

Abstract
The capacity of pancreatic β-cells to adapt to insulin resistance is critical to maintain glucose homeostasis and a factor in the development of Type 2 diabetes. The insulin receptor substrate (IRS-2/phosphoinositide 3-kinase (PI3K) pathway plays a critical role in regulating β-cell mass and function. The serine-threonine kinase Akt also known as protein kinase B is one of the major downstream targets of the PI3K pathway and is negatively regulated by phosphatase and tensin homologue deleted on chromosome 10 (PTEN). This Akt signaling pathway recently has been implicated in cell cycle progression and survival of pancreatic β-cells. Understanding the mechanisms that link Akt to modulation of β-cell mass, function and plasticity will positively affect treatment of human diabetes.
Pancreatic β-cell mass and diabetes
β-cell failure is a major component of diabetes. Type 1 diabetes is characterized by insulin absence as a result of autoimmune destruction of β-cells. In this form of diabetes, the capacity of β-cells to regenerate and their susceptibility to apoptosis determine the timing of hyperglycemia. In type 2 diabetes, failure of β-cells to adequately expand in settings of insulin resistance results in hyperglycemia and diabetes. The normal response of β-cells to conditions associated with insulin resistance is to increase their mass. Current evidence suggests that alterations in Akt signaling not only play an important role in insulin resistance but also in the capacity of β-cells to adapt to increased insulin demand. This review focuses on the importance of Akt and its major modulator PTEN in regulating β-cell mass (proliferation, apoptosis, cell size and neogenesis).
Akt/PKB: biological function and in vivo models of Akt signaling
Three highly conserved isoforms of Akt/PKB (PKBα/Akt1, PKBβ/Akt2, and PKBγ/Akt3) encoded by three separate genes have been identified [1] (Box 1). Akt activity is regulated by both PI3K dependent and independent mechanisms and requires multiple steps that involve membrane translocation and phosphorylation (Box 1 and Figure 1) [2]. Akt activation phosphorylates substrates that control various biological processes including insulin-mediated glucose transport, protein and glycogen synthesis, proliferation, cell growth, differentiation, and survival (Figure 2). Experiments in mouse models have assessed the role of Akt in glucose homeostasis (Table 1). Akt1-deficient mice show placental hypotrophy, growth retardation and reduced body weight; Akt2-deficient mice exhibit glucose intolerance and insulin resistance, with some developing diabetes; Akt3-deficient mice exhibit reduced brain size [3-7] (Table 1). This suggests that Akt2 is more important for modulating insulin sensitivity and carbohydrate metabolism. The lack of functional compensation observed in these animals suggests that different Akt isoforms might be associated with distinct biological responses. The significance of each Akt isoform in β-cell mass and function has not yet been assessed. Activation of Akt signaling in transgenic mice overexpressing constitutively active Akt/PKB in β-cells (caAktTg) results in increased β-cell mass, proliferation, neogenesis and cell size providing further evidence for the role of Akt in islet physiology [8, 9] (Table 1). In contrast, 80% reduction in Akt activity by overexpressing a kinase dead mutant in β-cells results in an insulin secretory defect [10]. The current evidence suggests that growth factors, insulin, incretins and glucose converge on PI3K/IRS2/Akt signaling to regulate β-cell mass [11-16].
Figure 1. Schematic representation of Akt/PKB activation and signaling.
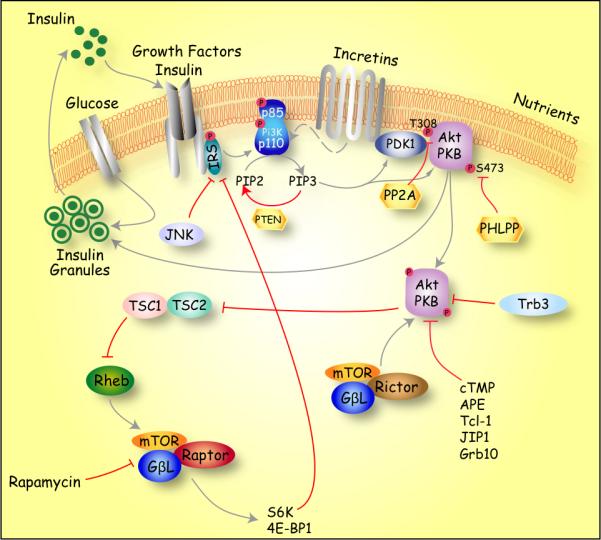
Activation of Akt signaling in β-cells occurs upon stimulation with Insulin growth factor (IGF)I, insulin, glucose, Hepatocyte growth factor (HGF), Fibroblast growth factor (FGF)21, Stromal derived factor (SDF), Obestatin and gut incretins such as Glucose-dependent insulinotropic polypeptide (GIP) and Glucagon like peptides (GLP), among others [90]. Akt activity is regulated at different levels via both Phosphoinositide 3-kinase (PI3K) dependent and independent mechanisms [72]. A critical step leading to activation of Akt is the generation of phosphatidylinositol (3,4,5) P3 phosphate (PIP3) by PI3K. PIP3 binding to the Pleckstrin homology-domain (PH) recruits Akt/PKB and phosphoinositide-dependent kinase-1 (PDK1) to the plasma membrane favoring phosphorylation of T308 (309 in AKT2 and 305 in AKT3). Phosphorylation of S473 by mTORC2 (mTor/Rictor/GβL) complex is necessary for full activation of Akt [91]. The inactivation of Akt/PKB signaling is mediated by protein phosphatase 2A (PP2A) and α isoform of PH-domain leucine-rich repeat phosphatase (PHLPP)-mediated dephosphorylation of T308 and S473 respectively. Another important mechanism that negatively regulates Akt/PKB activity is the dephosphorylation of PIP3 molecules by the phosphatase and Tensin homologue deleted on chromosome 10 (PTEN). Another level of regulation is achieved by binding to Akt-interacting proteins that lack significant kinase activity. These Akt interacting proteins include the mammalian homolog of Drosophila Tribbles (TRB3), and C-terminal modulator protein (cTMP) Akt phosphorylation enhancer (APE), Tcl-1 oncoprotein, c-Jun N-terminal kinase (JNK)–interacting protein 1 (JIP1), growth factor receptor–binding protein–10 (Grb10) among others. Akt activity can also be negatively regulated by c-Jun N-terminal kinase (JNK) after induction of oxidative stress, cytokines and ER stress, resulting in inhibition of Akt signaling and apoptosis. Recent evidence suggests that increased mTORC1 (mTOR/Raptor/GβL) signaling inhibits insulin signaling by phosphorylation of IRS1 and possibly IRS2 in a ribosomal S6 kinase (S6K)-dependent manner.
Figure 2. Schematic representation of various intracellular effects of Akt/PKB signaling.
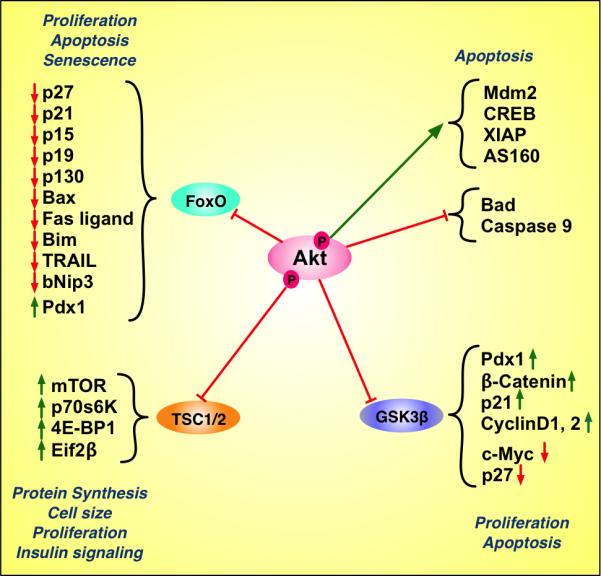
Upon activation, Akt phosphorylates numerous downstream targets that regulate diverse biological processes such as cell cycle progression, apoptosis, senescence, neogenesis, protein synthesis, cell size and insulin-mediated metabolism. This diagram illustrates a partial list of direct and indirect downstream effectors of Akt signaling. The green arrow demonstrates a direct stimulatory effect of Akt. The red horizontal bars demonstrate an inhibitory effect of Akt on the designated target. The arrows next to each molecule indicate upregulation (green) and downregulation (red).
Table 1.
Phenotypes of the different mouse models and Cre lines
| Genetic Mouse Model | Strategy | Phenotypes | References |
|---|---|---|---|
| Akt1-/- | Global Knock out | Placental hypotrophy, impaired fetal and postnatal growth with reduction of body weight | [2-4] |
| Akt2-/- | Global Knock out | Impaired glucose disposal, insulin resistance with diabetes in a subset of animals | [2][5][6] |
| Akt3-/- | Global Knock out | Reduced brain size | [2] |
| caAktTg | Transgenics overexpressing a constitutively active Akt in β-cells | Increase β cell mass, proliferation, neogenesis and cell size. | [8-9] |
| RIP-KdAkt | Transgenics expressing a kinase dead Akt in β cells | Insulin secretory defect | [10] |
| Irs2-/- | Global Knock out | Increase apoptosis, insulin resistance and diabetes | [17][21][45] |
| βPdk1-/- | Conditional deletion of Pdk1 in pancreatic β cells | Progressive hyperglycemia as a result of loss of β-cell mass | [19] |
| FoxO+/- | Deletion of one allele of FoxO | Increase β cell proliferation and expression of Pdx1 | [17][19] |
| FoxOS253A (Ttr305) | Transgenics overexpressing a constitutively nuclear FoxO1 in β cells | Early onset of diabetes due to inhibition of compensatory β-cell growth in response to peripheral insulin resistance through suppression of Pdx1 | [20-21] |
| RIP-GSK3βCA- | Transgenics overexpressing a constitutively active form of GSK3β (S9A) under the control of the rat insulin promoter | Impaired glucose tolerance and decreased β-cell mass and proliferation. Decrease cyclinD1 and Pdx1 levels | [25] |
| β-Tsc2-/- | Conditional deletion of Tsc2 in pancreatic β cells | Hypoglycemia, improved glucose metabolism and increase β-cell proliferation | [42-43] |
| S6K-/- | Global Knock out | Hypoinsulinemia, impaired insulin secretion, glucose intolerance and reduced β-cell size | [44] |
| β-Ir-/-;β-Igf1-/- | Conditional deletion of insulin receptor and insulin growth factor in pancreatic β cells | Diabetes due to decrease β-cell mass, β-cell proliferation and increase apoptosis | [45] |
| RIP-rtTA/tetOAkt1 | Doxycycline inducible transgenic mice expressing Akt in β cells | Increase β-cell proliferation | [41] |
| PTEN+/- | Deletion of one allele of PTEN | Development of spontaneous tumors | [72] |
| RIP-PTENLoxP/LoxP | Conditional deletion of PTEN in pancreatic β-cells | Mild fasting hypoglycemia, improved glucose tolerance and increased β-cell proliferation, cell size and mass | [73-74] |
| Pdx-PTENLoxP/LoxP | Conditional deletion of PTEN in pancreatic progenitors cells | Replacement of the exocrine tissue by ductal structures. Development of pancreatic ductal adenocarcinoma | [79] |
| Pdx1-Cre | Transgenic mice expressing Cre under the control of Pdx1 promoter | Specific expression in pancreatic progenitors | [80] |
| RIP-Cre | Transgenic mice expressing Cre under the control of the Rat Insulin promoter | Specific expression in pancreatic β-cells | [80] |
| Elastase-Cre | Transgenic mice expressing Cre under the control of the elastase promoter | Specific expression in acinar cells | [80] |
Akt and β-cell mass
β-cell mass is regulated by a dynamic balance of proliferation, cell size, apoptosis and neogenesis from other pancreatic cell types. The current data suggests that Akt regulates β-cell mass by modulating proliferation, cell size and apoptosis.
Akt and Proliferation
The maintenance of β-cell mass results predominantly from proliferation of pre-existing β-cells and depends on activation of the cyclin D/cdk4 complex [11-15]. Akt activates β-cell proliferation in a cdk4-dependent manner by inducing cyclin D1, D2 and p21Cip1 [16]. The regulation of the cell cycle by Akt signaling is not completely understood, but recent findings implicate FoxO1, glycogen synthase kinase 3β (Gsk3β) and TSC2/mTOR signaling in this process (Figure 2).
FoxO belongs to a large family of forkhead transcription factors with a common DNA-binding forkhead domain (Box 2). Akt inactivates FoxO1 by phosphorylation and nuclear exclusion. The restoration of β-cell mass and correction of diabetes in Irs2−/− and Pdk1−/− by placing them on a FoxO1 heterozygous background implicates this transcription factor in regulating β-cell mass [17-19] (Table 1). Moreover, overexpression of a constitutively active form of FoxO1 in β-cells prevents compensatory β-cell hyperplasia in insulin resistant models [20, 21]. Additional studies using cell lines derived from mouse insulinomas show that glucagon-like peptide (GLP-1) and glucose induce inactivation of FoxO1-mediated transcription by nuclear exclusion in a PI3K- and epidermal growth factor receptor (EGFR)-dependent manner. Overexpression of a constitutively nuclear FoxO1 in insulinoma cells and transgenic mice demonstrated that FoxO1 mediates proliferative and pro-survival effects of the GLP-1 agonist exendin-4 [22, 23] (Table 1). Experiments in mice with conditional gain or loss of FoxO1 function specifically in β-cells will provide further insight into the role of this transcription factor in β-cells.
The mechanism(s) responsible for modulation of the β-cell cycle by FoxO1 transcription factors are not completely understood, but it has been proposed that nuclear retention of FoxO1 (states of insulin resistance) reduces Pdx1 expression by competing with FoxA2 binding at the Pdx1 promoter [17]. Additionally, nuclear localization of FoxO1 induces transcription of the cell cycle inhibitors p27Kip1, p21cip1, p15INK4b and p19INK4d and could also be an important component in modulation of cell cycle by IRS2/Akt/FoxO1 signaling [24]. Some of these are further elaborated in Box 2.
One of the mechanisms involved in cell cycle regulation by Akt is the phosphorylation and inhibition of GSK3β. GSK3 is a ubiquitously expressed serine/threonine kinase initially identified as a regulator of glycogen synthesis (Box 2). Recent studies showed that GSK3β haploinsufficiency rescued the insulin resistance phenotype observed in Ir+/− mice and improved the diabetic phenotype of Irs2−/− mice in part by restoring β-cell proliferation [25]. More importantly, β-cell specific GSK3β deficiency improves hyperglycemia in Irs2−/− mice, indicating its importance for β-cell adaptation to insulin resistance [25]. The beneficial changes in β-cell mass observed in Irs2−/− mice lacking one allele of GSK3β were explained at least in part by restoration of Pdx1 expression and decreased p27Kip1 levels [25]. In contrast, overexpressing a constitutively active form of GSK3β in β-cells induced impaired glucose tolerance, decreased β-cell mass and proliferation that was associated with decreased cyclin D1 and Pdx1 levels [26] (Table 1). In vitro studies using isolated rat islets treated with small GSK3 inhibitors increased β-cell replication by 2–3-fold relative to controls, although the mechanisms responsible for this effect were not evaluated [27]. Taken together, these results suggest an important role of GSK3β in the Akt-mediated proliferative effect on β-cells.
The downstream mechanisms implicated in modulation of β-cell mass and functions by GSK3β have not been completely elucidated yet (Figure 2). GSK3β inhibition by Akt signaling stabilizes Pdx1 to prevent proteosomal degradation [28]. Modulation of the cell cycle by stabilization of cyclin Ds, p27Kip1 and c-myc could also be implicated [29]. GSK3β signaling also regulates cyclin D1 transcription in a β-catenin dependent manner [30], suggesting involvement of the Wnt/β-catenin pathway (canonical pathway) in β-cell cycle regulation (see Box 2) [31, 32].
In addition to modulating transcription factors and kinases, Akt can also regulate proliferation by phosphorylating cell cycle components (Box 2). Akt directly phosphorylates p27Kip1 on T157 and abolishes its inhibitory activity against Cdk2 [33]. This is critical because upregulation of p27Kip1 in states of decreased Akt signaling contributes to the pathogenesis of type 2 diabetes in mice [34]. Mice overexpressing constitutively active Akt displayed increased levels of the cell cycle inhibitor p21Cip1. However, in other systems, Akt phosphorylates p21Cip1, resulting in its exclusion from the nucleus and inhibition [35]. The effects of the modulation of p21Cip1 levels and cellular localization in β-cell cycle progression are currently under study.
Akt also affects mTOR signaling. Nutrients and growth factors regulate mTOR activity through the tuberous sclerosis complex (Tsc)-2 gene product (tuberin) as well as Tsc1 (hamartin) and the small G protein Ras homolog enriched in brain (Rheb). Akt phosphorylation of Tsc2 induces mTOR signaling by derepressing the Tsc2 GTPase protein activity toward Rheb [36] (Box 2 and Figure 1) [36, 37]. Mammalian TOR is found in two distinct complexes (mTORC1 and 2) [38]. mTORC1 is sensitive to rapamycin and comprises mTOR, Raptor (Regulatory-Associated Protein of mTOR), and mammalian lethal with sec18 protein 8 (mLST8) (also known as GβL) . mTORC2 is rapamycin-insensitive and comprises mTOR, Rictor, Sin1 and mLST8/GβL [38]. This latter complex is responsible for the phosphorylation/activation of Akt on Ser 473 and protein kinase C alpha (PKCα) [39]. Therefore, while not completely understood, it appears that mTOR lies both upstream (mTORC2) and downstream (mTORC1) of Akt. The mTORC1 complex phosphorylates eIF4 (eukaryotic initiation factor 4)-binding protein 4E-BP1/2 and S6K1/2 to induce mRNA translation and cell growth [40]. The importance of this pathway in regulating β-cell proliferation has been recently demonstrated in mice where inhibition of mTORC1 by rapamycin inhibits β-cell proliferation induced by Akt activation. This anti-proliferative effect of rapamycin was caused by alteration in the synthesis and stability of cyclin D2, a critical regulator of β-cell cycle [41]. Further confirmation of the importance of this pathway comes from mice with conditional deletion of Tsc2 in β-cells [42, 43]. These mice exhibit hypoglycemia, improved glucose metabolism and increase β-cell proliferation [42, 43], supporting an important role of mTOR signaling pathway in regulating β-cell mass [42]. However, the downstream events modulated by mTOR and the contribution of S6K and 4EBP in regulating β-cell proliferation remain unknown.
Akt and Cell size
Increased β-cell size contributes to the augmented β-cell mass observed in caAktTg mice [8, 9]. Recent evidence from mice with Tsc2 deletion demonstrates that mTORC1 is one factor that regulates β-cell size [42, 43]. Studies using S6K1 deficient mice suggest that S6K is one of the major candidates, although it is still not fully understood how downstream targets of S6K regulate cell size. S6K1 deficient mice, however, exhibit hypoinsulinemia, impaired insulin secretion, glucose intolerance and reduced β-cell size [44] (Table 1).
Akt and Apoptosis
Akt signaling is one of the critical pathways regulating cell survival, and its importance in β-cells has been suggested by increased apoptosis observed in different mouse models including Irs2−/−, β-cell specific Ir−/−/IgfI−/− and Pdk1−/− mice (Table 1) [45]. Further evidence of antiapoptotic effects of Akt comes from resistance of Akt-overexpressing β-cells to streptozotocin-induced apoptosis [8, 9]. In vitro experiments in insulinoma cell lines and isolated islets demonstrate that Akt activation by glucose, insulin, IGF-1 and GLP-1 is a major component for the anti-apoptotic effects of these molecules [46-49]. Activation of Akt signaling also has been shown to mediate the survival effect of decreased levels of thioredoxin-interacting protein (TXNIP) in mice [50]. Likewise, downregulation of Akt activity by cytokine, oxidative- and ER stress-mediated activation of JNK is also associated with apoptosis [51, 52]. Tribble-3 (TRB3), a mammalian homolog of Drosophila Tribbles, binds and inhibits the activation of Akt [53]. Modulation of TRB3 levels in INS1 cells altered susceptibility to high glucose- and ER stress-induced apoptosis, suggesting that TRB3 has major implications for β-cell mass and function [54]. Most recently, O-linked N-acetylglucosamine (O-GlcNAc) to Ser 473 decreased Akt activity and increased apoptosis induced by glucosamine treatment, suggesting that this Ser modification plays a major role in regulating Akt signaling in β-cells [55]. These data suggest that in addition to proliferative responses, Akt signaling is a major survival molecule in pancreatic β-cells by regulating a diverse group of downstream targets, one being FoxO1. The effects of FoxO1 in survival, however, appear to depend on experimental and metabolic conditions [56]. Phosphorylation and inactivation of FoxO1 activates genes involved in apoptosis like Fas ligand, TRAIL, Bim and bNIP3 [57]. Similarly, expression of nuclear constitutively active FoxO1 in cultured β-cells prevented the antiapoptotic actions of GLP-1 [22]. Akt/FoxO1 signaling also relays the survival signals from GIP by suppressing Bax [58]. More recently, expression of a dominant negative allele of FoxO in a β-cell line reduced expression of apoptotic and ER stress markers, promoting β-cell survival [59]. In contrast, exposure of cultured β-cells to oxidative stress induced nuclear FoxO1 localization and protected β-cells against oxidative damage [60]. This beneficial effect was associated with increased expression of NeuroD, MafA and insulin [60]. The transcriptional activation of FoxO1 by oxidative stress is self-limited and regulated by the balance between acetylation and deacetylation to prevent excessive and prolonged FoxO-dependent transcription that could result in apoptosis. In summary, FoxO1 can have inhibitory effects on β-cell mass expansion and protective effects under acute stress. Experiments using conditional deletion of FoxO isoforms exposed to different stresses will elucidate the role of this transcription factor in β-cell mass and function.
In addition to its important role in proliferation, Akt's inhibition of GSK3β activity can also relate survival signals. In vitro experiments in insulinoma cells demonstrated that inhibition of GSK3β signaling reduces apoptosis induced by ER stress, glucose or palmitate [27, 61]. This protective effect of Akt/GSK3β signaling could be mediated by the subcellular localization and stability of Pdx1 [28]. In other systems, GSK3β, and perhaps Akt then, has been suggested to contribute to cell death by modulating MLK3/JNK and phosphorylating transcription factors such as c-Myc and CREB [62-64].
Akt could also affect survival by direct phosphorylation of Bad, a member of the Bcl-2 family [65]. Caspase 9, another pro-apoptotic protein, is also inhibited by Akt in human tissues. CREB is also an important mediator of Akt-induced survival [66]. Activation of Mdm2 by Akt phosphorylation blocks p53 and protects from apoptosis in other systems [49]. Akt phosphorylation of XIAP (X-linked inhibitor of apoptosis protein) inhibits apoptosis induced by cytokine-mediated apoptosis and prolongs survival during islet transplantation [67]. Recent in vitro experiments using insulinoma cells demonstrated that the Akt target AS160 relates survival signals from Akt signaling [68]. These anti-apoptotic mechanisms induced by Akt have been evaluated in other systems, and further work is needed to determine the major anti-apoptotic signals of Akt in β-cells.
PTEN
The phosphatase and Tensin homologue deleted on chromosome 10 (PTEN) gene is a human tumor suppressor gene widely expressed throughout the body; PTEN hydrolyzes the 3-phosphate on PI3P resulting in PI3K inhibition and Akt signaling [69] (Figure 1 and Box 2). However, recent evidence also demonstrates some Akt-independent effects of PTEN such as activation of JNK signaling, p53 modulation and FAK phosphorylation [69, 70]. PTEN deletion regulates multiple biological processes that include proliferation, reduced apoptosis and enhanced migration.
Although the role of PTEN in a variety of cells has been well characterized [71, 72], its role in β-cell mass and function has been less studied. The first indirect evidence for a role of PTEN in β-cells was demonstrated by restoration of β-cell mass and function and normalization of blood glucose by introducing PTEN haploinsufficiency in Irs2−/− mice [73]. However, the improved insulin sensitivity made difficult to assess the direct effect of PTEN in β-cells. Conditional PTEN deletion in β-cells (RIP-cre;PTENloxP/loxP) results in mild fasting hypoglycemia, improved glucose tolerance and increased β-cell proliferation, cell size and mass [74] (Table 1). These mice also exhibit increased markers of Akt signaling, resistance to streptozotocin-induced diabetes and higher levels of Pdx1, Glut-2 and Irs2 [75]. Interestingly, specific deletion of PTEN in pancreatic β-cells using RIP-Cre mice is associated with augmented insulin sensitivity and decreased growth attributed to partial deletion of PTEN in the hypothalamus by aberrant expression of Cre recombinase in this tissue [75]. The changes in insulin sensitivity could also indirectly modulate the responses of β-cells to PTEN absence. It will be interesting to assess the phenotype of PTEN deletion in β-cells using a Cre line with more selective targeting to β-cells.
The differences between mice with deletion of PTEN and mice overexpressing a constitutively active form of Akt in β-cells highlight potential Akt-independent effects of PTEN deletion. In contrast to the normal islet morphology in the RIP-Cre;PTENloxP/loxP mice, the RIP-Cre;PTENloxP/loxP.exhibit enlarged islets with abnormal distribution of non-β-cells within the islet [74, 75]. In addition, the magnitude of β-cell mass changes described in caAktTg mice is significantly higher compared to RIP-Cre;PTENloxP/loxP.mice and one of these lines developed S6K1-dependent insulinomas [8, 9] [76]. While transgene insertion and copy number could explain this phenomenon, it is important to note that other factors could explain these differences. Deletion of PTEN results in activation of Akt, TSC/mTOR and S6K signaling activation that results in feedback inhibition of IRS and Akt signaling [77-79]. Therefore, Akt modulation by other signals through IRS proteins is impaired. In contrast, activation of Akt signaling in caAktTg is not susceptible to this feedback inhibition as the caAkt mutant acts downstream of IRS proteins. In addition, dephosphorylation of other proteins by PTEN (see above) could also explain the different phenotypes between RIP-Cre;PTENloxP/loxP and caAktTg mice. Therefore, the difference in the phenotypes of these two models could reveal Akt-dependent and independent pathways involved in PTEN regulation of β-cell mass and function.
Akt/PTEN in neogenesis
The studies of neogenesis in the pancreas have been limited by absence of markers for β-cell progenitors in adult tissues. However, the importance of Akt/PTEN in regulating β-cell differentiation has been assessed by activating Akt signaling in mice with conditional deletion of PTEN in pancreatic progenitors using the Pdx1-Cre transgenic line (Table 1). These mice exhibit progressive replacement of the exocrine tissue by ductal structures, presumably arising from expansion of centroacinar cells [80]. This important model demonstrates that PTEN deletion is dispensable for pancreas development and embryonic programs in ductal cells.
The importance of Akt signaling in regulating differentiation and plasticity of different pancreatic compartments has been recently demonstrated using a mouse model that combines a double reporter system with activation of a constitutively active form of Akt1 (caAkt) in a Cre-dependent manner [81][82]. This model allows to perform lineage tracing in cells that express the caAkt mutant upon Cre-mediated recombination [81]. Using this animal model, expression of mutant caAkt in the pancreatic epithelium, acinar or β-cells was performed by crossing to Pdx1-Cre, Elastase-Cre or RIP-Cre mice, respectively (Table 1). A major conclusion derived from these lineage tracing experiments was that activation of Akt signaling in acinar or β-cells induces acinar to ductal or β-cell to acinar/ductal transdifferentiation respectively. The acinar to ductal and β-cell to acinar/ductal transdifferentiation observed in Elastase-Cre;caAkt and RIP-Cre;caAkt mice suggest that the ductal epithelium could be a default fate after Akt signaling activation in acinar and β-cells. These studies also demonstrated that chronic activation of Akt signaling induces pre-malignant lesions and malignant transformation in old mice. In conclusion, the current work demonstrates for the first time that modulation of Akt signaling regulates the fate of differentiated pancreatic cells in vivo and suggests that modulation of Akt signaling could be used as a potential target to generate tissues for regenerative medicine.
It is important to mention several differences between the PTEN deletion model and the overexpression of Akt in pancreatic progenitors. A major difference is the presence of plasticity in acinar and β-cells following overexpression of Akt [81]. In addition to Akt activation levels, one possible explanation for the absence of transdifferentiation in the PTEN model is that PTEN is not expressed in the acinar compartment, so Akt signaling is not activated there [80]. It is also possible that Akt-independent effects could account for some of these different phenotypes.
Conclusion
It is clear the IRS2/PI3K/Akt signaling pathway is a critical regulator of β-cell mass and function. FoxO1 and GSK3β are important components mediating proliferative and survival signals in response to Akt activation, with the Tsc2/mTOR pathway another major component in regulating proliferation and cell size. Therefore, we can conclude that there is not a predominant downstream Akt pathway regulating β-cell mass; rather, each component contributes to the overall phenotype resulting from Akt activation. However, the molecular mechanisms responsible for the alterations of proliferation and apoptosis by different Akt downstream targets have not been completely elucidated. Experiments performed in mice with conditional deletions of different Akt signaling pathway components will allow us to understand their importance in regulating β-cell mass. A major gap in our knowledge regards the importance of Akt signaling in pancreas development, differentiation and cellular plasticity. A better understanding of the molecular mechanisms involved in these processes will provide pharmacological targets that can be used to expand β-cells in vitro and in vivo as an alternative therapy for diabetes.
Box 1. Akt/PKB: Structure.
Akt, also known as protein kinase B (PKB), is a serine/threonine kinase that acts as a major effector of the PI3Kinase signaling pathway. Akt kinases belong to the more general class of AGC kinases (related to AMP/GMP kinase and protein kinase C) and consist of two conserved domains: an N-terminal Pleckstrin homology (PH) domain, followed by a kinase domain that terminates in a regulatory hydrophobic motif. The N-terminal PH domain mediates binding to phosphatidylinositol (3,4,5) P3 phosphate (PIP3) and the catalytic domain contains a threonine residue (T308 for Akt1/PKBα) whose phosphorylation is necessary for activation of Akt/PKB [1]. The hydrophobic C-terminal tail is a characteristic feature of all AGC kinases (PKA, PKC, PDK1, p70 and p90 S6 kinase) and includes a second regulatory phosphorylation site (S473 in Akt1/PKBα) [1]. These two phosphorylation sites are required for the full activation of Akt/PKB [1]. Expression levels for the different isoforms vary in different tissues. Akt1 and 2 seem to be ubiquitously expressed at the highest levels in the brain, thymus, heart and lung, whereas Akt3 expression is mainly restricted to the brain and testis [1]. Expression of the three isoforms has been described in β-cells [83].
Box 2. Selective list of Akt signaling components.
FoxO
FoxO belongs to a large family of forkhead transcription factors that share a common DNA-binding forkhead domain [84]. Mammalian cells express FoxO1 (FKHR), FoxO3a (FKHRL1) and FoxO4 (AFX). Phosphorylation by Akt regulates the subcellular localization of FoxO proteins, translocating them from the nucleus to the cytoplasm where they are inactivated and ubiquinated. Therefore, Akt negatively regulates the transcriptional activity of FoxO proteins [85]. FoxO proteins regulate multiple biological processes that include cellular proliferation, apoptosis, differentiation, and metabolism. FoxO1 is the most abundant isoform in β-cells and is not expressed in acinar cells or other islet cells [84].
GSK3
Two isoforms (α and β) share a high degree of amino acid homology. Upon stimulation with growth factors or insulin, Akt mediates phosphorylation and inactivation of GSK3 on serine 23 in the α-isoform and serine 9 on the β-isoform [1]. GSK3 is part of a multiprotein complex with Axin, β-catenin and APC and is also regulated by the Wnt signaling pathway [86]. Inactivation of GSK3 by Wnt results in nuclear translocation of β-catenin where it acts as a transcriptional regulator of Lef/Tcf targets [87]. GSK3 is a converging point of the insulin and Wnt signaling pathways. GSK3 is involved in energy metabolism, cell fate determination, cell growth and transcriptional regulation [1].
Cell cycle components
Growth factor-mediated activation of Akt alters cell proliferation by triggering cells in G0 to enter the cell cycle (G1-S transition). D-type cyclin in association with cdk4 or cdk6 forms the first cyclin-cdk complex to be activated during the early G1-phase. Cyclin D/cdk4 phosphorylates the pocket proteins (Rb, p107, p130) and releases the transcription factor E2F. Cyclin E associates with cdk2 in late G1 and is essential for entry into S phase. Cell cycle is negatively regulated by the INK4 (p16INK4a, p15INK5b, p18INK4c, and p19INK4d) and Cip/Kip cell cycle inhibitors (p21Cip1, p27Kip1, and p57Kip2).
TSC2/mTOR
The TSC2/mTOR signaling integrates growth factors and nutrient signals and is essential for cell growth and proliferation [36, 37]. These signals regulate mTOR activity by regulating TSC gene products, TSC1 and TSC2, and the small G protein Rheb. TSC2 phosphorylation and inactivation releases the inhibition of Rheb, leading to activation of mTOR [40]. Recent experiments indicated that like yeast, mammalian TOR is associated with two distinct complexes (mTORC1 and 2)[40]. Activation of TSC/Rheb/mTOR/S6K by Akt signaling regulates cell growth, proliferation and ribosomal biogenesis.
PTEN
PTEN is one of the most frequently mutated genes in cancer, and germline mutations are found in Cowden disease, also termed Cowden syndrome and multiple hamartoma syndrome in humans [88]. This disease is characterized by hamartomatous neoplasms of the skin and mucosa, gastrointestinal tract, bones, central nervous system, eyes, and genitourinary tract. Skin is involved in 90-100% of cases, and the thyroid is involved in 66% of cases. Homozygous deletion of PTEN is embryonic lethal, and heterozygous mice develop tumors in multiple tissues [89]. Mutation of PTEN is observed in multiple tumors and overexpression of wild-type PTEN induces apoptosis and blocks cell-cycle progression, colony formation and cell migration [69].
Acknowledgements
This work was supported by grants from the National Institute of Health (R03 DK068028-01) and a Research Grant from the Juvenile Research Foundation. EBM is a recipient of a Career Development Award from the American Diabetes Association. We apologize to those whose work has not been covered and cited due to space constraints.
References
- 1.Woodgett JR. Recent advances in the protein kinase B signaling pathway. Current opinion in cell biology. 2005;17:150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Franke TF. Intracellular signaling by Akt: bound to be specific. Science signaling. 2008;1:pe29. doi: 10.1126/scisignal.124pe29. [DOI] [PubMed] [Google Scholar]
- 3.Chen WS, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes & development. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho H, et al. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. The Journal of biological chemistry. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 5.Cho H, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science (New York, N.Y. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 6.Garofalo RS, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. The Journal of clinical investigation. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bae SS, et al. Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. The Journal of biological chemistry. 2003;278:49530–49536. doi: 10.1074/jbc.M306782200. [DOI] [PubMed] [Google Scholar]
- 8.Tuttle RL, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nature medicine. 2001;7:1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- 9.Bernal-Mizrachi E, et al. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. The Journal of clinical investigation. 2001;108:1631–1638. doi: 10.1172/JCI13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernal-Mizrachi E, et al. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. The Journal of clinical investigation. 2004;114:928–936. doi: 10.1172/JCI20016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dor Y, et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 12.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. The Journal of clinical investigation. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kushner JA, et al. Cyclins D2 and D1 Are Essential for Postnatal Pancreatic {beta}-Cell Growth. Molecular and cellular biology. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rane SG, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nature genetics. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 15.Tsutsui T, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Molecular and cellular biology. 1999;19:7011–7019. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fatrai S, et al. Akt Induces {beta}-Cell Proliferation by Regulating Cyclin D1, Cyclin D2, and p21 Levels and Cyclin-Dependent Kinase-4 Activity. Diabetes. 2006;55:318–325. doi: 10.2337/diabetes.55.02.06.db05-0757. [DOI] [PubMed] [Google Scholar]
- 17.Kitamura T, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. The Journal of clinical investigation. 2002;110:1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura K, et al. PDK1 regulates cell proliferation and cell cycle progression through control of cyclin D1 and p27Kip1 expression. The Journal of biological chemistry. 2008;283:17702–17711. doi: 10.1074/jbc.M802589200. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto N, et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nature genetics. 2006;38:589–593. doi: 10.1038/ng1774. [DOI] [PubMed] [Google Scholar]
- 20.Nakae J, et al. The FoxO transcription factors and metabolic regulation. FEBS letters. 2008;582:54–67. doi: 10.1016/j.febslet.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 21.Okamoto H, et al. Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. The Journal of clinical investigation. 2006;116:775–782. doi: 10.1172/JCI24967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buteau J, et al. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass. Diabetes. 2006;55:1190–1196. doi: 10.2337/db05-0825. [DOI] [PubMed] [Google Scholar]
- 23.Martinez SC, et al. Glucose regulates Foxo1 through insulin receptor signaling in the pancreatic islet beta-cell. Diabetes. 2006;55:1581–1591. doi: 10.2337/db05-0678. [DOI] [PubMed] [Google Scholar]
- 24.Ho KK, et al. Many forks in the path: cycling with FoxO. Oncogene. 2008;27:2300–2311. doi: 10.1038/onc.2008.23. [DOI] [PubMed] [Google Scholar]
- 25.Tanabe K, et al. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008;6:307–318. doi: 10.1371/journal.pbio.0060037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, et al. Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia. 2008;51:623–631. doi: 10.1007/s00125-007-0914-7. [DOI] [PubMed] [Google Scholar]
- 27.Mussmann R, et al. Inhibition of glycogen synthase kinase (GSK)3 promotes replication and survival of pancreatic beta cells. The Journal of biological chemistry. 2007;282:12030–12037. doi: 10.1074/jbc.M609637200. [DOI] [PubMed] [Google Scholar]
- 28.Boucher MJ, et al. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. The Journal of biological chemistry. 2006;281:6395–6403. doi: 10.1074/jbc.M511597200. [DOI] [PubMed] [Google Scholar]
- 29.Kida A, et al. Glycogen synthase kinase-3beta and p38 phosphorylate cyclin D2 on Thr280 to trigger its ubiquitin/proteasome-dependent degradation in hematopoietic cells. Oncogene. 2007;26:6630–6640. doi: 10.1038/sj.onc.1210490. [DOI] [PubMed] [Google Scholar]
- 30.Welters HJ, Kulkarni RN. Wnt signaling: relevance to beta-cell biology and diabetes. Trends in endocrinology and metabolism: TEM. 2008;19:349–355. doi: 10.1016/j.tem.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Rulifson IC, et al. Wnt signaling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci U S A. 2007;104:6247–6252. doi: 10.1073/pnas.0701509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heiser PW, et al. Stabilization of {beta}-catenin impacts pancreas growth. Development. 2006;133:2023–2032. doi: 10.1242/dev.02366. [DOI] [PubMed] [Google Scholar]
- 33.Chu IM, et al. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nature reviews. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 34.Uchida T, et al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nature medicine. 2005;11:175–182. doi: 10.1038/nm1187. [DOI] [PubMed] [Google Scholar]
- 35.Zhou BP, et al. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature cell biology. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 36.Um SH, et al. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Avruch J, et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25:6361–6372. doi: 10.1038/sj.onc.1209882. [DOI] [PubMed] [Google Scholar]
- 38.Sarbassov DD, et al. Growing roles for the mTOR pathway. Current opinion in cell biology. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Sarbassov DD, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 40.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. The Biochemical journal. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balcazar N, et al. mTORC1 activation regulates beta -cell mass and proliferation by modulation of cyclin D2 synthesis and stability. The Journal of biological chemistry. 2009 doi: 10.1074/jbc.M807458200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rachdi L, et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A. 2008;105:9250–9255. doi: 10.1073/pnas.0803047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shigeyama Y, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Molecular and cellular biology. 2008;28:2971–2979. doi: 10.1128/MCB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pende M, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408:994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 45.Leroith D, Accili D. Mechanisms of disease: using genetically altered mice to study concepts of type 2 diabetes. Nature clinical practice. 2008;4:164–172. doi: 10.1038/ncpendmet0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srinivasan S, et al. Glucose promotes pancreatic islet beta-cell survival through a PI 3-kinase/Akt-signaling pathway. American journal of physiology. 2002;283:E784–793. doi: 10.1152/ajpendo.00177.2002. [DOI] [PubMed] [Google Scholar]
- 47.Ohsugi M, et al. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. The Journal of biological chemistry. 2005;280:4992–5003. doi: 10.1074/jbc.M411727200. [DOI] [PubMed] [Google Scholar]
- 48.Brubaker PL, Drucker DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. 2004;145:2653–2659. doi: 10.1210/en.2004-0015. [DOI] [PubMed] [Google Scholar]
- 49.Dickson LM, Rhodes CJ. Pancreatic beta-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? American journal of physiology. 2004;287:E192–198. doi: 10.1152/ajpendo.00031.2004. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, et al. Thioredoxin-interacting protein deficiency induces Akt/BclxL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008;22:3581–3594. doi: 10.1096/fj.08-111690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaneto H, et al. Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and insulin resistance. The international journal of biochemistry & cell biology. 2005;37:1595–1608. doi: 10.1016/j.biocel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 52.Eizirik DL, et al. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine reviews. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 53.Du K, et al. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science (New York, N.Y. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 54.Qian B, et al. TRIB3 is implicated in glucotoxicity- and oestrogen receptor-stress-induced beta-cell apoptosis. J Endocrinol. 2008;199:407–416. doi: 10.1677/JOE-08-0331. [DOI] [PubMed] [Google Scholar]
- 55.Kang ES, et al. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Experimental cell research. 2008;314:2238–2248. doi: 10.1016/j.yexcr.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 56.Buteau J, Accili D. Regulation of pancreatic beta-cell function by the forkhead protein FoxO1. Diabetes, obesity & metabolism. 2007;9(Suppl 2):140–146. doi: 10.1111/j.1463-1326.2007.00782.x. [DOI] [PubMed] [Google Scholar]
- 57.Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–2319. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim SJ, et al. Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. The Journal of biological chemistry. 2005;280:22297–22307. doi: 10.1074/jbc.M500540200. [DOI] [PubMed] [Google Scholar]
- 59.Martinez SC, et al. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes. 2008;57:846–859. doi: 10.2337/db07-0595. [DOI] [PubMed] [Google Scholar]
- 60.Kitamura YI, et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 61.Srinivasan S, et al. Endoplasmic Reticulum Stress-Induced Apoptosis Is Partly Mediated by Reduced Insulin Signaling Through Phosphatidylinositol 3-Kinase/Akt and Increased Glycogen Synthase Kinase-3{beta} in Mouse Insulinoma Cells. Diabetes. 2005;54:968–975. doi: 10.2337/diabetes.54.4.968. [DOI] [PubMed] [Google Scholar]
- 62.Mishra R, et al. Glycogen synthase kinase-3beta induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. The Journal of biological chemistry. 2007;282:30393–30405. doi: 10.1074/jbc.M705895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. Journal of neurochemistry. 2001;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clerk A, et al. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. Journal of cellular physiology. 2007;212:311–322. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- 65.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. The Biochemical journal. 2008;415:333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 66.Jhala US, et al. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes & development. 2003;17:1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Emamaullee J, et al. XIAP overexpression in islet beta-cells enhances engraftment and minimizes hypoxia-reperfusion injury. Am J Transplant. 2005;5:1297–1305. doi: 10.1111/j.1600-6143.2005.00891.x. [DOI] [PubMed] [Google Scholar]
- 68.Bouzakri K, et al. Rab GTPase-activating protein AS160 is a major downstream effector of protein kinase B/Akt signaling in pancreatic beta-cells. Diabetes. 2008;57:1195–1204. doi: 10.2337/db07-1469. [DOI] [PubMed] [Google Scholar]
- 69.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 70.Vivanco I, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer cell. 2007;11:555–569. doi: 10.1016/j.ccr.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 71.Cully M, et al. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nature reviews. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 72.Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–6488. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- 73.Kushner JA, et al. Phosphatase and tensin homolog regulation of islet growth and glucose homeostasis. The Journal of biological chemistry. 2005;280:39388–39393. doi: 10.1074/jbc.M504155200. [DOI] [PubMed] [Google Scholar]
- 74.Stiles BL, et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Molecular and cellular biology. 2006;26:2772–2781. doi: 10.1128/MCB.26.7.2772-2781.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nguyen KT, et al. Essential role of Pten in body size determination and pancreatic beta-cell homeostasis in vivo. Molecular and cellular biology. 2006;26:4511–4518. doi: 10.1128/MCB.00238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alliouachene S, et al. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. The Journal of clinical investigation. 2008;118:3629–3638. doi: 10.1172/JCI35237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Um SH, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 78.Harrington LS, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. The Journal of cell biology. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shah OJ, et al. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 80.Stanger BZ, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer cell. 2005;8:185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 81.Elghazi L, et al. Regulation of Pancreas Plasticity and Malignant Transformation by Akt Signaling. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.11.043. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Elghazi L, et al. Generation of a reporter mouse line expressing Akt and EGFP upon Cre-mediated recombination. Genesis. 2008;46:256–264. doi: 10.1002/dvg.20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holst LS, et al. Protein kinase B is expressed in pancreatic beta cells and activated upon stimulation with insulin-like growth factor I. Biochem Biophys Res Commun. 1998;250:181–186. doi: 10.1006/bbrc.1998.9166. [DOI] [PubMed] [Google Scholar]
- 84.Kitamura T, Ido Kitamura Y. Role of FoxO Proteins in Pancreatic beta Cells. Endocrine journal. 2007;54:507–515. doi: 10.1507/endocrj.kr-109. [DOI] [PubMed] [Google Scholar]
- 85.Kops GJ, Burgering BM. Forkhead transcription factors: new insights into protein kinase B (c-akt) signaling. Journal of molecular medicine (Berlin, Germany) 1999;77:656–665. doi: 10.1007/s001099900050. [DOI] [PubMed] [Google Scholar]
- 86.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 87.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 88.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 89.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 90.Elghazi L, et al. Emerging role of protein kinase B/Akt signaling in pancreatic beta-cell mass and function. The international journal of biochemistry & cell biology. 2006;38:157–163. doi: 10.1016/j.biocel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 91.Sarbassov DD, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science New York, N.Y. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]