to better inform decision making around DDI risk assessment and mitigation, a strategy that has largely evolved from the fact that chronic drug therapy and polypharmacy are commonplace in many patient populations. This is especially true in the cancer patient population, where a patient may be receiving multiple drugs for the primary disease, various comorbidities as well as palliative and supportive care 4. In addition, combination therapy wherein synergistic activity of two or more therapeutic agents is exploited to enhance disease or survival outcomes is an important drug development paradigm, either with standard-of-care agents or as novel combinations, the latter being exemplified with the recent FDA accelerated approval of dabrafenib and trametinib in combination, for the treatment of unresectable or metastatic melanoma with BRAF V600E or V600K mutations 5.
The primary focus in pharmacokinetic DDI is the CYP family of enzymes because of their promiscuity and prevalence in the metabolism of many drugs and other xenobiotics. In addition, the activity and expression level of various isoforms of this enzyme family can be modulated through inhibitory or inductive processes mediated by drugs and other xenobiotics. Within the CYP superfamily, CYP3A is the most important isoform since it is involved in the metabolism of approximately 50% of known drugs, is inducible by drugs and other small molecule agents (via the transactivation of the nuclear receptors, PXR and CAR) and is the major isoform expressed in the intestinal epithelium where it acts as a metabolic barrier to oral absorption.
In 2012, both the FDA and EMA issued revised guidance documents on DDI which describe a tiered risk assessment strategy for potential perpetrators of CYP DDI that utilizes basic approaches (R value) followed by the mechanistic static model and, where appropriate, dynamic physiologically-based pharmacokinetic (PBPK) models 6,7. For the evaluation of an investigational agent as a victim of drug interactions, a consensus threshold of ≥ 25% of systemic clearance is recommended based on results from in vitro enzyme phenotyping experiments, human pharmacokinetic studies following intravenous administration, a mass-balance study and/or pharmacokinetic studies in which renal/biliary clearances are determined.
The aim of this study was to investigate the CYP-mediated DDI profiles available for those therapies used in the oncology setting (excluding biologics) and evaluate how the basic and mechanistic static models performed in predicting DDI risk for these drugs. More specifically, (i) to identify the metabolic pathways typically implicated in drug interactions for oncology drugs, (ii) to assess the performance of the static models in predicting the potential of oncology drugs as perpetrators of CYP-mediated DDI and (iii) to assess the performance of the static models in predicting the potential of oncology drugs as victims of CYP-mediated DDI. Many previous works have established the utility of these models and approaches for addressing perpetrator interactions of CYP, but less so with respect to victim interactions 8–13. In these prior reports, importantly model validation was performed using large datasets of prototypic interacting drug pairs (e.g. the impact of ketoconazole and rifampicin on the kinetics of midazolam), historical data on older drugs or blinded proprietary data. The focus of this work was on anti-cancer drugs including many novel molecularly targeted agents, some with very recent approvals (e.g. ceritinib, ibrutinib, trametinib, dabrafenib, crizotinib), to gain further understanding of how these approaches perform on a dataset that is reflective of contemporary drug development.
Methods
Data collection
With a focus on small molecule oncology drugs, data on route of administration, approved dose, drug disposition, pharmacokinetics, in vitro DDI and clinical DDI studies was obtained from drug label information and CDER Clinical Pharmacology reviews from the drug approval history as listed on http://www.accessdata.fda.gov/scripts/cder/drugsatfda/. This was supplemented, where available, with data from literature reports 8,9,14,15. CYP inhibition, competitive and time-dependent, was obtained from in vitro studies using human liver microsomes. If Ki was not directly measured, Ki was assumed to follow, Ki = IC50/2 (for competitive inhibitors, assuming assay conditions of [S] = Km). CYP induction data were obtained from studies determining EC50 and Emax of mRNA as measured in cultured human hepatocytes. The CYP3A Ki values used for ketoconazole and itraconazole were 0.006 and 0.005 μ m respectively 12. For rifampicin a CYP3A EC50 value of 0.57 μ m and Emax of 33 were used 12,13. The AUC ratio (AUC in the presence of test perpetrator: AUC in the absence of test perpetrator; AUCR) from clinical DDI studies involving CYP3A probe substrates (midazolam, simvastatin, everolimus, verapamil), probe inhibitors (ketoconazole, itraconazole, diltiazem) and probe inducers (rifampicin, phenytoin, phenobarbital, carbamazepine) was collated. Dose and regimen, route of administration, systemic Cmax, free fraction in plasma or blood were obtained for all perpetrators.
Mechanistic static models
The mechanistic static models used in this work were as previously described 12 and outlined below, to calculate the net effect of competitive inhibition, inactivation and induction of CYP3A on the AUC ratio of the probe substrate:
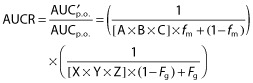 |
where A is the term for time-dependent inhibition (TDI) in the liver
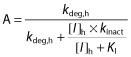 |
B is the term for induction in the liver
C is the term for reversible inhibition in the liver
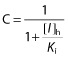 |
X is the term for TDI in the intestine
 |
Y is the term for induction in the intestine
Z is the term for reversible inhibition in the intestine
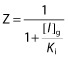 |
where [I]g and [I]h represent concentrations of perpetrator relevant for the intestine and liver, respectively. For the liver, the observed systemic unbound Cmax was used in the equations of time-dependent inhibition (term A) and induction (term B). A calculated portal unbound Cmax (based on the observed systemic Cmax) was utilized in equations for reversible inhibition (term C) as follows:
For the intestine, [I]g was used in terms X, Y and Z and was estimated using the following equation:
Values used for the degradation rates of CYP3A4 (kdeg) were 0.00032 min−1 and 0.00048 min−1 for the liver and intestine, respectively 13,16,17. Emax, EC50, and d represent the maximum-fold induction of mRNA observed in cultured human hepatocytes, the concentration of inducer associated with half-maximum mRNA induction, and a calibration factor d (0.3), as described previously 13. A midazolam fmCYP3A (fraction metabolised by CYP3A) of 0.93 and Fg (fraction of dose escaping gut first pass extraction) of 0.57 were used in this analysis 17. For the other CYP3A probe substrates, verapamil, simvastatin and everolimus fmCYP3A values of 0.3, 0.91 and 0.9 were used, respectively, and Fg was assumed to be 0.5 in all three cases. Enterocytic and hepatic blood flows of Qg 0.3 l min−1 and Qh 1.617 l min−1 were used throughout. When fraction absorbed (Fa) and absorption rate constant (ka) were not available, default values of 1 and 0.1 min−1 were used respectively, representing complete and rapid intestinal absorption.
Analysis of oncology drugs as perpetrators of CYP3A interactions
Prior to application of the mechanistic static models described above, the R values were calculated as outlined in the Food and Drug Administration (FDA) Guidance for Industry: Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labelling Recommendations, Draft Guidance, February 2012. For reversible inhibition and time-dependent inhibition an R value greater than 1.1 triggers further assessment as a potential perpetrator of CYP-mediated DDI based on:
for reversible inhibition and;
for time-dependent inactivation, where [I] is the total systemic Cmax. Additionally, for orally administered drugs the R value threshold of 11 was used to calculate interaction risk at the level of the intestine where [I] above is substituted with [I]gut (=molar dose/250 ml). For induction an R value less than 0.9 triggers further assessment as a potential perpetrator of CYP-mediated DDI based on:
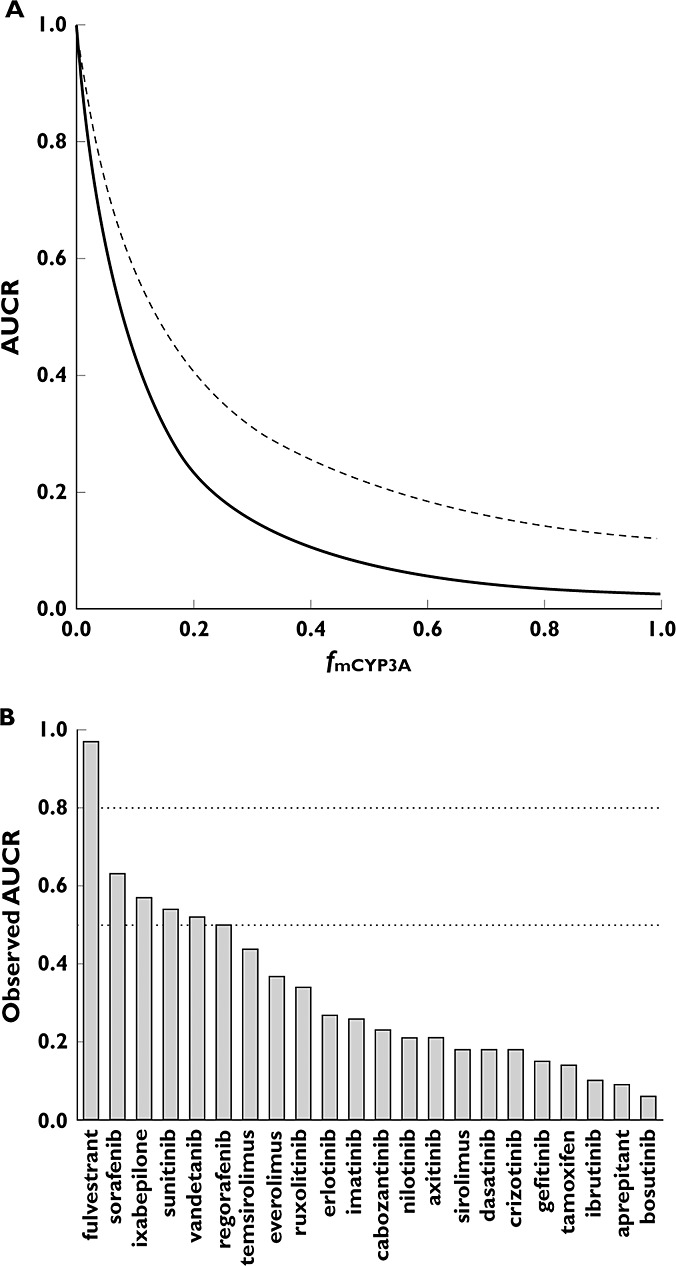
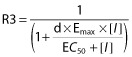 |
where [I] is total systemic Cmax. Furthermore, a mRNA increase above a predefined threshold is also proposed and a 4-fold absolute mRNA induction relative to vehicle control was used in this work, as recommended by Fahmi et al. 18, although this method only applied to one drug in the dataset, nilotinib. The mechanistic static model was also applied to the data, modelling both interactions at the level of the liver only or the combined effect of liver and intestine. The model performance was addressed using classification statistics as well as root mean square error (RMSE) and geometric mean fold error (GMFE) values.
Analysis of oncology drugs as victims of CYP3A interactions
An analogous approach was performed for oncology drugs as victims of CYP3A interactions using the mechanistic static model. The challenge in this case is defining the fractional clearance by CYP3A (fmCYP3A) based on the available data. This was achieved using the estimate of the in vivo fmCYP3A contribution based on ketoconazole clinical interaction data as below:
It should be noted that this relationship holds true under certain conditions. Firstly, for oral administration it assumes no change in F and therefore no change in Fg (CYP3A) or, for example, no inhibition of transporters that would affect the oral absorption or hepatic uptake of the victim drug. As described by Kirby et al. 19, AUCR is also impacted by changes in hepatic extraction even in cases where Fa and Fg do not change, which may lead to an underestimation of fm especially for high clearance drugs. However, this is not straightforward to address with this dataset since there is a paucity of hepatic extraction data, e.g. i.v. PK data are not available, or lack of quantitative information on hepatic clearance.
Importantly, complete inhibition of CYP3A is also assumed. This is a reasonable assumption given that ketoconazole elicits potent inhibition of CYP3A and the dosing paradigm of this agent in clinical DDI studies is well established. In addition, the impact on CYP3A activity has to be considered selective. If there was an effect on other enzymes involved in the metabolism of the victim drug then the fmCYP3A would be overestimated. These in vivo fmCYP3A estimates were then utilized in the mechanistic static model to predict the likely magnitude of an interaction on co-administration with rifampicin. The other challenge in modelling CYP3A victim interactions is deriving a reasonable estimate of Fg. In this study, a simple inverse linear relationship that is bounded by fmCYP3A of 0 equating to Fg = 1 and the fmCYP3A (0.93) and Fg (0.57) parameters for midazolam was used. The limitation with this perhaps overly simplistic relationship is as fm tends to unity, Fg tends to 0.5 which may not be reflective of the observed situation when other factors influence Fg such as permeability, active efflux, etc. Since accurate Fg values are not usually available in early drug development this was considered a reasonable assumption for the purposes of static modelling.
Results
Data were obtained on 125 of the most commonly used small molecule oncology therapeutics as shown in Table S1. These drugs represent a diverse range of pharmacological modalities including anti-metabolites, hormonal agents, kinase inhibitors, alkylating agents, differentiating agents, anti-tumour antibiotics and mitotic inhibitors that capture many of the traditional cytotoxic chemotherapeutics, molecularly targeted agents as well as adjuvant therapies. Amongst this group of drugs, route of administration is not limited to oral, with intravenous, subcutaneous and intramuscular dose routes also represented. A review of the routes of elimination and in vitro drug interaction data revealed a large number of the drugs to be substrates, inhibitors or inducers of CYP3A, perhaps not surprising given the promiscuity of this enzyme in the metabolism of drugs and other xenobiotics. In this dataset, 47% were substrates of CYP3A (or had CYP3A involvement in their disposition, e.g. elimination of active metabolites), whilst 22% had in vitro evidence for CYP3A inhibition (reversible or time-dependent) and 11% had in vitro evidence for CYP3A induction. In addition, 21% of the drug dataset showed both victim and perpetrator interaction behaviour with respect to CYP3A, and therefore based on in vitro data had the potential to impact their own metabolism (auto-induction or auto-inhibition). Based on these findings, a more detailed data analysis was conducted to evaluate in vitro–in vivo extrapolation and risk assessment approaches for drug interactions of anti-cancer medications.
Oncology drugs as perpetrators of CYP3A DDIs
A summary of the oncology drugs with evidence of CYP3A perpetrator behaviour is shown in Table 1, including in vitro inhibition (kinact, KI, and Ki where applicable) and induction (EC50 and Emax where applicable) data, the R value scores based on the Food and Drug Administration (FDA) Guidance for Industry: Drug Interaction Studies-Study Design, Data Analysis, Implications for Dosing, and Labelling Recommendations, Draft Guidance, February 2012, as well as the observed AUCR for the probe CYP3A substrate used. Within this dataset based on in vitro data, 10 drugs showed reversible and time-dependent inhibition, six drugs showed reversible inhibition only, four showed reversible inhibition and induction, three showed time-dependent inhibition only, two showed induction only and two showed reversible inhibition, time-dependent inhibition and induction.
Table 1.
Summary of oncology agents studied as perpetrators of CYP3A-mediated DDI
| Drug | Route of administration | CYP3A KI (μ m) | CYP3A kinact (min−1) | CYP3A Ki (μ m) | CYP3A mRNA Emax | CYP3A mRNA EC50 (μ m) | R1/R2 value for inhibition (systemic Cmax)* | R1/R2 value for inhibition (dose/250 ml)† | R3 value for induction | Observed AUCR | Clinical probe substrate |
|---|---|---|---|---|---|---|---|---|---|---|---|
| abiraterone | orally as acetate prodrug | 8.01 | 1.08 | 1428 | |||||||
| aprepitant | orally | 10 | 6.76 | 1.81 | 1.3 | 95 | 0.19 | 3.3 | midazolam | ||
| axitinib | orally | 8.3 | 1.01 | 7.2 | |||||||
| bicalutamide | orally | 2.33 | 1.77 | 201 | 1.9 | midazolam | |||||
| cabazitaxel | i.v. | 1.99 | 1.09 | n/a | |||||||
| carfilzomib | i.v. | 11 | 0.1 | 0.85 | 7.92 | n/a | 0.9 | midazolam | |||
| crizotinib | orally | 3 | 0.11 | 3.65 | 19.5 | 1.45 | 1.25 | 610 | 0.12 | 3.7 | midazolam |
| dabrafenib | orally | 7.89 | 30 | 12 | 1.36 | 147 | 0.15 | 0.26 | midazolam | ||
| dasatinib | orally | 1.9 | 0.022 | 18 | 1.01 | 33 | 1.2 | simvastatin | |||
| enzalutamide | orally | 42 | ‡ | 1.74 | 34 | 0.12 | midazolam | ||||
| erlotinib | orally | 8.2 | 0.057 | 44.62 | 178 | 0.76 | midazolam | ||||
| everolimus | orally | 0.9 | 0.022 | 2.3 | 5.48 | 19 | |||||
| fosaprepitant | i.v. | § | n/a | 1.6 | midazolam | ||||||
| fulvestrant | i.m. | 6600 | <1.1 | n/a | 1.11 | midazolam | |||||
| gefitinib | orally | 14.1 | 0.019 | 4.10 | 60 | n.f. | |||||
| imatinib | orally | 4.4 | 0.028 | 8 | 1.66 | 407 | 3.5 | simvastatin | |||
| ixabepilone | i.v. | 7.5 | 0.043 | 7.5 | 9.37 | n/a | n.f. | ||||
| lapatinib | orally | 29.2 | 0.031 | 1.1 | 6.00 | 7825 | 1.45 | midazolam | |||
| nilotinib | orally | 1.5 | 0.033 | 0.448 | 72 | 10.6 | 5064 | >4-fold threshold | 1.3¶ | midazolam | |
| pazopanib | orally | 2.9 | 0.021 | 6.25 | 140.25 | 1170 | 1.32 | midazolam | |||
| romidepsin | i.v. | 25.7 | 1.04 | n/a | |||||||
| sirolimus | orally | 0.9 | 0.027 | 2 | 2.47 | 34 | 1.5 | verapamil | |||
| sorafenib | orally | 0.881 | 0.045 | 27.6 | 1.60 | 126 | 0.85 | midazolam | |||
| sunitinib | orally | 31.5 | 0.02 | 1.20 | 60 | ||||||
| temsirolimus | i.v. | 0.6 | 0.021 | 3.1 | 1.19 | n/a | ** | ||||
| toremifene | orally | 8.6 | †† | 1.23 | 70 | 1.0 | midazolam | ||||
| trametinib | orally | 37.3 | 2.7 | n/a | n/a | 0.65 | 1.16 | everolimus | |||
| vemurafenib | orally | >50 | ‡‡ | n.d. | n.d. | 0.61 | midazolam |
Data obtained from drug label information and CDER Clinical Pharmacology reviews from the drug approval history as listed on http://www.accessdata.fda.gov, supplemented, where needed, with data from literature reports 8,9,14,15.
Threshold based on FDA guidance is R > 1.1. R value was calculated based on reversible inhibition data, followed by TDI data if reversible inhibition R ≤ 1.1.
Threshold based on FDA guidance is R > 11. R value was calculated based on reversible inhibition data, followed by TDI data if reversible inhibition R ≤ 11.
Concentration dependent effects up to 2.5 μm; < 28% 10 μm rifampin response.
Unstable in human in vitro test systems.
Following single dose administration of nilotinib.
Not performed, as no in vivo effect of CYP2D6 inhibition was observed.
15% of positive control response at 5 μm.
40% of positive control response. n/a; not applicable.
Using the R value approach for inhibition (reversible or time-dependent) based on total systemic Cmax, six drugs lead to R < 1.1 and 19 drugs gave R > 1.1. Using the R value approach for inhibition based on [I]gut (molar dose/250 ml), one oral drug gave an R value < 11 whilst 18 oral drugs gave R values > 11. Clinical data were available for 15 drugs where a formal DDI study with concomitant administration of a sensitive CYP3A substrate was performed. Using the R value approach for induction, four drugs with quantitative induction data (EC50 and Emax) gave rise to R < 0.9. Clinical data were available for all of these drugs where a formal DDI study with concomitant administration of a sensitive CYP3A substrate was performed. Where a clinical study was performed, midazolam was the most commonly used probe CYP3A substrate (15/19), whilst simvastatin (2/19), verapamil (1/19) and everolimus (1/19) were also used. Clinical interaction AUCR magnitude ranged from 0.12 with enzalutamide through to 3.7 with crizotinib. Using an AUCR threshold range of 0.8–1.25 for a negative drug interaction (and excluding complex interactions with both inhibitory and inductive potential), the R value approach lead to six true positives, two true negatives and five false positives. Obviously, it is difficult to assess fully classification accuracy in this situation since clinical studies were rarely conducted for compounds with R values falling outside the requisite range for further evaluation. Nevertheless, this is an encouraging result in that no false negatives were generated.
The performance of the mechanistic static model is illustrated in Figure 1. Using the full model including both interaction at the liver and intestine leads, in general, to over-prediction in the magnitude of the effect on a sensitive CYP3A substrate. This over-prediction is markedly reduced using the model incorporating interactions solely at the level of the liver. Using an AUCR threshold range of 0.8–1.25 for a negative drug interaction, the combined liver and intestine mechanistic static model led to nine true positives, one true negative, five false positives and two false negatives (Table 2). By comparison, the mechanistic static model for the liver alone led to six true positives, four true negatives, three false positives and four false negatives. Interestingly, the total number of misclassifications (false positives and false negatives) does not change between the liver alone and liver plus intestine models. Going from the combined model to the model for liver interactions alone does reduce the number of false positives but consequently increases the number of false negatives by a similar amount. Using a less stringent threshold (AUCR 0.5–2) does not necessarily improve the classification accuracy as shown in Table 2. A more quantitative indicator of predictive accuracy is illustrated in the RMSE and GMFE values for these two models. The liver and intestine combined model gives an RMSE of 4.67 and a GMFE of 3.00 whilst the liver alone model gives an RMSE and GMFE of 2.45 and 2.27 respectively. This suggests that modelling interactions solely at the level of the liver affords an improved predictive accuracy and that there may be confounding factors in the modelling of drug interactions at the level of the intestine. Focusing on a subset of the drugs, those with in vitro evidence for both inhibitory and inductive potential for CYP3A, it is interesting to see how the mechanistic static model performs for these compounds since conceptually a holistic approach is provided by this model. Compounds falling into this category include aprepitant, crizotinib, dabrafenib, enzalutamide, nilotinib and toremifene. For aprepitant, both models underpredicted the magnitude of the inhibitory effect (2.5–3.5-fold). For crizotinib and nilotinib, both models overpredicted the magnitude of the inhibitory effect (2–3.6 fold and 1.9–3.3 fold respectively), although it should be noted that the nilotinib–midazolam clinical interaction study was conducted with a single dose of nilotinib and so is likely an underestimate of the steady-state effect of nilotinib on midazolam kinetics. As a result of the clinical data being from a single dose study, the TDI component was excluded from the model. If TDI was included, the predicted AUCR for midazolam using the combined liver and intestine model was 14 (c.f. observed AUCR 1.3). For dabrafenib, both models did not predict that induction would predominate (3.8–4.4-fold error), whilst for enzalutamide both models underpredicted the induction effect (2.4–4-fold). For toremifene, the combined model was less accurate than the liver only model in predicting the lack of an interaction.
Figure 1.
Observed vs. predicted AUCR for oncology drugs as perpetrators of CYP3A DDI using the mechanistic static model for liver and intestine (A) and liver only (B). AUCR threshold of 0.8 and 1.25 shown as dotted lines as well as line of unity and 2-fold error lines
Table 2.
Statistics of the mechanistic static model for oncology drugs as perpetrators
| Model | Combined model | Liver only model | Combined model | Liver only model |
|---|---|---|---|---|
| Threshold | 0.8–1.25 | 0.5–2.0 | ||
| TN | 1 | 4 | 3 | 6 |
| TP | 9 | 6 | 3 | 3 |
| FP | 5 | 3 | 9 | 6 |
| FN | 2 | 4 | 2 | 2 |
| Correct classification rate (%) | 59 | 59 | 35 | 53 |
| RMSE | 4.67 | 2.45 | ||
| GMFE | 3.00 | 2.27 | ||
FN, false negative; FP, false positive; TN, true negative; TP, true positive.
Oncology drugs as victims of CYP3A DDI – inhibition
A summary of the data for the effect of CYP3A inhibition on the PK of oncology drugs is shown in Table S2, including route of administration of the victim oncology drug, the clinical probe inhibitor used and its dosing regimen as well as the observed AUCR of the oncology drug. A total of 33 interaction studies were obtained of which the vast majority used ketoconazole as the probe inhibitor (27/33), with itraconazole less commonly used (4/33) and single examples of other agents (diltiazem and corticosteroids). Ketoconazole was typically administered as 400 mg once daily although 200 mg twice daily was also used in some cases. Itraconazole was typically administered as 200 mg once daily. The magnitude of effect on AUC of the victim anti-cancer drug ranged from 0.89 with sorafenib at the lower end through to the highest observed AUCR of 24 with ibrutinib. Interestingly, for irinotecan and its inactive oxidative metabolite (APC), AUCR of 0.26 and 0.03, respectively, were observed on co-administration with ketoconazole. It has been reported that the 33-fold reduction in the inactive metabolite APC and inhibition of this pathway by ketoconazole leads to a shunting of irinotecan metabolism towards the esterase mediated route leading to elevated concentrations of the active metabolite, SN-38 20.
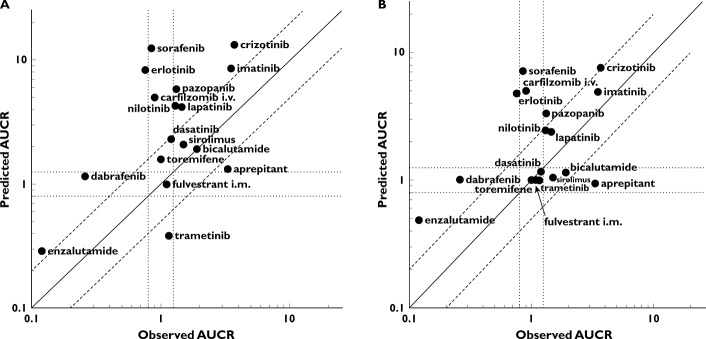
The relationship between fmCYP3A and AUCR for ketoconazole using the mechanistic static model is shown in Figure 2A. The sensitivity of prediction of DDI magnitude to fmCYP3A is clearly demonstrated in Figure 2A, especially at fmCYP3A of >0.7. The impact of including intestinal inhibition of CYP3A also has a much greater effect at fm > 0.7 as shown by the two simulated curves. Using the mechanistic static model with both liver and intestinal CYP3A inhibition by ketoconazole, the threshold AUCR values of 1.25 (no effect), 2 (moderate) and 5 (strong) correspond to fmCYP3A values of 0.15, 0.4 and 0.7, respectively. A comparison of the mechanistic static model for ketoconazole and itraconazole is shown in Figure 2B. This indicates that for weak and moderate CYP3A substrates the AUCR on co-administration of ketoconazole or itraconazole will be largely similar and will fall within the same AUCR thresholds of 1.25 and 2. Once a compound exhibits an fmCYP3A of > 0.7 (i.e. a sensitive substrate) the predicted AUCR would be categorized differently, strong for ketoconazole (AUCR > 5) and moderate for itraconazole (AUCR 2–5) (see Discussion). For comparative purposes, the observed AUCR on co-administration with a strong CYP3A inhibitor are shown in Figure 2C.
Figure 2.

(A) Relationship between fmCYP3A and AUCR for ketoconazole-mediated inhibition of CYP3A. Combined effect of liver and intestine (solid line) and liver only (dashed line). AUCR thresholds of 1.25, 2 and 5 shown as horizontal dotted lines. (B) Comparative relationship between AUCR and fmCYP3A for ketoconazole (solid line) and itraconazole (dashed line) using the liver and intestine model. (C) Observed AUCR on co-administration with strong or moderate CYP3A inhibitors. Diltiazem (dark grey), itraconazole (medium grey) and ketoconazole (light grey)
Oncology drugs as victims of CYP3A DDI – induction
A summary of the data for the effect of CYP3A induction on the PK of oncology drugs is shown in Table S3, including route of administration of the victim oncology drug, the clinical probe inducer used and its dosing regimen as well as the observed AUCR of the oncology drug. A total of 25 interaction studies were obtained, of which the vast majority used rifampicin as the probe inducer (23/25), with enzyme inducing anti-convulsants less commonly used (2/25). In all cases of rifampicin co-administration, a dosing regimen of 600 mg once daily was used. The magnitude of effect on AUC of the victim anti-cancer drug ranged from 0.06 for bosutinib at the lower end through to 0.97 for fulvestrant. The relationship between fmCYP3A and AUCR for rifampicin using the mechanistic static model is shown in Figure 3A. This also highlights the sensitivity of prediction of DDI magnitude to fmCYP3A as was shown for ketoconazole. In the case of rifampicin, the impact of including intestinal induction of CYP3A has a much greater effect at fm > 0.2 as shown by the two simulated curves in Figure 3A. Using the mechanistic static model with both liver and intestinal CYP3A induction by rifampicin, the fmCYP3A values of 0.15, 0.4, and 0.7 previously noted for ketoconazole simulate more marked changes in AUCR than the corresponding inhibition by ketoconazole. For example at fmCYP3A 0.4, a 2-fold change is predicted for ketoconazole with a 9-fold change predicted for rifampicin, and at fmCYP3A 0.7, a 5-fold change is predicted for ketoconazole with a 25-fold change predicted for rifampicin. For comparative purposes, the observed AUCR on co-administration with rifampicin are shown in Figure 3B.
Figure 3.
(A) Relationship between fmCYP3A and AUCR for rifampicin-mediated induction of CYP3A. Combined effect of liver and intestine (solid line) and liver only (dashed line). (B) Observed AUCR on co-administration with rifampicin
In order to establish a more quantitative assessment of victim interactions, there were 19 oncology drugs which had data from both ketoconazole and rifampicin interaction studies. The observed AUCR mediated by ketoconazole was used to derive an fmCYP3A estimate which was subsequently incorporated into projections of the likely AUCR magnitude on co-administration with rifampicin, as shown in Figure 4. The predicted AUCR when both liver and gut components are included leads to a marked overprediction of the observed effect of rifampicin, while the model of liver interaction alone gives a much improved prediction with most data points falling within the 2-fold limits. The combined liver and gut model gave rise to a RMSE of 0.24 and a GMFE of 3.30 while the liver model led to a RMSE of 0.20 and GMFE of 1.64.
Figure 4.
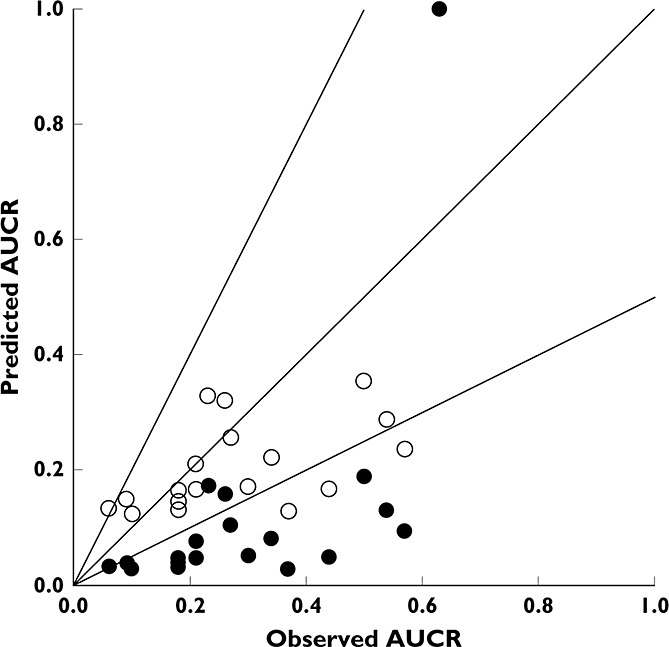
Observed vs. predicted AUCR for oncology drugs as victims of CYP3A DDI using the mechanistic static model for liver and intestine (closed circles) and liver only (open circles). Line of unity and 2-fold error lines shown for clarity
Discussion
The aim of this study was to investigate the CYP-mediated DDI profiles available for those therapies used in the oncology setting (excluding biologics) and evaluate how the basic and mechanistic static models performed in predicting DDI risk for these drugs. More specifically, (i) to identify the metabolic pathways typically implicated in drug interactions for oncology drugs, (ii) to assess the performance of the static models in predicting the potential of oncology drugs as perpetrators of CYP-mediated DDI and (iii) to assess the performance of the static models in predicting the potential of oncology drugs as victims of CYP-mediated DDI. Many previous works have established the utility of these models and approaches for addressing perpetrator interactions of CYP, but less so with respect to victim interactions 8–13. In these prior reports, importantly model validation was performed using large datasets of prototypic interacting drug pairs (e.g. the impact of ketoconazole and rifampicin on the kinetics of midazolam), historical data on older drugs or blinded proprietary data. The focus of this work was on oncology drugs including many novel molecularly targeted agents, some with very recent approvals (e.g. ceritinib, ibrutinib, trametinib, dabrafenib, crizotinib), to gain further understanding of how these approaches perform on a dataset that is reflective of contemporary drug development.
A review of the oncology drug landscape with regard to elimination pathways and CYP-mediated DDI potential revealed a prevalence of CYP3A substrates, inhibitors and inducers. This is perhaps not surprising given the importance of this enzyme in the clearance of drugs, and corroborates earlier observations in pharmacotherapy 21–23. However, it does raise the potential for complex DDIs both in considering combination therapeutic strategies, likely concomitant medications as well as time-dependent pharmacokinetics if an agent has the prospect of impacting its own metabolism by auto-inhibition or auto-induction. In addition to the frequency of co-medication use, the risk of a clinically relevant DDI will also be largely determined by the therapeutic window of the victim drug. Combination therapy is also typically employed in an attempt to decrease the likelihood of drug resistance, enhance efficacy and disease modification; particularly with novel therapies, the molecular basis for the concerted action of two or more agents being the selective and specific targeting of multiple nodes in an oncogenic-dependent transduction pathway. However, the judicious selection of molecularly targeted agents is needed to ensure there is no potential for clinically relevant DDI. The recent accelerated approval of trametinib and dabrafenib in combination is a good example. On co-administration there is a 23% increase in dabrafenib AUC, 33% increase in desmethyl-dabrafenib AUC and no change in trametinib or hydroxy- dabrafenib. Trametinib is not a CYP substrate, inhibitor or inducer and so despite the CYP3A induction potential shown for dabrafenib, the magnitude of interaction between the two agents is low and not clinically relevant.
Evaluating oncology drugs as precipitants of drug interactions based on basic and mechanistic static models revealed a number of important observations. The basic models generating separate R values for reversible inhibition, TDI and induction indicated no false negatives, which is reassuring as a conservative, initial risk assessment approach early in drug development. This is likely driven by the use of total concentration rather than free or unbound that is typically applied in the mechanistic models. Moving to the net effect mechanistic static model of liver and intestinal DDI also in general led to an over-prediction of the AUCR magnitude expected on co-administration with a sensitive CYP3A substrate. This was obviated to some extent by simplifying the model to interactions solely at the level of the liver, suggesting that there may be some confounding factors in modelling drug interactions in the gut for these compounds. Nevertheless, the predictive accuracy of this model for these compounds was very similar to that observed previously in larger analyses of prototypic CYP3A DDI data. A recent comprehensive review of static model prediction methods applied to a dataset of 119 CYP3A inhibitory interactions gave a RMSE and GMFE of 5.07 and 1.97, respectively, with the mechanistic static model using [I]h tailored to the specific interaction (unbound portal Cmax for reversible inhibition, unbound systemic Cmax for TDI) and RMSE 7.59, GMFE 2.82 when [I]h was set to unbound portal Cmax for both inhibitory mechanisms 8,9. This compares favourably with the model performance observed in this study (RMSE 4.67 and GMFE 3.00) albeit with a smaller dataset but nevertheless incorporating multiple modes of drug interaction. The liver only model led to further improvements in predictive accuracy (RMSE 2.45, GMFE 2.27) which is discussed in more detail below. Focusing on a subset of the drugs, those with in vitro evidence for both inhibitory and inductive potential of CYP3A, it is interesting to see how the mechanistic static model performs for these compounds since conceptually a holistic approach is provided by this model. For those drugs with dual inhibitory and inductive potential the model generally did well in correctly predicting the directionality of the interaction, with the exception of dabrafenib. Quantitatively there is probably some improvement needed although aprepitant may be underpredicted due to an absence of TDI kinetic data, while nilotinib is likely overpredicted due to AUCR derived from a single dose clinical study, highlighting the need for cautious and careful review of both in vitro and in vivo data when performing these types of analyses.
Modelling drug interactions from the perspective of the victim is clearly very dependent on the fractional clearance by the CYP isoform in question, and fmCYP is one of the most sensitive parameters to accurate AUCR predictions. From the regulatory perspective, evaluating an investigational agent as a victim of drug interactions, there is a consensus threshold of ≥25% of systemic clearance based on results from in vitro enzyme phenotyping experiments, human pharmacokinetic studies following intravenous administration, a mass-balance study and pharmacokinetic studies in which renal/biliary clearances are determined. However, early in drug development, fm estimation can be confounded by the lack of appropriate data (e.g. human radiolabel absorption, distribution, metabolism and excretion study) or is limited to simple in vitro reaction phenotyping data, at a stage when DDI risk assessment is deemed necessary or when clinical DDI studies are being considered. In this work, fm estimates derived from the ketoconazole interaction data were utilized to see how they would perform in modelling the interaction upon concomitant administration with the CYP3A inducer, rifampicin. Previous work looking at CYP3A-mediated victim interactions have tended to make use of in vitro CYP phenotyping data to derive fmCYP3A, and shown reasonable success 10. Using the ketoconazole-derived fmCYP3A, the liver only model performs soundly with most data points falling within the 2-fold limits, and with good predictive accuracy as judged by an RMSE of 0.20 and GMFE of 1.64. This demonstrates that with robust fm estimation based on clinical data, conceptually the mechanistic static model for liver provides quantitative predictions of CYP3A-mediated victim interactions, and is in good agreement with previous studies which have shown utility in predicting CYP3A induction-mediated DDIs for a wide range of drugs 11. This raises the possibility that modelling and simulation approaches could help prioritize clinical DDI studies during drug development. With the recent increased regulatory focus on DDI beyond CYP-mediated mechanisms (i.e. transport-mediated), there is potential for the number of clinical DDI studies per new molecular entity to rise with a consequent burden in development costs and resources. The application of static and dynamic modelling approaches to aid clinical DDI study prioritization during development warrants further investigation and is not unprecedented with the recent inclusion of PBPK modelling and simulation results to support ibrutinib drug interaction labelling statements.
One of the challenges in using fractional clearance inhibited by ketoconazole to estimate fmCYP3A is the potential concomitant impact on MDR1-mediated clearances (e.g. biliary and renal) since ketoconazole is a reasonably potent inhibitor of MDR1. In addition, the mechanistic static model assumes that oral bioavailability remains unchanged in the presence of the perpetrator and that may not be the case if there is appreciable inhibition of CYP3A and MDR1 at the gut wall mediated by ketoconazole. Interestingly, 74% of the victim oncology drugs in this analysis have in vitro evidence of MDR1-mediated transport, which may suggest that fmCYP3A is overestimated. However, it should be noted that rifampicin also induces MDR1 via PXR transactivation. Hence although this is not a straightforward system to deconvolute, fmCYP3A, in this instance, may represent a composite parameter including MDR1 and CYP3A mediated clearance.
On October 16 2013, the FDA advised against using oral ketoconazole in drug interaction studies due to serious potential side effects including liver injury and adrenal gland problems, and proposed the use of alternative strong CYP3A inhibitors for these studies, specifically mentioning clarithromycin and itraconazole. Since then, investigators have been evaluating the potential options since ketoconazole was considered the strong inhibitor of choice and was used in the vast majority of DDI studies of strong CYP3A inhibition. It has been noted that both itraconazole (midazolam AUCR 6.16–10.8, at 200 mg once daily for 4 days) and clarithromycin (maximal midazolam AUCR 8.4 at 500 mg twice daily for 7 days) are clinically less potent than the standard high dose ketoconazole regimen (AUCR 9.5–16.7 at 400 mg once daily for 4–10 days) and are more comparable with lower dose ketoconazole (AUCR 5.2–9.2 at 200 mg once daily for 3–5 days) 12,24,25. That trend between itraconazole and ketoconazole is borne out in simulations using the mechanistic static model (Figure 2C) where a midazolam fm of 0.93 predicts an AUCR of 4.6 and 18.4 for itraconazole and ketoconazole, respectively. The 2.3-fold underprediction of AUCR mediated by itraconazole is likely due to the absence of any contribution from the known inhibitory metabolites, hydroxy-itraconazole and N-desalkyl-itraconazole. Following multiple dosing, 40–50% of the overall CYP3A inhibition has been attributed to hydroxy-itraconazole (major) and N-desalkyl-itraconazole (minor) 26,27. Furthermore, the ketoconazole simulations indicate the AUCR thresholds of 1.25 (no effect), 2 (moderate) and 5 (strong) correspond to fmCYP3A values of 0.15, 0.4 and 0.7, respectively. Comparatively, the 25% of systemic clearance threshold proposed in recent DDI regulatory guidances, leads to an AUCR of 1.5, demonstrating reasonable congruence between the two approaches.
The mechanistic static model including both liver and intestinal components led to overprediction of AUCR magnitude in general. As suggested in previous works, this may be a limitation of the model in that the scenario of dynamic changes in drug concentrations of both perpetrator and victim are not incorporated as they are in PBPK approaches. It also does not capture the potential role of rate-limiting active transport processes in the disposition in liver or the gut wall. However, the major factor in limiting further improved predictive accuracy appears related to the estimation of [I]g and Fg. For [I]g calculations of many of the oncology drugs administered orally, Fa and ka were assumed to be 1 and 0.1 min−1. The oral oncology drugs with the highest fold-error in prediction (sorafenib, erlotinib, crizotinib and pazopanib) exhibit comparatively slow (tmax in the range of 3–8 h) and incomplete (Fa ≤ 0.5) absorption. Nonetheless, conservative assumptions around Fa and ka are appropriate when making prospective predictions early in development. Estimating Fg of an investigational agent can be challenging. In this study, a simple inverse linear relationship between fmCYP3A and Fg was assumed (bounded by fmCYP3A = 0, Fg = 1 and the reported values for midazolam, fmCYP3A = 0.93 and Fg = 0.57). This simple relationship appears suitable for most drug discovery and early development applications although further investigation is warranted especially in extrapolation as fmCYP3A tends to unity and Fg ∼0.54. Furthermore, it would be interesting to see if interactions at the level of the intestine are more accurately modelled based on the observed Cmax ratio, rather than AUCR.
The quantitative prediction of DDI potential has been enabled through modelling and simulation strategies that encompass static and dynamic systems. However, it is noteworthy that there is compelling evidence that CYP3A, in particular, is modulated through a number of mechanisms beyond inhibition and induction mediated by low molecular weight compounds. For example, certain biologics can modulate CYP3A expression levels via changes in cytokines related to their mechanism of action 28 and there have been in vitro observations of direct CYP3A5 activation with sorafenib and sunitinib 29. In addition, new therapeutic modalities such as antibody drug conjugates (ADC) can exhibit CYP3A mediated interactions, e.g. the cytotoxic component of the recently approved HER2 ADC, ado-trastuzumab emtansine, is metabolized by CYP3A 30. Furthermore, there are increasingly complex scenarios as illustrated above with irinotecan metabolism. The incidence of CYP3A interaction potential amongst molecularly targeted anti-cancer agents is further exemplified with the recent approval of idelalisib, a PI3K-delta inhibitor for relapsed CLL, FL and SLL (data not available at the time of this analysis). Labelling statements include avoidance of concomitant dosing with CYP3A inducers and substrates, based on AUCR of 0.25 observed in DDI studies with rifampicin and in addition Idelalisib is also characterized as a strong CYP3A inhibitor as it increased the AUC of midazolam 5.4-fold on co-administration 31.
A tiered DDI risk assessment approach that utilizes basic and mechanistic static models has shown good predictive performance for a set of contemporary oncology drugs with a multitude of CYP3A-mediated interactions. With robust fm estimates, the mechanistic static model also shows utility in predicting DDIs for victim drugs. The propensity for CYP3A involvement in the disposition of many anti-cancer agents as substrates, inhibitors and inducers, should be carefully evaluated from a patient population perspective but also in considering combination therapy approaches.
Competing Interests
The author has completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declares NJW had support from Epizyme, Inc. for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
The author gratefully acknowledges Dr Robert A. Copeland for review of this manuscript.
Supporting Information
Table S1
Dataset of anti-cancer drugs including therapeutic mechanism, route of administration, route of elimination, and DDI potential
Table S2
Summary of oncology agents studied as victims of CYP3A-mediated DDI: Inhibition
Table S3
Summary of oncology agents studied as victims of CYP3A-mediated DDI: Induction
Supporting infor item
Supporting infor item
Supporting infor item
References
- Jansman FGA, Reyners AKL, van Roon EN, Smorenburg CH, Helgason HH, Le Comte M, Wensveen BM, van den Tweel AMA, de Blois M, Kwee W, Kerremans AL, Brouwers JRBJ. Consensus-based evaluation of clinical significance and management of anticancer drug interactions. Clin Ther. 2011;33:305–14. doi: 10.1016/j.clinthera.2011.01.022. [DOI] [PubMed] [Google Scholar]
- Kohler GI, Bode-Boger SM, Busse R, Hoopmann M, Welte T, Boger RH. Drug–drug interactions in medical patients: effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000;38:504–513. doi: 10.5414/cpp38504. [DOI] [PubMed] [Google Scholar]
- Buajordet I, Ebbesen J, Erikssen J, Brors O, Hilberg T. Fatal adverse drug events: the paradox of drug treatment. J Intern Med. 2001;250:327–41. doi: 10.1046/j.1365-2796.2001.00892.x. [DOI] [PubMed] [Google Scholar]
- van Leeuwen RWF, Brundel DHS, Neef C, van Gelder T, Mathijssen RHJ, Burger DM, Jansman FGA. Prevalence of potential drug–drug interactions in cancer patients treated with oral anticancer drugs. Br J Cancer. 2013;108:1071–1078. doi: 10.1038/bjc.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies AM, Long GV. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin Cancer Res. 2014;20:2035–43. doi: 10.1158/1078-0432.CCR-13-2054. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration [FDA] 2012. Guidance for industry: drug interaction studies –study design, data analysis, implications for dosing, and labeling recommendations, draft guidance, February. Available at http://www.fda.gov.
- European Medicines Agency [EMA] 2012. Guideline on the investigation of drug interactions, 21 June. Available at http://www.ema.europa.eu.
- Vieira MLT, Kirby B, Ragueneau-Majlessi I, Galetin A, Chien JYL, Einolf HJ, Fahmi OA, Fischer V, Fretland A, Grime K, Hall SD, Higgs R, Plowchalk D, Riley R, Seibert E, Skordos K, Snoeys J, Venkatakrishnan K, Waterhouse T, Obach RS, Berglund EG, Zhang L, Zhao P, Reynolds KS, Huang S-M. Evaluation of various static in vitro–in vivo extrapolation models for risk assessment of the CYP3A inhibition potential of an investigational drug. Clin Pharmacol Ther. 2014;95:189–98. doi: 10.1038/clpt.2013.187. [DOI] [PubMed] [Google Scholar]
- Einolf HJ, Chen L, Fahmi OA, Gibson CR, Obach RS, Shebley M, Silva J, Sinz MW, Unadkat JD, Zhang L, Zhao P. Evaluation of various static and dynamic modeling methods to predict clinical CYP3A induction using in vitro CYP3A4 mRNA induction data. Clin Pharmacol Ther. 2014;95:179–88. doi: 10.1038/clpt.2013.170. [DOI] [PubMed] [Google Scholar]
- Youdim KA, Zayed A, Dickins M, Phipps A, Griffiths M, Darekar A, Hyland R, Fahmi OA, Hurst S, Plowchalk DR, Cook J, Guo F, Obach RS. Application of CYP3A4 in vitro data to predict clinical drug–drug interactions; predictions of compounds as objects of interaction. Br J Clin Pharmacol. 2008;65:680–92. doi: 10.1111/j.1365-2125.2007.03070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou M, Hayashi M, Pan Y, Xu Y, Morrissey K, Xu L, Skiles GL. Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction. Drug Metab Dispos. 2008;36:2355–70. doi: 10.1124/dmd.108.020602. [DOI] [PubMed] [Google Scholar]
- Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, Dickins M, Phipps A, Darekar A, Hyland R, Obach RS. Comparison of different algorithms for predicting clinical drug–drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab Dispos. 2009;37:1658–66. doi: 10.1124/dmd.108.026252. [DOI] [PubMed] [Google Scholar]
- Fahmi OA, Maurer TS, Kish M, Cardenas E, Boldt S, Nettleton D. A combined model for predicting CYP3A4 clinical net drug-drug interaction based on CYP3A4 inhibition, inactivation, and induction determined in vitro. Drug Metab Dispos. 2008;36:1698–708. doi: 10.1124/dmd.107.018663. [DOI] [PubMed] [Google Scholar]
- Kenny JR, Mukadam S, Zhang C, Tay S, Collins C, Galetin A, Khojasteh SC. – potential of marketed oncology drugs: in vitro assessment of time-dependent cytochrome P450 inhibition, reactive metabolite formation and drug–drug interaction prediction. Pharm Res. 2012;29:1960–76. doi: 10.1007/s11095-012-0724-6. [DOI] [PubMed] [Google Scholar]
- Filppula AM, Neuvonen PJ, Backman JT. In vitro assessment of time-dependent inhibitory effects on CYP2C8 and CYP3A activity by fourteen protein kinase inhibitors. Drug Metab Dispos. 2014;42:1202–9. doi: 10.1124/dmd.114.057695. [DOI] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K, Houston JB, Tremaine LM. In vitro cytochrome P450 inhibition data and the prediction of drug–drug interactions: qualitative relationships, quantitative predictions, and the rank-order approach. Clin Pharmacol Ther. 2005;78:582–92. doi: 10.1016/j.clpt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K. Mechanism-based inactivation of human cytochrome P450 enzymes and the prediction of drug–drug interactions. Drug Metab Dispos. 2007;35:246–55. doi: 10.1124/dmd.106.012633. [DOI] [PubMed] [Google Scholar]
- Fahmi OA, Kish M, Boldt S, Obach RS. Cytochrome P450 3A4 mRNA is a more reliable marker than CYP3A4 activity for detecting pregnane X receptor-activated induction of drug-metabolizing enzymes. Drug Metab Dispos. 2010;38:1605–11. doi: 10.1124/dmd.110.033126. [DOI] [PubMed] [Google Scholar]
- Kirby BJ, Unadkat JD. Impact of ignoring extraction ratio when predicting drug–drug interactions, fraction metabolized, and intestinal first-pass contribution. Drug Metab Dispos. 2010;38:1926–33. doi: 10.1124/dmd.110.034736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer DFS, Mathijssen RHJ, Verweij J, de Bruijn P, Sparreboom A. Modulation of irinotecan metabolism by ketoconazole. J Clin Oncol. 2002;20:3122–29. doi: 10.1200/JCO.2002.08.177. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 2nd edn. New York: Plenum Press; 1995. [Google Scholar]
- Bloomer J, Derimanov G, Dumont E, Ellens H, Matheny C. Optimizing the in vitro and clinical assessment of drug interaction risk by understanding co-medications in patient populations. Expert Opin Drug Metab Toxicol. 2013;9:737–51. doi: 10.1517/17425255.2013.781582. [DOI] [PubMed] [Google Scholar]
- Teo Yi L, Ho Han K, Chan A. Metabolism-related pharmacokinetic drug–drug interactions in tyrosine kinase inhibitors: current understanding, challenges and recommendations. Br J Clin Pharmacol. 2014 doi: 10.1111/bcp.12496. doi: 10.1111/bcp.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke AB, Zamek-Gliszczynski MJ, Higgins JW, Hall SD. Itraconazole and Clarithromycin as ketoconazole alternatives for clinical CYP3A inhibition studies. Clin Pharmacol Ther. 2014;95:473–76. doi: 10.1038/clpt.2014.41. [DOI] [PubMed] [Google Scholar]
- Han B, Mao J, Chien JY, Hall SD. Optimization of drug–drug interaction study design: comparison of minimal physiologically based pharmacokinetic models on prediction of CYP3A inhibition by ketoconazole. Drug Metab Dispos. 2013;41:1329–38. doi: 10.1124/dmd.112.050732. [DOI] [PubMed] [Google Scholar]
- Templeton IE, Thummel KE, Kharasch ED, Kunze KL, Hoffer C, Nelson WL, Isoherranen N. Contribution of itraconazole metabolites to inhibition of CYP3A4 in vivo. Clin Pharmacol Ther. 2008;83:77–85. doi: 10.1038/sj.clpt.6100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton I, Peng CC, Thummel KE, Davis C, Kunze KL, Isoherranen N. Accurate prediction of dose-dependent CYP3A4 inhibition by itraconazole and its metabolites from in vitro inhibition data. Clin Pharmacol Ther. 2010;88:499–505. doi: 10.1038/clpt.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease– involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2011;89:735–40. doi: 10.1038/clpt.2011.35. [DOI] [PubMed] [Google Scholar]
- Sugiyama M, Fujita K-, Murayama N, Akiyama Y, Yamazaki H, Sasaki Y. Sorafenib and sunitinib, two anticancer drugs, inhibit CYP3A4-mediated and activate CY3A5-mediated midazolam 1'-hydroxylation. Drug Metab Dispos. 2011;39:757–62. doi: 10.1124/dmd.110.037853. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration. 2014. KADCYLA Product label information. Available at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. (last accessed August )
- Food and Drug Administration. 2014. ZYDELIG Product label information. Available at http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. (last accessed August )
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting infor item
Supporting infor item
Supporting infor item