

Abstract
Background
Progressive muscular atrophy (PMA) is a rare type of degenerative motor neuron disease (MND) of which the onset happens in adult period. Despite its well-defined clinical characteristics, its neuropsychological profile has remained poorly understood, considering the consensus of cognitive and behavioral impairment reached in amyotrophic lateral sclerosis (ALS).
Methods
We conducted a cross-sectional evaluation of Chinese PMA patients with a series of comprehensive batteries emphasizing the executive and attention function, and covering other domains of memory, language, visuospatial function, calculation and behavior as well. Their performances were compared with those of age- and education-matched ALS and healthy controls (HC).
Results
21 patients newly diagnosed with PMA were consecutively enrolled into our ALS and other MND registry platform, accounting for 14.7% of all the incident MND cases registered during the same period. 20 patients who completed the neuropsychological batteries were included into analysis. Compared with HC, PMA performed significantly worse in maintenance function of attention, while they exhibited quantitative similarity to ALS in all behavioral inventories and neuropsychological tests except the time for Stroop interference effect.
Conclusion
PMA could display mild cognitive dysfunction in the same frontal-mediated territory of ALS but in a lesser degree, whereas they did not differ from ALS behaviorally.
Introduction
Progressive muscular atrophy (PMA) is an adult onset progressive neurological disease, clinically manifested by prominent lower motor neuron (LMN) involvement and the absence of upper motor neuron (UMN) features, yet pathologically appearing as a variant of amyotrophic lateral sclerosis (ALS) [1]. It has been shown that ALS is subject to a neurodegenerative spectrum beyond motor neurons: sharing similar genetic and molecular basis with frontotemporal lobe degeneration (FTLD), up to 50% of patients with ALS have demonstrated cognitive and/or behavioral impairment, especially in frontal-mediated territory, and 10% to 15% of the patients could display frank dementia [2–4]. Thus it is a reasonable hypothesis that patients with PMA could exhibit the same pattern of deficit with that of ALS if they belong to the same pathological category. However, most relevant researches for ALS adopted 1998 revised El Escorial criteria [5] which excluded the category of “suspected ALS”[6], and consequently PMA was not included into analysis. Bias might arise for the aim of depicting the neuropsychological profile in the context of motor neuron disease (MND), considering PMA accounting for around 10% of MND [7] and their potential of turning into ALS with UMN signs appearing [8]. Besides, clarifying this issue would in turn assist to confirm whether ALS and PMA are heterogeneous or not.
These facts contributed to the need of expanding investigations to the relative less frequent subtype of MND. Hence, our object is to characterize cognitive and behavioral status in PMA, and to testify whether PMA and ALS belong to a same entity by comparing them with ALS and healthy controls (HC). By this effort, we hope to provide more comprehensive description of Chinese patients with MND.
Methods
Inclusion and exclusion criteria
All newly diagnosed PMA patients would be consecutively enrolled into our registry platform for ALS and other MND [7, 9] and neuropsychological investigations began in September, 2013. In this present study, PMA patients fulfilled the inclusion criteria proposed by Visser J et al as follows [8]: (1) a disease duration of less than 5 years from the time of symptom onset; (2) clinical and electrophysiological evidence of LMN involvement in two or more of four regions (bulbar, cervical, thoracic and lumbosacral); (3) no conduction blocks on nerve conduction studies; and (4) no clinical UMN signs and symptoms, including forced yawning, crying and laughing, clonus of masseter reflex, (sub)clonic myotatic reflexes, Hoffmann-Trömner sign, extensor plantar response or spasticity. The data of those with history of other neurological conditions which could have an impact on neuropsychological assessments (major stroke, traumatic brain injury, learning disability and severe active epilepsy), alcohol-dependence, drug-dependence, severe active mental illness, use of high-dose psychoactive medication, other mother language instead of Chinese Mandarin and illiteracy were excluded from analysis. 20 ALS patients registered in the platform during the same period and 20 HC were recruited to our hospital, and they were selected in accordance with the same exclusion criteria as PMA patients. Patients with PMA, ALS and HC were matched for age and education level. This study was approved by the Research Ethics Committee of Peking Union Medical College Hospital. Patients visited our clinic accompanied by their legal guardians, and all the patients and HC were included after informed written consent had been obtained from them or their guardians, as set forth by the Declaration of Helsinki.
Demographic and clinical assessment
Demographic and clinical information including age, gender, level of education, site of symptom onset, disease duration (defined as time lapse between symptom onset and time of neuropsychological evaluation) were collected. Disease severity was assessed by the revised ALS functional rating scale (ALSFRS-R) [10]. To further analyze bulbar function, we then applied swallowing subscale of ALS Severity Scale (ALSSS) [11].
Neuropsychological assessment
We’ve chosen a series of standardized batteries covering the cognitive domains of executive function, memory, language, attention, visuospatial function and calculation, especially emphasizing the detection of executive and attention dysfunction. Selected tests are as follows: the Mini-Mental State Examination (MMSE) [12]; category and phonemic verbal fluency [13–15]; the Stroop Color-Word Test (SCWT) [16]; the Clock Drawing Test [17] (4 point method was adopted and lower than 4 point was defined as abnormality); paired associate word learning of the Clinical Memory Test (CMT) [18]; episodic memory of modified Wechsler Memory Scale (WMS) [19]; the Symbol Digit Modalities Test [20]; digit span and calculations of the Wechsler Adult Intelligence Scale (WAIS) [21]; copy and repetition subsets from Aphasia Battery of Chinese (ABC) [22]. Stroop interference effect (SIE) was assessed by time (Stroop C time-Stroop B time) and correct number (Stroop B correct number- Stroop C correct number), respectively. The neurobehavioral evaluation was conducted through the interview with caregivers about the patients’ daily performance and the administration of the Frontal Behavioral Inventory-ALS version (FBI-ALS) [23, 24]. It is a 24-item scale originally designed to screen behavioral variant FTLD (bvFTD) and afterwards revised for availability to patients with ALS by adding scripted questions to distinguish behavioral symptoms from physical disability. Depression and anxiety were assessed by Hamilton Depression Rating Scale (HAMD) [25] and Hamilton Anxiety Scale (HAMA) [26].
The batteries were administered within a week after the diagnosis being made, and it required about 2 hours to complete them. So, in order to reduce the impact of fatigue, subjects could choose to take a short break or finish it at the same time next day if necessary, but the tests must be completed in a given sequence to avoid the possible interference of one test over the following ones.
Case-by-case analysis
By comprehensively analyzing medical history, cognitive and behavioral performance, we obtained a conclusion about whether the patient or the control was demented or not. The Neary criteria for frontotemporal dementia were adopted to diagnose FTLD [27], and the diagnosis of non-FTLD dementia was based on the criteria of the Diagnostic and Statistical Manual of Mental Disorders-IV [28]. To further analyze behavioral change, we adopted he consensus criteria proposed by Strong et al [29] to diagnose ALS/MND with behavioral impairment. We did not make the diagnosis of mild cognitive impairment because several batteries lack normative value for Chinese population.
Statistical Methods
Continuous values were shown in the form of mean (standard deviation, SD) or median and those categorical ones were in proportion.
Concerning intergroup comparison, χ2 test was employed to analyze nominal variables and non-parametric tests were adopted for continuous variables because none of these variables were normally distributed. Specifically, Kruskal-Wallis tests were adopted for the comparison of clinical and demographical characteristics between ALS, PMA and HC. Mann-Whitney tests were used to analyze cognitive performances of PMA vs. ALS, PMA vs. HC and ALS vs. HC, respectively. In this section, we aimed to display in detail intergroup differences of cognitive data, so we chose multiple 2 sample comparison instead of a 3 group test and Mann-Whitney as post hoc. And afterwards Bonferroni correction was made to adjust α value. For the comparison of continuous variables of behavioral and mood status between PMA and ALS, Mann-Whitney tests was also employed.
The level of significance was set at p<0.05. Statistical analyses were carried out using SPSS 11.5 (SPSS Inc).
Results
From September 1st, 2013 to August 31st, 2014, 21 patients newly diagnosed with PMA were consecutively enrolled into our ALS and MND registry platform, accounting for 14.7% of all the incident MND cases registered during the same period (Fig 1). None of them had a family history of MND, dementia or psychosis. The mean age of PMA patients at the time of diagnosis was 52.6±13.0 years (22.0–74.0 years). 4 patients (19.0%) had bulbar-onset PMA, while the remaining 17 patients (81.0%) had spinal onset disease. Median time from symptom onset to diagnosis was 12 months (range 5–40). At baseline, median ALSFRS-R score was 42 (range 28–47), and median ALSSS swallowing score was 10 (range 7–10). At the time of diagnosis, no one had gastrostomy or non-invasive ventilation. All of them underwent neuropsychological tests, but the data of a 69 year-old female patient was excluded due to illiteracy, thus there were 20 patients in the final analysis. Recruited ALS patients fulfilled the revised El Escorial criteria for clinically definite (2 patients), probable (11 patients), and lab-supported probable ALS (7 cases). Age, education level and gender distribution were not significantly different among PMA, ALS and HC group. Differences of clinical variables, including onset type, function score, disease duration and progression rate were also proved to be not significant between PMA and ALS (Table 1).
Fig 1. Spectrum of MND registered in Peking Union Medical College Hospital (n = 143).
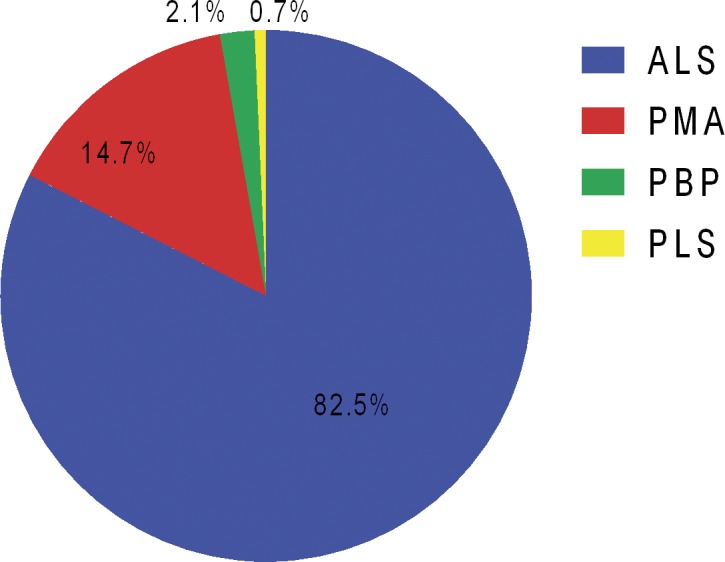
ALS, amyotrophic lateral sclerosis; MND, motor neuron disease; PBP, progressive bulbar palsy; PLS, progressive lateral sclerosis; PMA, progressive muscular atrophy.
Table 1. Demographic and clinical characteristics of patients and controls.
| PMA (n = 20) | ALS (n = 20) | HC (n = 20) | p Value | |
|---|---|---|---|---|
| Age at symptom onset (years) | 50.6±12.6 | 50.4±10.5 | 0.871 | |
| Age at evaluation (years) | 51.8±12.7 | 51.6±10.4 | 51.7±10.7 | 0.951 |
| Education level (years) | 10.4±3.8 | 10.8±3.7 | 10.5±3.1 | 0.876 |
| Gender (male, %) | 70 | 65 | 45 | 0.233 |
| Onset type (bulbar, %) | 20 | 15 | 0.677 | |
| Swallowing subscale of ALSSS | 9.7±0.9 | 9.8±0.8 | 0.655 | |
| ALSFRS-R score | 40.6±4.8 | 42.2±3.3 | 0.313 | |
| Disease duration (months) | 14.1±9.3 | 13.8±11.7 | 0.464 | |
| Progression rate | 0.6±0.5 | 0.7±0.5 | 0.766 |
ALS, amyotrophic lateral sclerosis; ALSFRS-R, revised ALS functional rating scale; ALSSS, ALS severity scale; HC, healthy controls; PMA, progressive muscular atrophy; the disease progression rate was calculated according to the formula of (48-ALSFRS-R score)/disease duration (month).
Data were means±SD.
In the aspect of cognitive performances, compared to HC, PMA patients performed significantly worse in episodic memory and attention maintenance detected by time of Stroop A and B, whereas ALS patients had significantly longer time for SIE not only than HC but also than PMA. And significant difference also occurred between ALS patients and HC in the MMSE and Stroop B time. However, only difference of Stroop A time between PMA patients and HC, and longer time for SIE of ALS patients than HC and PMA patients survived Bonferroni correction (α = 0.05/3 = 0.017). All three groups did not significantly differ in the cognitive domains of language, visuospatial function and calculation (Table 2).
Table 2. Comparison of neuropsychological performances among patients with PMA, ALS and HC.
| PMA | ALS | HC | p Value | |||
|---|---|---|---|---|---|---|
| PMA vs. HC | ALS vs. HC | PMA vs.ALS | ||||
| MMSE | 27.8±1.3 | 26.7±1.8 | 28.1±1.1 | 0.558 | 0.020 | 0.062 |
| Executive function | ||||||
| Phonemic verbal fluency | 6.0±2.6 | 5.4±1.8 | 5.1±2.7 | 0.257 | 0.303 | 0.773 |
| Category verbal fluency | 15.8±4.1 | 17.7±4.2 | 18.1±4.3 | 0.097 | 0.946 | 0.114 |
| Backward digital span of the WAIS | 5.1±1.5 | 4.9±1.6 | 5.2±1.3 | 0.725 | 0.421 | 0.615 |
| Correct for SIE | 1.2±2.3 | 2.9±3.5 | 2.1±2.8 | 0.274 | 0.520 | 0.117 |
| Time for SIE | 42.7±12.9 | 62.15±26.4 | 42.4±15.0 | 0.819 | 0.001 * | 0.003 * |
| the CDT raw score | 3.8±0.6 | 3.7±0.6 | 3.8±0.4 | 0.938 | 0.655 | 0.724 |
| Abnormal CDT (%) | 20 | 25 | 20 | 1.000 | 0.705 | 0.705 |
| Attention | ||||||
| Symbol Digit Modalities Test | 41.1±9.9 | 38.2±11.9 | 42.3±8.6 | 0.603 | 0.255 | 0.346 |
| Forward digital span of the WAIS | 5.1±1.5 | 4.9±1.6 | 5.2±1.3 | 0.832 | 0.102 | 0.150 |
| Stroop A time | 55.0±13.9 | 51.3±12.0 | 44.8±9.1 | 0.007 * | 0.101 | 0.626 |
| Stroop B time | 81.1±17.9 | 81.9±21.2 | 69.8±12.0 | 0.049 | 0.035 | 0.819 |
| Memory | ||||||
| Paired associate word learning of the CMT | 9.4±2.5 | 9.3±4.6 | 12.3±3.9 | 0.125 | 0.076 | 0.478 |
| Episodic memory of modified WMS | 4.8±1.5 | 5.4±1.8 | 6.0±1.5 | 0.041 | 0.307 | 0.450 |
| Language | ||||||
| Repetition of ABC | 35.5±1.6 | 35.5±1.7 | 35.5±1.5 | 0.496 | 0.846 | 0.625 |
| Visuospatial function | ||||||
| Copy of ABC | 8.7±2.1 | 9.2±1.7 | 9.4±1.0 | 0.520 | 0.814 | 0.764 |
| Calculation | ||||||
| Calculations of the WAIS | 13.0±1.6 | 12.6±3.4 | 12.3±3.9 | 0.662 | 0.849 | 0.841 |
3 PMA patients did not complete Stroop Color-Word Task; 2 PMA in copy tests and 1 PMA in digit span and Symbol Digit Modalities Test. ABC, Aphasia Battery Chinese; ALS, amyotrophic lateral sclerosis; CDT, clock drawing test; CMT, Clinical Memory Test; HC, healthy controls; PMA, progressive muscular atrophy; SIE, Stroop interference effect; time for SIE was calculated according to the formula of (Stroop C time-Stroop B time) and correct number for SIE was calculated according to the formula of (Stroop B correct number- Stroop C correct number); WAIS, Wechsler Adult Intelligence Scale; WMS, Wechsler Memory Scale.
Data were means±SD.
*p value remained to be significant after Bonferroni correction (α = 0.05/3 = 0.017).
The FBI-ALS scores were separated into two parts, negative and disinhibitive. ALS patients were not more susceptible to behavioral dysfunction than PMA patients in either subscale, no matter whether it was analyzed quantitatively or proportionately. As shown by HAMA, ALS patients are significantly more prone to anxiety than PMA patients, yet with similar degree in depression shown in HAMD (Table 3). In case-by-case analysis, none of the patients or the HC reached the diagnosis criteria of FTLD or non-FTLD dementia, and one ALS patients and 2 PMA patients could be diagnosed as behavioral impairment.
Table 3. Behavioral impairment and mood differences between PMA and ALS.
| PMA (n = 20) | ALS (n = 20) | p Value | |
|---|---|---|---|
| Behavioral impairment | |||
| FBI-ALS negative | 0.8±2.9 | 1.0±2.4 | 0.758 |
| FBI-ALS disinhibitive | 0.5±1.1 | 0.1±0.4 | 0.414 |
| FBI-ALS negative (score≥1, %) | 15 | 20 | 0.677 |
| FBI-ALS disinhibitive (score≥1, %) | 20 | 5 | 0.151 |
| Mood | |||
| HAMD | 2.5±3.1 | 2.7±3.2 | 0.495 |
| HAMA | 1.9±2.3 | 4.0±4.1 | 0.049 |
ALS, amyotrophic lateral sclerosis; FBI, frontal behavioral inventory; HAMA, Hamilton Anxiety Scale; HAMD, Hamilton Depression Rating Scale; PMA, progressive muscular atrophy.
Data were means±SD.
Discussion
The present study is the first one to describe cognitive and behavioral profile of PMA in a Chinese population. From the perspective of epidemiology, the proportion of PMA in our registry platform represents its actual ranking in MND spectrum [7], although it is a clinic-based design of small sample size. The demographic variables of included patients are similar to those published before [7, 9], and the high response rate has effectively minimized the selection bias. All of these have guaranteed the credibility of this cohort to truly reflect the characteristics of Chinese patients. In the diagnosis link, evidence of 2 LMN regions involved was set as the threshold of inclusion, but indeed, it was shown that all patients were neurophysiologic abnormal in at least 3 regions in electromyography test, which ensured the diagnosis accuracy.
Occupying the largest portion of neurodegenerative MND, ALS was regarded as the key to understanding the relationship between MND and FTLD [30–33]. However, it does not mean that other types of MND are free from cognitive or behavioral impairment. It has been revealed that primary lateral sclerosis (PLS), a purely UMN involved MND with relatively mild disease course, could display the similar pattern of cognitive decline to that of ALS [34], while disputes concerning PMA still remained. Research by Raaphorst J et al indicated that PMA patients showed cognitive deficit especially in executive function and attention/working memory [35], corresponding to their following neuroimaging results of prefrontal cortex activation abnormalities [36], while Wick P et al failed to identify any significant differences between PMA patients and HC on neuropsychological level [37]. In the present investigation, we have also observed several variables implying attention decline in PMA and ALS with only one survival after Bonferroni correction (PMA vs. HC in Stroop A time). Even if the data of patients with dysarthria were excluded from comparison, this difference remained to be significant (p = 0.01). This phenomenon detected by SCWT has seldom been reported in PMA, but in PLS [34], and it possibly indicates that these different phenotypes could be traced to a common origin. The importance of SCWT is not confined to this, although it is not applicable to patients with severe bulbar dysfunction. The only significant difference between PMA and ALS just in the executive area–resistance to interference- was revealed by it, supporting the previous finding of more extensive deficit in ALS [35]. The completion of SCWT required participation of prefrontal cortical areas, especially the dorsolateral prefrontal cortex which was just the vulnerable area of patients with ALS/MND [38, 39]. Also demanding involvement from multiple frontostriatal circuits, verbal fluency has been repeatedly proved to be a sensitive task for executive dysfunction in ALS [30–33]. However, this result was not replicated in the present study.
Intermediate status between cognitive intactness and FTLD also presented as behavioral changes. Compared by mean score of the FBI-ALS, PMA patients did not exhibit a significant milder extent of involvement than ALS in this aspect. Our conclusion might be challenged for its being established on an assumption that HC would nearly score 0 in the FBI-ALS, so we carried out the case-by-case analysis: one ALS could be diagnosed as ALS-behavioral impairment, whereas 2 PMA would receive the same diagnosis if the criteria were applied to them, which was still in favor of the occurrence of frontotemporal syndrome in PMA. Taking into consideration these results and their cognitive performance, none of them could be diagnosed as FTLD or dementia of other types. However, it is inappropriate to regard PMA as a special type resistant to dementia due to equally low rate of comorbid FTLD in ALS of Eastern Asian cohort, probably resulting from C9ORF72 mutation rarely seen in this population [30].
Clinically, ALS and PMA displayed unexpected comparabilities in such variables as onset type, function score, progression rate and so on, considering ALS patients were selected to match PMA ones with age and education in this study. Ruling out the possibility that their neuropsychological comparison was influenced by physical disability, this result might hint the homogeneity of these 2 subtypes. Disease progression rate before diagnosis we presented might be impacted by recall bias, though, it was still of predictive value because of suggested linear progression of ALS [40]. Besides, failing to be proved as an independent protective factor for prognosis, PMA phenotype could have relentless disease course as ALS [8]. Moreover, bulbar-onset patients seemed vulnerable to cognitive impairment and dementia, correlating with relevant cortex hypometabolism in Positron Emission Tomography [33]. We noticed relatively higher prevalence of bulbar-onset PMA in our cohort than previous ones [35, 37], and consequently the similar distribution of this type might serve as an explanation to almost the same neuropsychological performance between ALS and PMA.
Our study has several limitations. Firstly, single center-based investigation would be influenced by possible referral bias to some extent, making it necessary to testify our conclusion in future multicenter-based or population-based design of large sample size. Moreover, neuropsychological tests still waited to be perfected: considering the small proportion of patients with dysarthria, we did not make adjustment to the time-dependent tests which might potentially lower the score of patients with bulbar palsy. The assessment of premorbid IQ was not included in our neuropsychological batteries for avoiding patients’ fatigue. Last but not least, there is a possibility that PMA could turn into ALS with disease progression, and the cross-sectional observation would be inherently incapable of resolving this issue. Future follow-up study and comprehensive analysis of cognitive profile and disease turnover would further substantiate the relationship between ALS and PMA.
In conclusion, we depicted the cognitive and behavioral features of Chinese patients with PMA. They performed significantly worse in Stroop time than HC, providing evidence of attention impairment. When compared to ALS patients, they displayed cognitive dysfunction confined in a relatively smaller area but with similar pattern of behavioral impairment.
Acknowledgments
The authors thank all the patients, caregivers and healthy controls for their participation in this study.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by Sino-Germany Science Research Foundation (GZ876, http://www.sinogermanscience.org.cn) to BC LC ML. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ince PG, Evans J, Knopp M, Forster G, Hamdalla HH, Wharton SB, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60:1252–8. [DOI] [PubMed] [Google Scholar]
- 2. Murphy JM, Henry RG, Langnlore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:530–4. [DOI] [PubMed] [Google Scholar]
- 3. Phukan J, Pender NP, and Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol.2007;6:994–100. [DOI] [PubMed] [Google Scholar]
- 4. Lomen-Hoerth C. Clinical phenomenology and neuroimaging correlates in ALS-FTD.J Mol Neurosci. 2011;45:656–62. 10.1007/s12031-011-9636-x [DOI] [PubMed] [Google Scholar]
- 5. Brooks BR, Miller RG, Swash M, Musat M, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9. [DOI] [PubMed] [Google Scholar]
- 6. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors. J Neurol Sci. 1994;124 Suppl: 96–107. [DOI] [PubMed] [Google Scholar]
- 7. Cui F, Liu M, Chen Y, Huang X, Cui L Fan D, et al. Epidemiological characteristics of motor neuron disease in Chinese patients. Acta Neurol Scand. 2014;130:111–7. 10.1111/ane.12240 [DOI] [PubMed] [Google Scholar]
- 8. Visser J, van den Berg-Vos RM, Franssen H, van den Berg LH, Wokke JH, de Jong JM, et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol. 2007;64:522–8 [DOI] [PubMed] [Google Scholar]
- 9. Liu MS, Cui LY, Fan DS, Chinese ALS Association. Age at onset of amyotrophic lateral sclerosis in China. Acta Neurol Scand. 2014;129:163–7. 10.1111/ane.12157 [DOI] [PubMed] [Google Scholar]
- 10. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169:13–21. [DOI] [PubMed] [Google Scholar]
- 11. Hillel AD, Miller RM, Yorkston K, McDonald E, Norris FH, Konikow N. Amyotrophic lateral sclerosis severity scale. Neuroepidemiology. 1989;8:142–50. [DOI] [PubMed] [Google Scholar]
- 12. Zhang ZX, Hong X, Li H, Zhao JH, Huang JB Wei J, et al. [The Mini-Mental State Examination in population aged 55 years and over in urban and rural areas of Beijing]. Chin J Neurol. 1993;32:149–53. Chinese. [Google Scholar]
- 13. Butters N, Granholm E, Salmon DP, Grant I, Wolfe J. Episodic and semantic memory: a comparison of amnesic and demented patients. J Clin Exp Neuropsychol. 1987;9:479–97. [DOI] [PubMed] [Google Scholar]
- 14. Shi LL, Hong X, Ni J, Wu LY, Zhang ZX, Wei J. [Validity of verbal fluency in diagnosis of Alzheimer Disease]. Chin J Ment Health. 2009;23:701–5.Chinese [Google Scholar]
- 15. Abrahams S, Goldstein LH, Kew JJ, Brooks DJ, Lloyd CM, Frith CD, et al. Frontal lobe dysfunction in amyotrophic lateral sclerosis. A PET study. Brain. 1996;119:2105–20. [DOI] [PubMed] [Google Scholar]
- 16. Guo QH, Hong Z, Lv cz, zhou y, lu jc, ding d. [Application of Stroop color-word test on Chinese elderly patients with mild cognitive impairment and mild Alzheimer’s dementia]. Chin J Neuromed, 2005;4:701–4.Chinese. [Google Scholar]
- 17. Meng C, Zhang XQ, Wang H, Sun HL Liu HJ Tang Z, et al. [the Clock Drawing Test for detecting cognitive impairment]. Chin J Nerv Ment Dis. 2004;30:452–4. Chinese [Google Scholar]
- 18. The cooperation group for the construction of “The Clinical Memory Test”. [The construction of “The Clinical Memory Test”]. Acta Psychologica Sinica. 1986;1:100–8. Chinese. [Google Scholar]
- 19. Gong YX. Manual of modified Wechsler Memory Scale (WMS) Changsha, China: Hunan Med College; 1989. 19. [Google Scholar]
- 20. Sheridan LK, Fitzgerald HE, Adams KM, Nigg JT, Martel MM, Puttler LI, et al. Normative Symbol Digit Modalities Test performance in a community-based sample. Archives of Clinical Neuropsychology. 2006;21:23–8. [DOI] [PubMed] [Google Scholar]
- 21. Gong YX. Manual of modified Wechsler Adult Intelligence Scale (WAIS-RC) Changsha, China: Hunan Med College; 1982. p.45–48,52–53. [Google Scholar]
- 22. Gao S, He J, Li Y. Aphasia Beijing, China: Peking University Medical Press; 2006. p.439. [Google Scholar]
- 23. Murphy J, Ahmed F, Lomen-Hoerth C. The UCSF screening exam effectively screens cognitive and behavioral impairment in patients with ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;10:1–7. [DOI] [PubMed] [Google Scholar]
- 24.the National Institute of Neurological Disorders and Stroke Common Data Elements. http://www.commondataelements.ninds.nih.gov/
- 25. Moberg PJ, Lazarus LW, Mesholam RI, Bilker W, Chuy IL, Neyman I, et al. Comparison of the standard and structured interview guide for the Hamilton Depression Rating Scale in depressed geriatric inpatients. Am J Geriatr Psychiatry. 2001;9:35–40. [PubMed] [Google Scholar]
- 26. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959; 32:50–5. [DOI] [PubMed] [Google Scholar]
- 27. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–54. [DOI] [PubMed] [Google Scholar]
- 28. Association AP. Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington: American Psychiatric Association; 1994. [Google Scholar]
- 29. Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2009;10:131–46. [DOI] [PubMed] [Google Scholar]
- 30. Oh SI, Park A, Kim HJ, Oh KW, Choi H, Kwon MJ, et al. Spectrum of Cognitive Impairment in Korean ALS Patients without Known Genetic Mutations. PloS One. 2014;9:e87163 10.1371/journal.pone.0087163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. 2012;83:102–8. 10.1136/jnnp-2011-300188 [DOI] [PubMed] [Google Scholar]
- 32. Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology.2005;65:586–90. [DOI] [PubMed] [Google Scholar]
- 33. Montuschi A, Iazzolino B, Calvo A, Moglia C, Lopiano L, Restagno G, et al. Cognitive correlates in amyotrophic lateral sclerosis:a population-based study in Italy. J Neurol Neurosurg Psychiatry. 2015;86:168–73. 10.1136/jnnp-2013-307223 [DOI] [PubMed] [Google Scholar]
- 34. Piquard A, Le Forestier N, Baudoin-Madec V, Delgadillo D, Salachas F, Pradat PF,et al. Neuropsychological changes in patients with primary lateral sclerosis. 2006;7:150–60. [DOI] [PubMed] [Google Scholar]
- 35. Raaphorst J, de Visser M, van Tol MJ, Linssen WH, van der Kooi AJ, de Haan RJ, et al. Cognitive dysfunction in lower motor neuron disease: executive and memory deficits in progressive muscular atrophy. J Neurol Neurosurg Psychiatry. 2011;82:170–5. 10.1136/jnnp.2009.204446 [DOI] [PubMed] [Google Scholar]
- 36. Raaphorst J, van Tol MJ, Groot PF, Altena E, van der Werf YD, Majoie CB, et al. Prefrontal involvement related to cognitive impairment in progressive muscular atrophy. Neurology. 2014;83:818–25. 10.1212/WNL.0000000000000745 [DOI] [PubMed] [Google Scholar]
- 37. Wicks P, Abrahams S, Leigh PN, Williams T, Goldstein LH. Absence of cognitive, behavioral, or emotional dysfunction in progressive muscular atrophy. Neurology. 2006;67:1718–9. [DOI] [PubMed] [Google Scholar]
- 38. Vendrell P, Junque C, Pujol J, Jurado MA, Molet J, Grafman J. The role of prefrontal regions in the Stroop task. Neuropsychologia. 1995;33:341–52. [DOI] [PubMed] [Google Scholar]
- 39. Quinn C, Elman L, McCluskey L, Hoskins K, Karam C, Woo JH. Frontal lobe abnormalities on MRS correlate with poor letter fluency in ALS. Neurology. 2012;79:583–8. 10.1212/WNL.0b013e3182635720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Armon C, Graves MC, Moses D, Forte DK, Sepulveda L, Darby SM, et al. Linear estimates of disease progression predict survival in patients with amyotrophic lateral sclerosis. Muscle Nerve. 2000;23:874–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.