

Abstract
The molecular circadian clock in mammals is generated from transcriptional activation by the bHLH-PAS transcription factor CLOCK–BMAL1 and subsequent repression by PERIOD and CRYPTOCHROME (CRY). The mechanism by which CRYs repress CLOCK–BMAL1 to close the negative feedback loop and generate 24-hour timing is not known. Here we show that CRY1 competes for binding with coactivators to the intrinsically unstructured C-terminal transactivation domain (TAD) of BMAL1 to establish a functional switch between activation and repression of CLOCK–BMAL1. Mutations within the TAD that alter affinities for coregulators change the balance of repression and activation to consequently change intrinsic circadian period or eliminate cycling altogether. Our results suggest that CRY1 fulfills its role as an essential circadian repressor by sequestering the TAD from coactivators and highlight regulation of the BMAL1 TAD as a critical mechanism for establishing circadian timing.
Keywords: Negative feedback, transactivation domain, CBP(p300), intrinsically disordered, NMR spectroscopy
INTRODUCTION
Mammalian circadian rhythms integrate behavior, physiology and metabolism into daily cycles that are coordinated with the solar day1. Circadian timing arises from a genetically encoded oscillator that uses transcriptional feedback loops to generate a self-sustaining clock and control the output of clock-controlled genes with near 24-hour periodicity2. At the core of this molecular circadian clock, the heterodimeric basic helix-loop-helix PER-ARNT-SIM (bHLH-PAS) transcription factor CLOCK–BMAL1 drives expression of the Period (Per) and Cryptochrome (Cry) genes. PER and CRY proteins form complexes that are posttranslationally modified to control stability and translocation into the nucleus where they repress CLOCK–BMAL1-mediated transcription, thereby closing the transcriptional feedback loop3-5. However, the mechanisms by which PER and CRY fulfill their roles as negative transcriptional regulators are still not fully understood. In addition to its role in early repressive PER–CRY complexes, CRY1 also interacts directly with CLOCK–BMAL1 in vitro6 and on chromatin later in the circadian cycle7,8, and it can potently repress CLOCK–BMAL1 activity in the absence of PER9,10. Therefore, identifying how CRY1 regulates CLOCK–BMAL1 will provide valuable insight into its essential role in the molecular circadian clock.
Because many factors contribute to robustness of circadian behavior at the organismal level (e.g., intercellular coupling in the suprachiasmatic nucleus and other systemic cues), delineating the biochemical mechanisms needed to establish circadian cycling from in vivo studies has been challenging11,12,13. For example, BMAL1 (also known as ARNTL) and its partner CLOCK interact through their N-terminal bHLH and tandem PAS domains14; these core bHLH-PAS domains are largely conserved in the paralog BMAL2 (also known as ARNTL2). However, Bmal1–/– mice lack all behavioral and molecular circadian rhythms, establishing its essential role in the molecular circadian clock15. While overexpression of Bmal2 in Bmal1–/– mice can partially rescue circadian behavior, paradoxically, it does not restore circadian rhythms in most tissues16. Therefore, we still lack a fundamental understanding of how core clock proteins function to generate the transcription-based circadian clocks in mammals.
The distinct functions of the two Bmal paralogs offer a heuristic opportunity to uncover properties of CLOCK–BMAL1 that are needed to generate circadian oscillations. To define the circadian function of BMAL1, we employed cell-based genetic complementation in Bmal1–/– fibroblasts derived from mice harboring the Per2Luc transgenic reporter17. Domain-based swapping of Bmal1 and Bmal2 revealed the oscillatory function of the BMAL1 C-terminus that functionally distinguishes it from its non-circadian paralog, BMAL2. We found that CRY1 binds to the transactivation domain (TAD) of BMAL paralogs at sites that overlap with binding of the coactivator, CBP (CREB-binding protein) and its homolog, p300. Mutations within the TAD markedly altered affinities for CRY1 and caused large changes in period length. These findings provide new insights into transcriptional regulation by CRY1 and demonstrate that the BMAL1 TAD plays an important role in establishing 24-hour timing.
Only BMAL1 can generate cell-autonomous circadian rhythms
Fibroblasts derived from Bmal1–/– Per2Luc mice are completely arrhythmic, but circadian rhythms can be restored through genetic complementation of Bmal1 under control of the constitutive UBC promoter17. Bmal2 was unable to rescue circadian rhythms in these cells, even when its transcript and protein were expressed to similar levels as Bmal1 (Fig. 1a-c and Supplementary Fig. 1). Moreover, Bmal2 was neither upregulated in Bmal1–/– cells nor upon ectopic expression of Bmal1 (Fig. 1c), displaying a lack of paralog compensation that is a common network feature of genes involved in molecular circadian clocks18. Compared to high amplitude oscillations of Per2 and Rev-erbα mRNA expression in Bmal1-rescued cells, we observed constitutive expression of Per2 and Rev-erbα in cells expressing Bmal2 but with lower overall mRNA levels, perhaps caused by increased expression of Cry1 in these cells (Fig. 1c). Overall, these data demonstrate that E-box-mediated circadian transcription is substantially attenuated in the absence of Bmal1. Therefore, Bmal1 and Bmal2 are not functionally redundant; while Bmal1 is an essential clock component, Bmal2 is not sufficient to support the circadian clock at the cellular level.
Figure 1.

Only BMAL1 can restore circadian rhythms in Bmal1–/– Per2Luc fibroblasts. (a) Luminescence records from Bmal1–/– Per2Luc fibroblasts expressing EGFP, Bmal1 or Bmal2. Traces from replicate cell cultures are shown from one representative experiment (n = 3 clonal lines). (b) Western blot of Flag-tagged Bmal paralog expression in Bmal1–/– Per2Luc fibroblasts. Time, hours after synchronization. Uncropped images can be found in Supplementary Data Set 1. (c) Expression of clock-controlled genes in Bmal1–/– Per2Luc fibroblasts determined by quantitative reverse transcription PCR. Values are expressed as percentage of maximum expression, set to 100% for each gene except for Bmal2, which was normalized to Bmal1 based on PCR amplification efficiency to reflect their relative levels. Points represent mean expression ± s.d. of triplicate measurements from two independent timecourses. Time, hours after synchronization. (d) Luciferase-based mammalian two-hybrid assay in HEK293T cells transiently transfected with the indicated plasmid pairs. pBind, fusion with Gal4 DNA-binding domain; pAct, fusion with VP16 transactivation domain. Luminescence values are expressed as mean ± s.d. of triplicate measurements from one representative experiment (n = 3). Relative activity normalized to pAct-Clock–pBind-Bmal1 set to 100. (e) Per1-Luc assay of CLOCK–BMAL activity in HEK293T cells transiently transfected with indicated plasmids. Vec, vector; Bm, Bmal1 or Bmal2; Clk, Clock. Luminescence values are expressed as mean ± s.d. of triplicate measurements from one representative experiment (n = 3). Relative activity normalized to Clock–Bmal1 set to 100.
BMAL paralogs have similar steady-state activities
To begin to explore differences in the BMAL paralogs, we compared biochemical activities known to be critical for circadian function. Using a mammalian two-hybrid assay, we showed that BMAL2 had a slightly higher intrinsic affinity for CLOCK than BMAL1 (Fig. 1d). Steady-state transcription assays showed that CLOCK–BMAL2 also activated transcription of the Per1-luciferase (Per1-Luc) reporter to higher levels than CLOCK–BMAL1 (Fig. 1e), all consistent with previous observations19,20. Increased transcriptional activation by BMAL2 may correlate with its higher affinity for CLOCK, as mutations that decrease affinity of BMAL1 for CLOCK lead to decreased transactivation14. Moreover, complexes of CLOCK with either BMAL1 or BMAL2 were both capable of further activation by CBP or p300, and repression by CRY1 (Fig. 1e). Therefore, even though BMAL2 is unable to support the molecular circadian clock in Bmal1–/– cells, its steady-state activities appear to be comparable to BMAL1. These apparently paradoxical data suggest that a balance of affinities for transcriptional coregulators may be important for establishing the appropriate strength of activation and repression needed to establish circadian oscillations.
The C-terminus of BMAL1 is needed for circadian cycling
To identify specific sequences in BMAL1 that confer circadian function, we generated a series of domain-swapping chimeras by systematically substituting regions of Bmal1 with corresponding sequences from Bmal2 (Fig. 2a). To minimize issues with protein misfolding, we used highly conserved or identical sequences at swap junctions (Supplementary Fig. 2) and tested the ability of chimeras to express full-length proteins (Supplementary Fig. 3). We then examined the ability of the chimeras to restore circadian rhythms in Bmal1–/– Per2Luc fibroblasts following lentiviral transduction and stable selection.
Figure 2.

The BMAL1 C-terminus is needed for circadian function. (a) Domain organization of BMAL proteins and diagram of chimeric constructs. Chimera boundaries can be found in Supplemental Figure 2. (b) Luminescence records from Bmal1–/– Per2Luc fibroblasts expressing WT Bmal1 or Bmal1 chimeras with Bmal2 substitutions of the N-terminal core domains. One trace per chimera is shown from a representative experiment (n = 3 clonal lines with 2 cell culture replicates each). (c) Mean period of rescued circadian luminescence rhythms ± s.d. (n = 3 clonal lines with 2 cell culture replicates each). (d) Luminescence records from Bmal1–/– Per2Luc fibroblasts expressing WT Bmal1 or PAS-swapping chimeras. One trace per chimera is shown from a representative experiment (n = 3 clonal lines with 2 cell culture replicates each). (e) Luminescence records from Bmal1–/– Per2Luc fibroblasts expressing WT Bmal1 or C-terminal chimeras. One trace per chimera is shown from a representative experiment (n = 3 clonal lines with 2 cell culture replicates each). (f) Mean period of rescued circadian luminescence rhythms ± s.d. (n = 3 clonal lines with 2 cell culture replicates each). *** P < 0.001 compared to WT Bmal1, two-tailed paired t test.
The Bmal1-A2 mutant, harboring substitution of the distal N-terminus of Bmal2 into Bmal1, restored circadian rhythms of the same period (Fig. 2b,c) albeit with significantly lower amplitude than wild type Bmal1 (Supplementary Fig. 3) possibly arising from the lack of an NLS in the BMAL2 N-terminus21. Chimeras based on swaps of other individual regions within the core bHLH-PAS domains of BMAL2 (i.e., Bmal1-B2, -C2, -D2, -E2 and -F2) were all able to rescue circadian rhythms, albeit with some changes in period (-D2) or amplitude (-D2, -E2) depending on the individual region (Fig. 2b,c and Supplementary Fig. 3). A Bmal1 chimera containing the intact, tandem PAS domains from Bmal2 was also able to rescue circadian rhythms (Fig. 2d). By contrast, substitution of the PAS domain core of Bmal1 into Bmal2 (the Bmal2-PAS1 mutant) did not confer the ability to generate rhythms to Bmal2 (Fig. 2d). Together, these data demonstrate that the distal N-terminus and bHLH-PAS core domains do not define the distinct ability of BMAL1 to generate circadian rhythms.
Next we examined the divergent C-termini, focusing on the H region that encompasses a transcriptional activation domain (TAD) at the distal C-terminus22, and an upstream G region that contains the intervening sequence after the PAS domains (Fig. 2a). Swaps of either the G or H region (Bmal1-G2 and Bmal1-H2) rescued circadian rhythms, but exhibited a significantly shorter period and lower amplitude compared to Bmal1 (Fig. 2e,f and Supplementary Fig. 3). Strikingly, a Bmal1 chimera fused to the entire C-terminus of Bmal2 (Bmal1-G2H2) was unable to restore circadian rhythms of the Per2Luc reporter (Fig. 2e) or mRNA expression of clock-controlled genes (Supplementary Fig. 3). Therefore, the sequence determinants that distinguish the circadian functions of the two BMAL paralogs lie in their divergent and poorly characterized C-termini.
BMAL1 C-terminal TAD determines circadian period
To begin to dissect how the BMAL1 C-terminus regulates circadian function, we focused our attention on the transactivation domain of the H region. The ability of the TAD to interact with both negative22-25 and positive26 transcriptional coregulators provides a clear first step towards understanding how the BMAL1 C-terminus may control protein interactions and progression of circadian transcription. We began by confirming that the TAD is both required for transcriptional activation by CLOCK–BMAL122 and necessary to rescue circadian cycling in Bmal1–/– Per2Luc fibroblasts (Supplementary Fig. 3).
In our chimera studies, substitution of the Bmal2 TAD (H domain) into Bmal1 gave rise to a circadian period that was more than three hours shorter than wild-type (WT) Bmal1 (Fig. 2f). Nearly half of the C-terminal 30 residues in the TAD are divergent between BMAL1 and BMAL2 (Supplementary Fig. 2), with many in the predicted α-helix (Fig. 3a). To identify the sequence determinants that give rise to this dramatic change in period length, we performed site-directed mutagenesis to mutate groups of Bmal1 TAD residues to their corresponding sequences in Bmal2 and tested their ability to rescue circadian rhythms in Bmal1–/– Per2Luc fibroblasts. Two Bmal1 mutants, E597S A598I A599D and V602A I603F, exhibited significantly shorter period lengths (~22.5 and ~22.0 h, respectively) compared to WT Bmal1 (~23.5 h) (Fig. 3b,e). The effect of these mutations was nearly additive, as the combined Bmal1 mutant (E597S A598I A599D V602A I603F) led to a substantially shorter period length (~20.6 h) (Fig. 3c,e).
Figure 3.

A helical motif within the BMAL1 H domain controls circadian period length. (a) Comparison of BMAL paralog sequence and predicted secondary structure near the H domain α-helix. Underlined, KIX-binding IxxLL motif. Residue coloring: red, conserved; orange, similar; black, non-conserved. Cylinder, predicted α-helix. (b-d) Luminescence records from Bmal1–/– Per2Luc fibroblasts expressing WT Bmal1 or Bmal1 chimeras with substitutions from the Bmal2 H domain α-helix, including mutants of the N-terminal (b-c) or C-terminal half of the α-helix (d). One trace per chimera is shown from a representative experiment (n = 3 clonal lines with 2 cell culture replicates each). (e) Mean period of rescued circadian luminescence rhythms ± s.d. (n = 3 clonal lines with 2 cell culture replicates each). ** P < 0.01, *** P < 0.001 compared to WT Bmal1, two-tailed paired t test.
The Bmal1 V602A I603F mutant may interfere with function of the TAD IxxLL recognition motif (Fig. 3a), a helical sequence known to recruit proteins associated with transcriptional regulation such as the KIX domain of CBP or p300 (refs. 27,28). Mutation of the first leucine within the IxxLL motif in the Bmal1 S605N L606Y mutant led to a significantly longer period and higher amplitude cycling compared to Bmal1 (Fig. 3d,e). Therefore, we observed large changes in period length of the molecular circadian clock (~5 h from shortest to longest) upon incorporation of BMAL2 residues into the BMAL1 TAD α-helix. These data suggest that modulation of TAD structure and/or interactions with transcriptional coregulators can fine-tune the balance of activation and repression to control progression of the circadian clock.
The BMAL1 TAD is intrinsically unstructured
Striking changes in circadian period elicited by TAD mutations prompted us to investigate the biochemical basis by which the TAD regulates CLOCK–BMAL1 function. The last 30 residues of the TAD are highly conserved between vertebrates and insects that possess a vertebrate-like clock (Supplementary Fig. 4), in which transcriptional repression is driven by CRY29. Aside from the single α-helix, the BMAL1 TAD (residues 579-626) is predicted to be intrinsically disordered, typical of transactivation domains found in other transcription factors30. Given the predicted flexibility of this essential domain, we turned to solution NMR spectroscopy to study the TAD. A heteronuclear single quantum coherence (HSQC) spectrum of the 15N-labeled TAD protein backbone displayed the modest chemical shift dispersion expected for a disordered protein. The lack of structure was verified by chemical shift analyses of Cα and Cβ assignments31,32, which confirmed modest helical propensity localized only to the predicted α-helical region (Supplementary Fig. 4).
CRY1 and CBP(p300) compete for BMAL1 TAD binding
To examine binding of transcriptional coregulators on the BMAL1 TAD, we used the CRY1 CC peptide representing a monomeric, surface-exposed α-helix on CRY1 needed for CLOCK–BMAL1 repression33, or the isolated CBP(p300) KIX domain, one of several TAD-binding modular domains from CBP and p300 proteins (Supplementary Fig. 4). Both the CRY1 CC and CBP(p300) KIX domain have been reported to interact with the BMAL1 TAD24,26. To identify binding sites on the TAD, we collected 15N HSQC spectra of 15N-labeled TAD alone or in the presence of coregulators. Titration of 15N TAD with either the CRY1 CC, CBP or p300 KIX domains led to chemical shift perturbations at similar residues, confirming their interaction and further demonstrating that their binding sites overlap on the TAD (Supplementary Fig. 4). Marked differences in the linewidth of 15N TAD crosspeaks occurred upon binding to coregulators, particularly at residues in or near the IxxLL motif (e.g. V602 and S605, Supplementary Fig. 4). As other TADs can undergo a coupled folding and binding process to form helical complexes with KIX domains34, the observed changes in linewidth upon coregulator binding reflect different rates of chemical exchange that could arise from changes in affinity, conformational exchange, or both.
Chemical shift mapping revealed that the shared binding sites center on the canonical KIX-binding IxxLL motif within the TAD α-helix, as well as a second cluster of seven residues in the distal C-terminus (Figs. 4a,b). The CRY1 binding sites we identify here agree with previous studies that indicate 1:1 stoichiometry of the complex23-25. To determine if CRY1 and p300 could compete for binding to the TAD, we performed a 15N HSQC titration with 15N TAD and CRY1 CC to a 1:1 stoichiometric ratio, and then added an equivalent amount of the p300 KIX domain (Fig. 4c). 15N TAD crosspeaks are expected to fall somewhere between the binary 15N TAD–CRY1 or 15N TAD–KIX complexes, representing a population-weighted average of binary complexes present in solution. We found that 15N TAD crosspeaks moved near the 15N TAD–KIX complex (Fig. 4c), reporting that the p300 KIX domain can effectively compete CRY1 CC off the TAD in vitro.
Figure 4.

CBP(p300) and CRY1 interact with BMAL1 TAD. (a-b) Backbone chemical shift perturbations of 15N BMAL1 TAD with stoichiometric p300 KIX (a) or CRY1 CC (b). p.p.m., parts per million. Dashed line, ΔδTOT significance cutoff = 0.04 p.p.m. *residues broadened beyond detection. (c) Highlighted regions of 15N HSQC spectra showing titration of 15N BMAL1 TAD with CRY1 CC to 1:1 stoichiometry (light to dark purple; solid arrow); at 1:1 stoichiometry with p300 KIX (green); or in the presence of both CRY1 CC and p300 KIX (brown). Dashed line shows transition of 15N TAD–CRY1 to 15N TAD–p300 KIX upon competition. (d-e) Relative15N TAD–EDTA peak intensities from PRE experiments as ratio of Mn2+ chelation by the C-terminal EDTA (black circle) in the free protein (gray), or upon complex formation with p300 KIX (green) (d) or CRY1 CC (purple) (e). (f) Relative15N TAD 619X–EDTA peak intensities from PRE experiments as ratio of Mn2+ chelation by the C-terminal EDTA (black circle) in the free protein (gray), or upon complex formation with CRY1 CC (purple).
Dynamic interactions of TAD with CRY1 and CBP(p300)
To investigate coupling between the TAD α-helix and distal C-terminus, we took advantage of paramagnetic relaxation enhancement (PRE) with manganese, a paramagnetic cation that induces loss of peak intensity when brought into close proximity (~20-25 Å) to NMR-active nuclei. We acquired 15N HSQC spectra of 15N TAD possessing a C-terminal cysteine covalently coupled to EDTA (15N TAD–EDTA) in the presence and absence of chelated Mn2+. Covalent modification of the TAD with EDTA via the added cysteine does not impact complex formation with p300 KIX or CRY1 CC (Supplementary Fig. 5). We found no evidence that the distal C-terminus interacts appreciably with the α-helix in the free TAD, as Mn2+-dependent broadening was confined to residues proximal to the labeling site (Fig. 4d), consistent with localized PRE effects seen in other disordered proteins35.
To determine if the TAD undergoes dynamic conformational rearrangement upon complex formation, we collected 15N HSQC spectra of the 15N TAD–EDTA ± Mn2+ titrated with either p300 KIX or CRY1 CC (Supplementary Fig. 5). Quantitative analysis of Mn2+-dependent changes in peak intensity identified regions near the TAD helix that are decreased upon formation of both 15N TAD–p300 KIX (Fig. 4d) and 15N TAD–CRY1 CC complexes (Fig. 4e). We observed a striking, near complete loss of signal intensity at the TAD α-helix upon binding to CRY1 CC (Fig. 4e), suggesting that the distal C-terminus is brought into close proximity to the TAD α-helix in a CRY1-dependent manner. A 15N TAD mutant lacking the seven residues at the distal C-terminus lost this CRY1- and Mn2+-dependent broadening of the TAD α-helix (Fig. 4f), demonstrating the dependence of the conformational rearrangement on the distal C-terminus. Thus, the BMAL1 TAD is highly dynamic in its interactions with transcriptional coregulators.
TAD mutations alter affinity for CRY1 and CBP(p300)
To explore functional differences in the TADs of BMAL1 and BMAL2, we generated a suite of mutant proteins to quantitatively compare against WT TAD interactions with transcriptional coregulators. Like BMAL1, the BMAL2 TAD (residues 540-579) is intrinsically disordered with a weakly formed α-helix, and it interacts with both p300 KIX and CRY1 CC at the α-helix as well as the C-terminal seven residues (Supplementary Fig. 6). We focused on two substitution mutants of the BMAL1 TAD α-helix (the short period V602A I603F mutant and long period S605N L606Y mutant) (see Fig. 3), and two structure-based mutants, the L606A L607A mutant, designed to interfere with the IxxLL motif, and the 619X truncation mutant that removed the C-terminal seven residues. In all cases, mutation-induced backbone chemical shift perturbations localized to the site of mutation, demonstrating that we can selectively perturb either the α-helix or the distal C-terminus (Supplementary Fig. 6).
We determined affinities of the TADs for the CBP KIX domain or CRY1 CC using isothermal titration calorimetry (ITC, Supplementary Figure 7). Due to relatively low heats evolved in the TAD–CRY1 CC ITC experiments, we also determined affinities for CRY1 CC using a fluorescence polarization (FP) assay (Supplementary Fig. 7) and obtained comparable affinities to our ITC data (Table 1). The L606A L607A mutant disrupted interaction with the CBP KIX domain or CRY1 CC under our assay conditions (Fig. 5a,b). We found that the other TADs had relatively similar affinities for the CBP KIX domain while affinities for CRY1 CC varied by an order of magnitude. Notably, the short period V602A I603F mutant decreased affinity for CRY1 CC by about three-fold compared to the WT BMAL1 TAD, while the long period S605N L606Y mutant increased affinity by about three-fold (Table 1). These opposing phenotypes result from substitution of different residues from BMAL2; however, affinity of the intact BMAL2 TAD was similar to BMAL1 (Table 1), suggesting a complicated interplay of sequence context and structural dynamics in recognition of transcriptional coregulators by BMAL paralog TADs. Moreover, truncation of the C-terminal seven residues (619X) decreased affinity for CRY1 CC to an extent similar to the V602A I603F mutation. Collectively, these data suggest that the IxxLL motif in the TAD α-helix is the major binding site for both CRY1 CC and CBP(p300) KIX domains, although the distal C-terminus also makes an important contribution. The preferential effect of mutations on CRY1 binding relative to CBP(p300) suggests that modulating affinity for CRY1 may elicit the changes in circadian periodicity we measured in the mutants.
Figure 5.
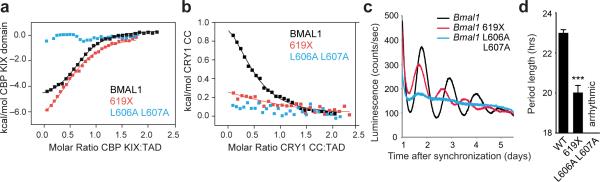
BMAL1 TAD mutations decrease affinity for CBP(p300) and CRY1 and influence circadian period. (a) Isothermal titration calorimetric (ITC) profiles for the interaction of CBP KIX domain with WT or mutant BMAL1 TADs. Line, one-site binding model representing best fit to data; see Table 1. (b) ITC profile for the interaction of CRY1 CC peptide with WT or mutant BMAL1 TADs. (c) Luminescence records from Bmal1–/– Per2Luc fibroblasts expressing WT or mutant Bmal1. One trace per chimera is shown from a representative experiment (n = 3 clonal lines with 2 cell culture replicates each). (d) Mean period of rescued circadian luminescence rhythms ± s.d. (n = 3 clonal lines with 2 cell culture replicates each). *** P < 0.001 compared to WT Bmal1, two-tailed paired t test.
TAD mutations perturb circadian timekeeping
To test our prediction that BMAL1 TAD interactions play an important role in period determination, we complemented Bmal1–/– Per2Luc cells with either the C-terminal truncation (619X) or the L606A L607A mutant of Bmal1. Truncation of the C-terminus shortened the intrinsic period by nearly three hours, while the L606A L607A mutant abolished cycling (Fig. 5c,d and Supplementary Fig. 7). Given that these TAD mutations substantially reduce affinity for CRY1 (Table 1), we sought to measure CRY1 repression of CLOCK–BMAL1 mutants in Per1-Luc reporter assays. CLOCK–BMAL1 complexes harboring either the L606A L607A or 619X mutation exhibited decreased steady-state activity, although BMAL1 proteins were expressed to similar levels (Fig. 6a and Supplementary Fig. 8). Repression by CRY1 was significantly reduced with both mutants, but to a greater extent with the L606A L607A mutant (Fig. 6b), consistent with our observation that this mutation leads to a greater reduction in apparent affinity for CRY1 (Table 1).
Figure 6.

Distinct sites on CLOCK and BMAL1 facilitate repression by CRY1. (a) Per1-Luc assay in HEK293T cells transiently transfected with plasmids encoding Clock WT and Bmal1 WT, 619X or L606A L607A. Relative light units (RLU) are scaled down by 103 and expressed as mean ± s.d. of triplicate measurements from one representative experiment (n = 3). (b) Per1-Luc assay with increasing amounts of plasmids encoding Cry1 with Clock WT and Bmal1 WT, 619X or L606A L607A. Relative activity normalized to Clock WT–Bmal1 WT set to 100. (c) Residues in the Hβ-Iβ (HI) loop on CLOCK PAS-B (PDB 4F3L)14 that reduce CRY1 repression when mutated (yellow25 or orange37). (d) Coimmunoprecipitation (IP) of Flag-CLOCK WT or HI (Q361P W362R) and Flag-BMAL1 by CRY1-myc from HEK293T cells with anti-myc antibody. Western blots (IB) were performed using indicated antibodies. Uncropped images can be found in Supplementary Data Set 1. (e) Per1-Luc assay with plasmids encoding Clock WT and Bmal1 WT, 619X or L606A L607A. RLU represented as in panel (a). (f) Per1-Luc assay with increasing amounts of plasmid encoding Cry1 with Clock HI and Bmal1 WT, 619X or L606A L607A. Relative activity normalized to Clock HI–Bmal1 WT set to 100. (g) Tryptophan indole region of 15N HSQC spectra showing 15N CLOCK PAS-B in the absence (black) and presence (orange) of CRY1 CC. The full spectrum can be found in Supplementary Fig. 8. ** P < 0.01; ***P < 0.001 compared to Bmal1 WT by two-tailed paired t test.
CRY1 binds to multiple sites on CLOCK–BMAL1
Formation of a stable ternary complex consisting of CLOCK, BMAL1, and CRY1 lies at the heart of CRY1-dependent negative feedback in the clock6,7,10,36. Given the modest affinities that we measured for the BMAL1 TAD and isolated CRY1 CC peptide, we reasoned that there are likely other interactions between the CLOCK–BMAL1 complex and CRY1. Mutations that localize in or adjacent to the solvent-exposed HI loop of the CLOCK PAS-B domain (named for its location between the Hβ and Iβ strands of the PAS domain; Fig. 6c)14 also reduce repression by CRY1 (refs. 22,25,37). To determine if mutation of the CLOCK HI loop (Q361P W362R) affects complex formation with CRY1, we performed coimmunoprecipitation assays to examine interaction of CRY1-myc with WT or HI mutant Flag-tagged CLOCK and WT BMAL1. We showed that, while WT CLOCK and BMAL1 coimmunoprecipitated with CRY1-myc, mutation of the CLOCK HI loop rendered CLOCK and BMAL1 unable to stably interact with CRY1-myc (Fig. 6d and Supplementary Fig. 8). Taken together, complex formation between CRY1 and CLOCK–BMAL1 occurs through interactions with both the BMAL1 C-terminus and CLOCK PAS-B.
To explore how the interaction of CRY1 with CLOCK PAS-B impacts regulation of CLOCK–BMAL1, we assayed Per1-Luc activity. Transcriptional activation by CLOCK HI–BMAL1 complexes is similar to WT CLOCK–BMAL1 (Fig. 6a, e), but repression by CRY1 is significantly reduced (Fig. 6f). Strikingly, we found that mutation of the CLOCK PAS-B HI loop in conjunction with BMAL1 L606A L607A or 619X eliminated repression by CRY1 (Fig. 6f), even when CRY1 was overexpressed to suprastoichiometric levels relative to CLOCK–BMAL1 (Supplementary Fig. 8). We could not detect a direct interaction between CRY1 CC and 15N CLOCK PAS-B by NMR spectroscopy (Fig. 6g and Supplementary Fig. 8), indicating that the CLOCK PAS-B domain mediates interactions with CRY1 through a site distinct from the CRY1 CC α-helix. Therefore, simultaneous disruption of CRY-interacting interfaces on CLOCK PAS-B and the BMAL1 TAD eliminates CRY1 regulation of CLOCK–BMAL1, demonstrating the importance of multivalent interactions for CRY1 repression.
DISCUSSION
Our study leveraged the finding that although BMAL1 and its close paralog BMAL2 share similar steady-state biochemical activities, BMAL2 is unable to sustain circadian cycling15,17, allowing us to discover that the C-terminus of BMAL1 holds the key to generating circadian oscillations by BMAL paralogs. By integrating data from biophysical and cell-based assays, we now identify the important role the BMAL1 TAD plays in the clock and highlight two distinct binding sites on CLOCK–BMAL1 that are required for CRY1 function. The first site on the CLOCK PAS-B domain presumably functions as a docking site to localize CRY1 to the complex, and the second site on the BMAL1 TAD controls the circadian activity of CLOCK–BMAL1. From the mutations studied here, we find that affinity of the TAD for CRY1 appears to correlate with period; decreasing affinity leads to shorter periods while increasing affinity leads to longer periods (Fig. 7a). We find it compelling that substitution of only a few residues within the BMAL1 TAD α-helix can elicit circadian rhythms with shorter or longer than normal periods (Fig. 7b), demonstrating that the TAD plays an important role in establishing intrinsic circadian timekeeping. Our observation of opposing period phenotypes encoded by adjacent sequences within the α-helix of the BMAL2 TAD may explain why it is an outlier when comparing CRY1 affinity and period (Fig. 7a). Additional studies of BMAL1 and BMAL2 TADs that also take into consideration the G region identified in this study will be needed to understand the biochemical properties of BMAL1 that enable the dynamic interactions with coregulators that underlie generation of self-sustaining circadian rhythms.
Figure 7.

Regulation of the BMAL1 TAD by CRY1 contributes to determination of circadian period. (a) Correlation of coregulator affinities for isolated TADs with period lengths from circadian rescue experiments. Not shown: Bmal1 L606A L607A mutant, which has no observable affinity for either CBP(p300) KIX or CRY1 CC and does not rescue circadian rhythms in Bmal1–/– Per2Luc fibroblasts. (b) Location of long and short period substitution mutants in the TAD α-helix. (c) Model of CRY1 interactions with the CLOCK–BMAL1 complex. (d) Model for regulation of the circadian transcriptional feedback loop by the ternary CLOCK–BMAL1–CRY1 complex. Black and white bars underneath indicate night or day, respectively. See text for more details.
CRY1 appears to fulfill part of its essential role as a negative regulator by interacting directly with CLOCK–BMAL1 to sequester the BMAL1 TAD from coactivators (Fig. 7c). Here we examined interactions of the BMAL1 TAD with the CBP(p300) KIX domain as a proxy for coactivator recruitment. We expect that our proposed mechanism for CRY1, linking inhibition to TAD sequestration, will hold true for other coactivators and other domains from CBP(p300) that may interact with the BMAL1 TAD. It is worth noting that the related bHLH-PAS transcription factor HIF (hypoxia-inducible factor) uses both TAD-dependent and -independent modes to recruit transcriptional regulators38-40, suggesting the potential for additional regulatory mechanisms that control CLOCK–BMAL1 activity. In this context, we note that our model is compatible with studies showing that CRY1 may also interact with additional epigenetic regulators to help achieve complete repression in vivo41.
The CRY1 mechanism we propose here occurs independently of the direct involvement of PER proteins (Fig. 7d). Recent studies show that PER2 titrates CRY1 away from direct interactions with CLOCK–BMAL1 in vitro and in cells6,42,43. Two recent CRY structures provide evidence for a possible CLOCK–BMAL1 binding site in the antenna chromophore-binding pocket, distinct from the PER2 binding site centered on the CRY1 CC α-helix23,44. Because both PER2 and BMAL1 interact with the CRY1 CC α-helix23,24,33,44, our data suggest that a timely handoff between early repressive PER2–CRY1 complexes3-5,45 to the late repressive CLOCK–BMAL1–CRY1 complexes7,8 may represent a critical control mechanism for the molecular circadian clock. We do not discount the importance of PER2 in regulating the stability and timing of nuclear entry of the PER2–CRY1 complexes and resultant temporal progression of circadian transcriptional repression46,47, or roles that CRY1 may play in the PER2–CRY1 complexes10.
Precedence for regulation of protein function based on competition for mutually exclusive binding sites exists throughout biology38,48. Competitive mechanisms can make the conversion between inactive and active states more switch-like instead of graded49, and switch-like responses that exhibit a high degree of cooperativity amplify rhythms in the molecular circadian clock, preventing them from reaching an equilibrium that terminates oscillation50. Furthermore, computational models suggest that tight binding of repressors and activators along with control over protein stoichiometry is key for generation of a robust circadian clock51. The remarkable decrease in CRY1 repression we observed upon disruption of the stable CLOCK–BMAL1–CRY1 complex (Fig. 6f) illustrates how important ternary complex formation is for repression when CRY is expressed to near stoichiometric levels with the CLOCK–BMAL1 complex in vivo52.
It is well recognized that changes in clock protein abundance, posttranslational modifications and subcellular localization play critical roles in circadian timekeeping36. Our data suggest that conformational dynamics also play a critical role in period determination, as disordered regions can easily coordinate protein interactions with diverse coregulators by coupling folding to binding34,53. Modest changes in the intrinsic secondary structure of other TADs affect the thermodynamics of binding to CBP(p300) to influence the temporal basis (i.e., transient or constitutive) of coactivation54,55. Therefore, the structural dynamics we observe within the BMAL1 TAD could play an interesting role in setting the timescale of activation and repression to ultimately contribute to circadian timing.
ONLINE METHODS
Lentiviral DNA constructs, preparation and transduction
Bmal1 and UBC promoters were cloned into the pENTR-5’-TOPO vector (Life Technologies) as before17. Mouse Bmal1, Bmal2 and various chimeras or mutants were first subcloned to p3xFlag-CMV-10 or -14 vectors to obtain 3xFlag tags at the N- or C-terminus, respectively, and then cloned to the pENTR/D-TOPO vector (Life Technologies). The promoter and cDNA pENTR vectors were recombined with pLV7 destination vector56 using Clonase (Life Technologies) to generate the lentiviral expression constructs. All constructs were verified by sequencing of the entire open reading frame.
Recombinant lentiviral particles were produced by transient transfection in human embryonic kidney HEK293T cells (ATCC) using the calcium-phosphate method as previously described56,57. Bmal1–/– Per2Luc mouse fibroblast cell line was generated in our previous studies17. Cells were cultured in DMEM supplemented with 10% FBS and 1x penicillin-streptomycin-glutamine mixture. All cell culture reagents were from HyClone. For infection of Bmal1–/– Per2Luc fibroblasts, culture medium containing viral particles (~106 viral particles/ml) were harvested at 48 hr post-transfection and used for subsequent infection of cells. Transduced cells were selected with 10 μg/ml blasticidin (InvivoGen) as previously done17.
Luminescence recording and data analysis
We used a LumiCycle luminometer (Actimetrics) for luminescence recording as previously described56. Cells were grown to confluence in 35 mm dishes prior to recording. The recording medium contained 1x DMEM, 25 mM HEPES (pH 7.4), 1% FBS, 1x B-27 and 1 mM luciferin as previously17. Three independent clonal lines and 2-3 dishes for each clone were tested to validate phenotypes. Raw luminescence data (counts/sec) as a function of time (days) in culture were analyzed using the LumiCycle Analysis program (version 2.53, Actimetrics) to determine circadian parameters. Due to high transient luminescence upon medium change, the first cycle was excluded from analysis. Briefly, raw data were fitted to a linear baseline, and the baseline-subtracted data were fitted to a damped sine wave, from which period length, goodness of fit, and damping constants were determined. A goodness-of-fit of >80% was usually achieved from samples that showed persistent rhythms. Damping rate is described by the value of 1/damping constant. For amplitude analysis, raw data from days 3-5 were fitted to a linear baseline (polynomial number = 1), and the baseline-subtracted data were fitted to a sine wave, from which amplitude was determined.
Transient transfection and reporter assays
Reporter assays investigating steady-state differences between Bmal1 and Bmal2 function were performed using HEK293T cells in triplicate on 384-well plates. For mammalian two-hybrid assays, 12.5 ng of pGL4P-4xUAS::Luc, 2.5 ng Renilla luciferase (RLuc), 25 ng pACT and 25 ng pBIND plasmids were used in each well as previously described25. The Per1-Luc reporter assay was performed as reported elsewhere with minor modifications25. For each well, 12.5 ng Per1-Luc reporter, 25 ng pLV7-P(UBC)-mouse Bmal, 25 ng pLV7-P(CAG)-mouse Clock, 25 ng pcDNA-CMV-mouse p300 or Cbp, 25 ng pLV156-P(CMV)-mouse Cry1, and 2.5 ng RLuc were used. When needed, empty vectors were included to make up to 115 ng of total plasmid amount. RLuc was used for reporter normalization. In assays with Cry1, indicated amounts of pLV156-P(CMV)-Cry1 were compensated with empty pLV7 destination vector to a total of 50 ng. Lipofectamine 2000 (Life Technologies) were used with a reverse transfection protocol. Reporter assay was performed 24 hr post transfection using DualGlo luciferin reagent (Promega) and luciferase reporter activity was normalized with Renilla luciferase.
To compare expression of Flag-tagged Bmal1 genes, cells were lysed in RIPA buffer containing complete protease and phosphatase inhibitors (Sigma). Immunoblotting was done using the following primary antibodies: mouse anti-Flag antibody (M2) (Sigma cat. # F3165) and goat anti-β actin (C-11) (Santa Cruz Biotechnology cat. # sc-1615), and the following secondary antibodies: anti-mouse IgG-HRP (Santa Cruz Biotechnology cat. # sc-2005) and anti-goat IgG-HRP (Santa Cruz Biotechnology cat. # sc-2020). SuperSignal West Pico substrate (Pierce) was used for chemiluminescent detection on autoradiograph film.
For coimmunoprecipitation assays, plasmids were transfected into HEK293T cells in a 35 mm dish using LT-1 transfection reagent (Mirus) with the indicated plasmid ratios: 1.5 μg each pSG5 mouse Flag-Bmal1 and pSG5 mouse His6Flag-Clock, plus 50 ng pcDNA3 mouse Cry1 (no tag; used for anti-Flag IP) or 250 ng pcDNA4 Cry1 (C-terminal Flag, myc, His6-tagged; used for anti-myc IP). Cells were harvested 48 hours later for coimmunoprecipitation with anti-myc (9E10) (distributed by the Developmental Studies Hybridoma Bank at the University of Iowa, IA, cat. # 9E 10-a) or anti-Flag antibody (Sigma). Briefly, cells were lysed on ice for 15 min in 250 μl 50 mM HEPES pH 7.5, 150 mM NaCl, 5% (v/v) glycerol, 0.5% (v/v) NP-40 and EDTA-free protease inhibitors (RPI Biochemicals). Clarified extracts were added to 300 μl lysis buffer containing 1 μg of anti-myc monoclonal antibody plus 15 μl Protein A/G resin (Santa Cruz biotechnology) or 15 μl anti-Flag M2 affinity resin (Sigma cat. #A2220) after retaining 25 μl as the input sample. Tubes were rotated end over end for 4 hours, and resin was washed three times with 400 μl lysis buffer. Proteins were eluted from resin by addition of 30 μl 2X SDS Laemmli buffer and boiled for 3 minutes. Complexes were resolved by 7.5% SDS-PAGE after transfer to nitrocellulose. Proteins were detected by immunoblotting using the following primary antibodies: mouse anti-myc, mouse anti-Flag, rat anti-CRY1 (a gift from A. Sancar)58, mouse anti-BMAL1 (B-1) (Santa Cruz Biotechnology cat. # sc-365645), or rabbit anti-CLOCK (H-276) (Santa Cruz Biotechnology cat. # sc-25361), and secondary antibodies: anti-mouse IgG-HRP (Sigma cat. # A9917), anti-rabbit IgG-HRP (Sigma cat. # A0545), and anti-rat IgM-HRP (Thermo Fisher cat. # 31476). Clarity reagent (Bio-Rad) was used for chemiluminescent detection on a ChemiDoc XRS+ imager (Bio-Rad). All data shown are representative from three independent experiments.
For Per1-Luc reporter gene assays investigating CRY1 repression, plasmids were transfected in duplicate into HEK293T cells in a 48-well plate using LT-1 transfection reagent (Mirus) with the indicated plasmids: 5 ng pGL3 Per1-Luc reporter59, 100 ng each pSG5 Flag-Bmal1 and pSG5 His6Flag-Clock, and pcDNA3 Cry1 (untagged) in increasing amounts as indicated; empty pcDNA4 vector was used to normalize total plasmid to 800 ng/well. Cells were harvested 30 hours after transfection using Passive Lysis Buffer (NEB) and luciferase activity assayed with Bright-Glo luciferin reagent (Promega). Each reporter assay was repeated at least three independent times. To compare relative expression of CLOCK, BMAL1, and CRY1 proteins, a 12-well plate of HEK293T cells was transfected with 400 ng each pSG5 His6Flag-Clock and Flag-Bmal1 with pcDNA4B Cry1 (C-terminal Flag, myc, His6 epitope tags)60 as indicated, using empty pcDNA4 vector to normalize plasmid concentration to 1 μg total DNA. Cells were harvested 48 hours later for immunoblotting with anti-Flag antibody, anti-BMAL1 (B-1), and anti-CLOCK (H-276) antibodies. Immunoblot of relative expression in Supplementary Figure 8 shown is representative of two independent experiments.
All cell lines used in the study were routinely checked for mycoplasma contamination. Original images of autoradiographs and blots used in this study can be found in Supplementary Data Set 1.
qPCR analysis
Cells were synchronized with 200 nM dexamethasone and the first time point was collected 24 hours later as previously described17. Total RNAs were prepared using the RNeasy 96 kit (Qiagen). High-capacity RNA to cDNA kit (Applied Biosystems) was used for reverse transcription, and qPCR was performed using SYBR Green PCR master mix (Thermo Scientific) on an iCycler thermal cycler (BioRad). Transcript levels for each gene were normalized to Gapdh and values were expressed as percentage of expression as indicated.
Expression and purification of recombinant proteins
Proteins were expressed in E. coli Rosetta2 (DE3) cells from a pET22b vector backbone (EMD Millipore). Mouse BMAL1 TAD (residues 579-626), BMAL2 TAD (residues 540-579), and p300 KIX (residues 565-653) domains all possessed an N-terminal TEV-cleavable His6-GST or His6-SUMO tag. Mutations were introduced by site-directed mutagenesis and confirmed by sequencing. The plasmid encoding untagged mouse CBP KIX was a gift from Peter Wright. CBP KIX protein was expressed in Rosetta (DE3) cells overnight at 18°C after IPTG induction and the soluble fraction was purified on Ni-NTA resin as described above using native His residues on CBP KIX. Cells were either grown in LB medium or M9 minimal medium containing 1 g/L 15NH4Cl for uniform 15N labeling as described before40; for triple resonance experiments, uniformly 13C, 15N-labeled samples were made using 1 g/L 15NH4Cl and 3g/L 13C-glucose as the sole nitrogen and carbon sources in M9 medium. Protein expression was induced at OD600 ~0.8-1.0 with 0.5 mM IPTG for ~16 h at 18°C. Cells were lysed in buffer containing 50 mM Tris (pH 7.5), 300 mM NaCl and 20 mM imidazole. Affinity purification was carried out with Ni-NTA resin (Qiagen), and eluted protein was buffer exchanged to a low imidazole buffer by desalting column or by diafiltration with an Amicon stirred cell concentrator under nitrogen pressure, followed by proteolysis with TEV protease overnight at 4°C. Cleaved protein was retained from flow-through from an Ni-NTA column and further purified by size exclusion chromatography on Superdex 75 16/600 (GE Life Sciences) in NMR buffer (10 mM MES pH 6.5, 50 mM NaCl). Site-specific labeling of the BMAL1 TAD with the added C-terminal cysteine was performed with S-methanethiosulfonylcysteaminyl-EDTA (MTS-EDTA; Toronto Research Chemicals) as previously indicated40. Protein identities and/or covalent labeling were confirmed by ESI-MS on a Micromass ZMD mass spectrometer (Wythenshawe, UK).
Peptide synthesis and purification
The mouse CRY1 CC peptide (residues 470-503, sequence MVNHAEASRLNIERMKQIYQQL SRYRGLGLLASV) or a CC peptide containing an N-terminal cysteine (N-Cys CRY1 CC; residues 471-505, sequence CNHAEASRLNIERMKQIYQQLSRYRGLGLLASVPS) were synthesized on a CEM Liberty1 microwave peptide synthesizer using standard Fmoc chemistry. Amino acids were purchased from NovaBiochem and assembled on Rink-amide-MBHA resin; all other reagents were purchased from Sigma-Aldrich. Fmoc deprotection was achieved using 20% piperidine with 0.1 M hydroxybenzotriazole (HOBt) in dimethylformamide (DMF). Couplings used 5 molar equivalents of Fmoc-amino acid, 5 molar equivalents of Diisopropylcarbodiimide and 10 molar equivalents of HOBt in DMF. Fully synthesized peptide resins were washed with dichloromethane and dried. Cleavage of the peptide from the resin was performed in a trifluoroacetic acid/thioanisole/ethanedithiol/phenol (90:4:4:2) solution for 90 minutes. Peptides were purified by RP-HPLC (Vydac) on preparative C18 columns. Fractions were collected and analyzed by ESI-MS on a Micromass ZMD mass spectrometer to confirm the correct molecular weight. In each case the major peak was found to be the peptide, and fractions that contained the peptide as a major constituent (≥95% purity) were combined and lyophilized.
NMR spectroscopy
NMR experiments were conducted at 25°C on a Varian INOVA 600-MHz spectrometer equipped with 1H, 13C, 15N triple resonance, Z-axis pulsed field gradient probes. All NMR data were processed using NMRPipe and NMRDraw61. Chemical shift assignments were made with SPARKY62 using NMR data obtained from standard 3D triple resonance experiments acquired on 300 μM uniformly 13C,15N labeled TAD protein, including: HNCO, HNCACB, CBCA(CO)NH, H(CCO)NH-TOCSY, (H)C(CO)NH-TOCSY and HCCH-TOCSY spectra. Chemical shifts of mutant TADs were re-assigned by using triple resonance data in NMRViewJ RunAbout or, when warranted from minor chemical shift perturbations, by minimal chemical shift analysis in NMRViewJ63. Secondary structure predictions were validated by TALOS+64 and by comparison to the Chemical Shift Index31,32. 15N HSQC titration of 100 μM 15N TAD proteins was done by stepwise addition of CRY1 CC peptide, CBP or p300 KIX domain in 10 mM MES pH 6.5, 50 mM NaCl. Samples were concentrated to 300 μl final volume and adjusted to a final concentration of 10% (v/v) D2O. 15N HSQC titration data were analyzed with NMRViewJ63 using chemical shift perturbations defined by the equation: ΔδTOT [(Δδ1H)2 = + (χ(Δδ15N)2]½ and normalized with the scaling factor χ = 0.17, established from estimates of atom-specific chemical shift ranges in a protein environment65. For PRE NMR experiments, peak intensities from 15N HSQC spectra were determined using the Rate Analysis tool in NMRViewJ to extract normalized peak intensities. Minor broadening at negatively charged residues throughout the TAD is likely due to interaction with trace amounts of unchelated Mn2+, as acquisition of NMR data under moderately higher salt conditions (100 mM vs. 50 mM NaCl) alleviated this non-specific broadening (data not shown).
Isothermal titration calorimetry (ITC)
Proteins were extensively dialyzed at 4°C in 10 mM MES pH 6.5, 50 mM NaCl using 2 kDa molecular weight cutoff filter dialysis tubing (Spectrum Labs) prior to collecting ITC data. CBP KIX was used for analysis of TAD binding by ITC due to increased stability over p300 KIX, which was slightly more prone to precipitation under our ITC experimental conditions. ITC was performed on a MicroCal VP-ITC calorimeter at 25°C with a stir speed of 177 rpm, reference power of 10 μCal/sec and 10 μL injection sizes. Protein ratios for the cell and syringe for the ITC assays (2 or 3 independent ITC experiments were performed for each complex) are as follows: 223 μM CBP KIX titrated into 23 μM BMAL1 TAD (Stoichiometry, N = 0.74), 225 μM CBP KIX titrated into 18 μM BMAL2 TAD (N = 0.70), 223 μM CBP KIX titrated into 18 μM BMAL1 SL/NY (N = 1.06), 223 μM CBP KIX titrated into 22 μM BMAL1 VI/AF (N = 0.82), 252 μM CBP KIX titrated into 29 μM LL/AA TAD (N = not determined), 221 μM CBP KIX titrated into 25 μM 619X TAD (N = 0.78), or 225 μM CRY1 CC titrated into 22 μM BMAL1 TAD (N = 0.68), 220 μM CRY1 CC titrated into 25 μM BMAL2 TAD (N = 0.7), 225 μM CRY1 CC titrated into 18 μM BMAL1 SL/NY (N = 0.90), 225 μM CRY1 CC titrated into 20 μM BMAL1 VI/AF (N = not determined), 230 μM CRY1 CC titrated into 22 μM LL/AA TAD (N = not determined), and 221 μM CBP KIX titrated into 25 μM 619X TAD (N = 0.78). All data were best fit by a one-site binding model using Origin software.
Fluorescence polarization (FP)
The tetramethylrhodamine 5-maleimide fluorophore (Molecular Probes) was reacted with N-Cys CRY1 CC peptide at a 2-5 fold molar excess for 1.5 – 4 hours at 4°C in 25 mM MES, pH 5.0 with 25 mM NaCl and 10 % DMSO. The low pH of the labeling reaction was essential to avoid non-specific attachment of the TMR group to the lysine amide in the CRY CC peptide as determined by mass spectrometry. Fluorescently labeled TMR-CRY1 CC was purified by reverse phase HPLC and electrospray ionization mass spectrometry was used to verify the labeling and assess purity. Samples with greater than 95% purity were lyophilized and reconstituted in 10 mM MES pH 6.5, 50 mM NaCl and stored at −80 °C until needed.
Equilibrium binding assays between TMR-CRY1 CC and BMAL TADs were performed in 40 mM Tris, pH 8.0, 150 mM NaCl, 0.05% (v/v) Tween-20, 1 mM DTT at 25°C. Binding was monitored by changes in fluorescence polarization using an Perkin Elmer EnVision 2103 Multilabel plate reader with excitation at 531 nm and emission at 595 nm. 10 nM TMR-CRY1 CC peptide was preincubated with buffer alone or increasing concentrations of BMAL TAD constructs (27 nM-300 μM) for 150 minutes at room temperature in the dark prior to FP analysis. The equilibrium dissociation constant (KD) was calculated by fitting the dose-dependent change in milli-polarization level (Δmp) to a one-site specific binding model in the GraphPad Prism software package, using averaged Δmp values from quadruplicate assays. Data shown are from one representative experiment of three independent assays.
Supplementary Material
TABLE 1.
Dissociation constants for the interaction of BMAL transactivation domains with transcriptional coregulators
| CBP KIX | CRY1 CC | ||
|---|---|---|---|
| ITC* | ITC* | FP** | |
| BMAL1 | 2.05 ± 0.11 | 6.13 ± 0.82 | 6.80 ± 1.00 |
| BMAL1 V603A I604F | 1.17 ± 0.09 | nd | 17.3 ± 0.30 |
| BMAL1 S605N L606Y | 0.98 ± 1.18 | 2.16 ± 0.28 | 2.9 ± 1.50 |
| BMAL1 619X | 4.44 ± 0.23 | 19.67 ± 2.57 | nd |
| BMAL1 L606A L607A | nd | nd | nd |
| BMAL2 | 5.59 ± 0.81 | 6.10 ± 1.50 | 6.00 ± 0.80 |
KD from a representative ITC experiment with standard error for fit to one-site binding model (n = 2 or 3)
Mean KD with s.d. (calculated from n = 4 replicates) from a representative FP experiment (n = 3)
nd, no detectable binding under experimental conditions
ACKNOWLEDGMENTS
We thank P.E. Wright (The Scripps Research Institute) and A. Sancar (University of North Carolina, Chapel Hill) for sharing reagents. We thank Jillian Miller, Rafael Palomino and Glenn Millhauser (University of California, Santa Cruz) for technical assistance and access to instrumentation for automated peptide synthesis and purification, S.M. Rubin (University of California, Santa Cruz) for access to calorimetry instrumentation, critical discussions and advice, and the Chemical Screening Center (University of California, Santa Cruz) for access to other instrumentation. This work was supported by the US National Science Foundation (grant IOS-0920417 to A.C.L.), the US National Institutes of Health (grant GM107069 to C.L.P) and start-up funds from the University of California, Santa Cruz (to C.L.P.).
Footnotes
AUTHOR CONTRIBUTIONS
H.X., C.L.G., and P.J.S. designed the project with A.C.L. and C.L.P. H.X., C.L.G., P.J.S., S.K.K., C.R., H.-W.L. and C.L.P. performed experiments. N.C.P. contributed new reagents and materials. H.X., C.L.G., P.J.S., A.C.L. and C.L.P. analyzed data. The manuscript was written by H.X., C.L.G., A.C.L. and C.L.P. All authors contributed to editing the manuscript and support the conclusions.
ACCESSION CODES
Chemical shift assignments have been deposited with the Biological Magnetic Resonance Bank for the BMAL1 TAD (25280) and BMAL2 TAD (25549).
REFERENCES
- 1.Bass J. Circadian topology of metabolism. Nature. 2012;491:348–356. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- 2.Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends in cell biology. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown S, et al. PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science. 2005;308:693–696. doi: 10.1126/science.1107373. [DOI] [PubMed] [Google Scholar]
- 4.Duong H, Robles M, Knutti D, Weitz C. A molecular mechanism for circadian clock negative feedback. Science. 2011;332:1436–1439. doi: 10.1126/science.1196766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee C, Etchegaray J, Cagampang F, Loudon A, Reppert S. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 6.Ye R, Selby C, Ozturk N, Annayev Y, Sancar A. Biochemical analysis of the canonical model for the mammalian circadian clock. J Biol Chem. 2011;286:25891–25902. doi: 10.1074/jbc.M111.254680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koike N, et al. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stratmann M, Stadler F, Tamanini F, van der Horst G, Ripperger J. Flexible phase adjustment of circadian albumin D site-binding protein (DBP) gene expression by CRYPTOCHROME1. Genes Dev. 2010;24:1317–1328. doi: 10.1101/gad.578810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shearman L, et al. Interacting molecular loops in the mammalian circadian clock. Science. 2000;288:1013–1019. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- 10.Ye R, et al. Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 2014;28:1989–1998. doi: 10.1101/gad.249417.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu A, et al. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell. 2007;129:605–616. doi: 10.1016/j.cell.2007.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans J, Pan H, Liu A, Welsh D. Cry1-/- circadian rhythmicity depends on SCN intercellular coupling. J Biol Rhythms. 2012;27:443–452. doi: 10.1177/0748730412461246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan S, et al. Identification of a novel cryptochrome differentiating domain required for feedback repression in circadian clock function. J Biol Chem. 2012;287:25917–25926. doi: 10.1074/jbc.M112.368001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang N, et al. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science. 2012;337:189–194. doi: 10.1126/science.1222804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunger M, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi S, et al. Circadian clock gene Bmal1 is not essential; functional replacement with its paralog, Bmal2. Curr Biol. 2010;20:316–21. doi: 10.1016/j.cub.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu AC, et al. Redundant function of REV-ERBalpha and beta and non-essential role for Bmal1 cycling in transcriptional regulation of intracellular circadian rhythms. PLoS Genet. 2008;4:e1000023. doi: 10.1371/journal.pgen.1000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baggs JE, et al. Network features of the mammalian circadian clock. PLoS Biol. 2009;7:e52. doi: 10.1371/journal.pbio.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogenesch JB, et al. The basic helix-loop-helix-PAS protein MOP9 is a brain-specific heterodimeric partner of circadian and hypoxia factors. J Neurosci. 2000;20:RC83. doi: 10.1523/JNEUROSCI.20-13-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoenhard JA, et al. Regulation of the PAI-1 promoter by circadian clock components: differential activation by BMAL1 and BMAL2. J Mol Cell Cardiol. 2003;35:473–81. doi: 10.1016/s0022-2828(03)00051-8. [DOI] [PubMed] [Google Scholar]
- 21.Kwon I, et al. BMAL1 shuttling controls transactivation and degradation of the CLOCK/BMAL1 heterodimer. Mol Cell Biol. 2006;26:7318–30. doi: 10.1128/MCB.00337-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiyohara Y, et al. The BMAL1 C terminus regulates the circadian transcription feedback loop. Proc Natl Acad Sci U S A. 2006;103:10074–10079. doi: 10.1073/pnas.0601416103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czarna A, et al. Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell. 2013;153:1394–1405. doi: 10.1016/j.cell.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Czarna A, et al. Quantitative analyses of cryptochrome-mBMAL1 interactions: mechanistic insights into the transcriptional regulation of the mammalian circadian clock. J Biol Chem. 2011;286:22414–22425. doi: 10.1074/jbc.M111.244749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato T, et al. Feedback repression is required for mammalian circadian clock function. Nature genetics. 2006;38:312–319. doi: 10.1038/ng1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahata S, et al. Transactivation mechanisms of mouse clock transcription factors, mClock and mArnt3. Genes to cells. 2000;5:739–747. doi: 10.1046/j.1365-2443.2000.00363.x. [DOI] [PubMed] [Google Scholar]
- 27.Heery D, Kalkhoven E, Hoare S, Parker M. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 28.Radhakrishnan I, et al. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 29.Yuan Q, Metterville D, Briscoe A, Reppert S. Insect cryptochromes: gene duplication and loss define diverse ways to construct insect circadian clocks. Molecular biology and evolution. 2007;24:948–955. doi: 10.1093/molbev/msm011. [DOI] [PubMed] [Google Scholar]
- 30.Wells M, et al. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci U S A. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein science. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wishart D, Sykes B. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 33.Chaves I, et al. Functional evolution of the photolyase/cryptochrome protein family: importance of the C terminus of mammalian CRY1 for circadian core oscillator performance. Mol Cell Biol. 2006;26:1743–1753. doi: 10.1128/MCB.26.5.1743-1753.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugase K, Dyson H, Wright P. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 35.Felitsky D, Lietzow M, Dyson H, Wright P. Modeling transient collapsed states of an unfolded protein to provide insights into early folding events. Proc Natl Acad Sci U S A. 2008;105:6278–6283. doi: 10.1073/pnas.0710641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustafson CL, Partch CL. Emerging models for the molecular basis of mammalian circadian timing. Biochemistry. 2015;54:134–49. doi: 10.1021/bi500731f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao W-N, et al. CIPC is a mammalian circadian clock protein without invertebrate homologues. Nature cell biology. 2007;9:268–275. doi: 10.1038/ncb1539. [DOI] [PubMed] [Google Scholar]
- 38.Freedman S, et al. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nature structural biology. 2003;10:504–512. doi: 10.1038/nsb936. [DOI] [PubMed] [Google Scholar]
- 39.Partch C, Card P, Amezcua C, Gardner K. Molecular basis of coiled coil coactivator recruitment by the aryl hydrocarbon receptor nuclear translocator (ARNT). J Biol Chem. 2009;284:15184–15192. doi: 10.1074/jbc.M808479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Partch C, Gardner K. Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc Natl Acad Sci U S A. 2011;108:7739–7744. doi: 10.1073/pnas.1101357108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Etchegaray J-P, et al. The polycomb group protein EZH2 is required for mammalian circadian clock function. J Biol Chem. 2006;281:21209–21215. doi: 10.1074/jbc.M603722200. [DOI] [PubMed] [Google Scholar]
- 42.Akashi M, et al. A positive role for PERIOD in mammalian circadian gene expression. Cell reports. 2014;7:1056–1064. doi: 10.1016/j.celrep.2014.03.072. [DOI] [PubMed] [Google Scholar]
- 43.Chen R, et al. Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol Cell. 2009;36:417–430. doi: 10.1016/j.molcel.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nangle SN, et al. Molecular assembly of the period-cryptochrome circadian transcriptional repressor complex. eLife. 2014 doi: 10.7554/eLife.03674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Padmanabhan K, Robles M, Westerling T, Weitz C. Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science. 2012;337:599–602. doi: 10.1126/science.1221592. [DOI] [PubMed] [Google Scholar]
- 46.Etchegaray J-P, et al. Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol Cell Biol. 2009;29:3853–3866. doi: 10.1128/MCB.00338-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee H.-m., et al. The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc Natl Acad Sci U S A. 2011;108:16451–16456. doi: 10.1073/pnas.1107178108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirschi A, et al. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nature structural & molecular biology. 2010;17:1051–1057. doi: 10.1038/nsmb.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rossi F, Kringstein A, Spicher A, Guicherit O, Blau H. Transcriptional control: rheostat converted to on/off switch. Mol Cell. 2000;6:723–728. doi: 10.1016/s1097-2765(00)00070-8. [DOI] [PubMed] [Google Scholar]
- 50.Forger D. Signal processing in cellular clocks. Proc Natl Acad Sci U S A. 2011;108:4281–4285. doi: 10.1073/pnas.1004720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim J, Forger D. A mechanism for robust circadian timekeeping via stoichiometric balance. Molecular systems biology. 2012;8:630. doi: 10.1038/msb.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee Y, Chen R, Lee H.-m., Lee C. Stoichiometric relationship among clock proteins determines robustness of circadian rhythms. J Biol Chem. 2011;286:7033–7042. doi: 10.1074/jbc.M110.207217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuxreiter M, et al. Malleable machines take shape in eukaryotic transcriptional regulation. Nature chemical biology. 2008;4:728–737. doi: 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parker D, et al. Analysis of an activator:coactivator complex reveals an essential role for secondary structure in transcriptional activation. Mol Cell. 1998;2:353–359. doi: 10.1016/s1097-2765(00)80279-8. [DOI] [PubMed] [Google Scholar]
- 55.Parker D, et al. Role of secondary structure in discrimination between constitutive and inducible activators. Mol Cell Biol. 1999;19:5601–5607. doi: 10.1128/mcb.19.8.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramanathan C, Khan S, Kathale N, Xu H, Liu A. Monitoring cell-autonomous circadian clock rhythms of gene expression using luciferase bioluminescence reporters. Journal of visualized experiments : JoVE. 2012;67:e4234. doi: 10.3791/4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levin RD, et al. Circadian function in patients with advanced non-small-cell lung cancer. Br J Cancer. 2005;93:1202–8. doi: 10.1038/sj.bjc.6602859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ozturk N, Lee J, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2841–2846. doi: 10.1073/pnas.0813028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gekakis N, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 60.Partch C, Shields K, Thompson C, Selby C, Sancar A. Posttranslational regulation of the mammalian circadian clock by cryptochrome and protein phosphatase 5. Proc Natl Acad Sci U S A. 2006;103:10467–10472. doi: 10.1073/pnas.0604138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 62.Goddard TD, Keneller DG. Sparky3. University of California; San Francisco: 2006. [Google Scholar]
- 63.Johnson B. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods in molecular biology. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 64.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farmer B, et al. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nature structural biology. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 66.Ozber N, Baris I, Tatlici G, Kilinc S, Unal EB, Kavakli IH. Identification of two amino acids in the C-terminal domain of mouse CRY2 essential for PER2 interaction. BMC Mol Biol. 2010;11:69. doi: 10.1186/1471-2199-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.