

Abstract
Although poorly understood, androgen receptor (AR) signaling is sustained despite treatment of prostate cancer with antiandrogens and potentially underlies development of incurable castrate-resistant prostate cancer. However, therapies targeting the AR signaling axis eventually fail when prostate cancer progresses to the castrate-resistant stage. Stat5a/b, a candidate therapeutic target protein in prostate cancer, synergizes with AR to reciprocally enhance signaling of both proteins. In this work, we demonstrate that Stat5a/b sequesters antiandrogen-liganded (MDV3100, Bicalutamide, Flutamide) AR in prostate cancer cells and protects it against proteasomal degradation in prostate cancer. Active Stat5a/b increased nuclear levels of both unliganded and antiandrogen-liganded AR, as demonstrated in prostate cancer cell lines, xenograft tumors and clinical patient-derived prostate cancer samples. Physical interaction between Stat5a/b and AR in prostate cancer cells was mediated by the DNA-binding domain of Stat5a/b and the N-terminal domain of AR. Moreover, active Stat5a/b increased AR occupancy of the Prostate Specific Antigen promoter and AR-regulated gene expression in prostate cancer cells. Mechanistically, both Stat5a/b genetic knockdown and antiandrogen treatment induced proteasomal degradation of AR in prostate cancer cells, with combined inhibition of Stat5a/b and AR leading to maximal loss of AR protein and prostate cancer cell viability. Our results indicate that therapeutic targeting of AR in prostate cancer using antiandrogens may be substantially improved by targeting of Stat5a/b.
Keywords: Stat5a/b, androgen receptor, antiandrogen, protein stability, proteasomal degradation, prostate cancer
Introduction
Although curative therapies exist for organ-confined or locally advanced prostate cancer, treatment options are limited for metastatic castrate-resistant prostate cancer (CRPC). Progression to CRPC is defined by resistance to androgen deprivation therapy, which typically occurs less than three years after initiation of androgen deprivation therapy (1-3). The transition from androgen-dependent prostate cancer to CRPC is not fully understood. Androgen receptor (AR) signaling is known to be maintained despite low levels of circulatory androgens (1), which has been attributed to numerous mechanisms, including: 1) AR gene amplification (4); 2) AR ligand-binding domain mutations conferring ligand promiscuity (5, 6); 3) intracrine androgen biosynthesis within prostate cancer cells from adrenal steroids and cholesterol (7, 8); 4) ligand-independent, non-canonical AR transactivation by kinase signaling pathways (9, 10); 5) upregulation of constitutively active, AR splice variants which do not require ligand to support prostate cancer growth (11, 12).
Most therapeutic strategies for prostate cancer are directed at the AR signaling axis, with established antiandrogens such as Flutamide and Bicalutamide being joined by recently U.S. Food and Drug Administration-approved agents such as the androgen biosynthesis inhibitor abiraterone acetate (Zytiga®) (13, 14) and the second-generation antiandrogen MDV3100 (Enzalutamide, Xtandi®)(14). The first-generation antiandrogens function by competitive inhibition of androgen binding to AR (15, 16) and by altering coactivator and corepressor recruitment to induce formation of a transcriptionally inactive AR complex (16). The second-generation antiandrogen MDV3100 induces a distinct conformational change in AR which impairs AR nuclear localization and DNA binding (17).
Signal transducer and activator of transcription 5a/b (Stat5a/b) provides a non-AR therapeutic target protein in prostate cancer (18-26). Transcription factor Stat5a/b is composed of two highly homologous isoforms, Stat5a and Stat5b, which display >90% amino acid identity and function as both signaling proteins and nuclear transcription factors. Activation of Stat5a/b occurs by phosphorylation of a conserved C-terminal residue by an upstream kinase, most commonly Jak2 in prostate cancer, which induces Stat5a/b dimerization, nuclear translocation and target gene regulation (27, 28). Stat5a/b promotes growth of prostate cancer and tumor progression, critically sustaining viability of prostate cancer cells in vitro (18, 21) and xenograft tumor growth in vivo (19). Stat5a/b is active in 95% of clinical CRPCs (29), with the Stat5a/b gene locus amplified in 29% of distant CRPC metastases (30). Additionally, high active Stat5a/b expression predicts early disease recurrence (24, 26) and prostate cancer-specific death (26), and promotes metastatic behavior of prostate cancer cells in vitro and in vivo (22), suggesting Stat5a/b involvement in clinical progression of prostate cancer. In further support of this concept, pharmacological targeting of Stat5a/b signaling blocked growth of not only primary (19, 31) but also CRPC xenograft tumors in nude mice (31).
Stat5a/b regulation of prostate cancer cell viability involves both AR-dependent and AR-independent mechanisms (18, 19, 21, 25, 29). We have shown previously that Stat5a/b and AR functionally synergize in prostate cancer to enhance nuclear localization and transcriptional activity of both proteins (29). At the same time, it is well established that AR activity in prostate cancer is regulated not only at transcriptional level but also by translational and post-translational mechanisms (32). Recently,Stat5a/b was proposed to be involved in up-regulation of AR levels in prostate cancer cells when AR is liganded by androgens (25). Here, we sought to investigate mechanistically if Stat5a/b regulates AR when liganded by antiandrogens, or when AR is unliganded during androgen deprivation of prostate cancer cells. In this work, we show for the first time that Stat5a/b protects both unliganded and antiandrogen-liganded AR from proteasomal degradation in prostate cancer. Antiandrogens induce proteasomal degradation of AR, which can be accelerated by disruption of Stat5a/b activity. Maximal loss of AR protein and inhibition of AR-driven prostate cancer cell growth is achieved through a combination of antiandrogen treatment and Stat5a/b inhibition. Our results highlight a novel means of exploiting AR proteasomal degradation by targeting of Stat5a/b to potentially improve efficacy of antiandrogens in prostate cancer.
Materials and Methods
Cell lines and reagents
Human prostate cancer cell lines LNCaP, CWR22Rv1 and PC-3 (ATCC, Manassas, VA) were cultured in RPMI 1640 (Mediatech, Herndon, VA) containing 10% fetal bovine serum (FBS; Quality Biological, Gaithersburg, MD) and penicillin/streptomycin (Mediatech, Inc., 50 IU/ml and 50 μg/ml, respectively). LAPC-4 cell line (Provided by Dr. Charles Sawyers in 2012, Sloan-Kettering Memorial Cancer Center, NYC) was maintained in IMDM (Mediatech) supplemented with 1% penicillin/streptomycin and L-glutamine and 10% FBS. LNCaP and LAPC-4 cells were cultured in the presence of 0.5 nM dihydrotestosterone (DHT; Sigma-Aldrich, St. Louis, MO). Human prolactin (Prl) was obtained from the National Hormone and Peptide Program (Harbor-UCLA Medical Center, Torrance, CA), Flutamide and Bicalutamide from Selleck Chemicals (Houston, TX), MDV3100 (enzalutamide)from MedChem Express (Princeton, NJ), and IST5-002 was synthesized by Fox Chase Chemical Diversity Center (Doylestown, PA). All cell lines included in this study have been authenticated on a regular basis in the users' laboratory. The testing has been conducted by observation of characteristic cell morphology, androgen-responsiveness and the expression of cell lines specific markers such as PSA, androgen receptor, Stat3/Stat5, Erk1/2Protein. CWR22Rv1 cells were obtained in 2005 from Dr. Thomas Pretlow (Casewestern Reserve Univ.) and LNCaP and DU145 cells in 2009 from ATCC.
Protein solubilization and immunoblotting
Cell pellets were solubilized in lysis buffer [10 mM Tris-HCl (pH 7.6), 5 mM EDTA, 50 mM sodium chloride, 30 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 1 μg/ml pepstatin A and 2 μg/ml leupeptin] (19, 21, 22, 29). Protein concentrations of were determined by simplified Bradford method (Bio-Rad). The primary antibodies used were: anti-Stat5a/b mAb (1:1000; BD Biosciences), anti-androgen receptor (AR) mAb (1:1000, BioGenex Laboratories, Fremont, CA), anti-prostate-specific antigen (PSA) pAb (1:1000; Dako, Glostrup, Denmark), anti-β-actin pAb (1:2000; Sigma-Aldrich).
Protein coimmunoprecipitation
LNCaP cells were infected with adenovirus expression PrlR (MOI:4), serum-starved overnight in 0.5% FBS, and stimulated with or without 10 nM Prl and 1 nM DHT, 10 μM Bicalutamide, 10 μM Flutamide, or 10 μM MDV3100 for 1 h.Stat5a and Stat5b were immunoprecipitated from whole cell lysates with anti-Stat5a or anti-Stat5b (4 μl/ml; Millipore) vs. IgG from normal rabbit serum (4 μl/ml; Sigma). Antibodies were captured by incubation for 60 min with protein A-sepharose beads (Amersham Pharmacia Biotech). Primary antibodies used for immunoblotting were anti–AR mAb (1:1000, Biogenex Laboratories) and anti-Stat5a/b mAb (1:1000; BD Biosciences). The cell lysates were separated on 4-12% SDS-PAGE (Life Technologies) and transferred electrophoretically to polyvinylidene fluoride membrane (Millipore). For immunoblotting, blocking buffer used was Tris-Buffered Saline and Tween 20 (TBST, 0.15 M NaCl; 0.1% Tween 20; 50 mM Tris, pH 8.0) with 3% Bovine Serum Albumin (BSA). The immunoreaction was detected by horseradish peroxidase-conjugated goat anti-mouse secondary antibodies (1:2000; BD Biosciences).
Double immunofluoresence cytochemistry of Stat5a/b and AR
PC-3 cells were infected with AdWTStat5a, AdDNStat5a, AdStat5a(Y694F), AdWTPrlR, and AdWTAR each at MOI 4, as indicated (Fig. 1A, B, D). LNCaP cells were infected with AdPrlR at MOI 4 (Fig. 1C). The cells were serum-starved overnight before stimulation with 10 nM Prl (30 min) and/or 1 nM DHT (60 min). In Fig. 1D, PC-3 cells were treated with 1 nM DHT, 10 μM Flutamide, 10 μM Bicalutamide or 10 μM MDV3100 for 1 h prior to stimulation with 10 nM Prl (30 min). Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 (Sigma) and incubated with anti-Stat5 pAb (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) and anti-AR mAb (1:200; Santa Cruz) followed by goat anti-rabbit fluorescein IgG (1:200; Vector Laboratories, Burlingame, CA) and horse anti-mouse Texas Red IgG (1:200; Vector Laboratories), respectively. Immunofluorescence staining was detected by a Zeiss LSM 510 laser scanning microscope with an Apochromat X63/1.4 oil immersion objective (Zeiss, Thornwood, NY).
Figure 1. Activation of Stat5a/b by tyrosine phosphorylation is required for Stat5a/b-induced nuclear translocation of unliganded and antiandrogen-liganded AR in prostate cancer cells.
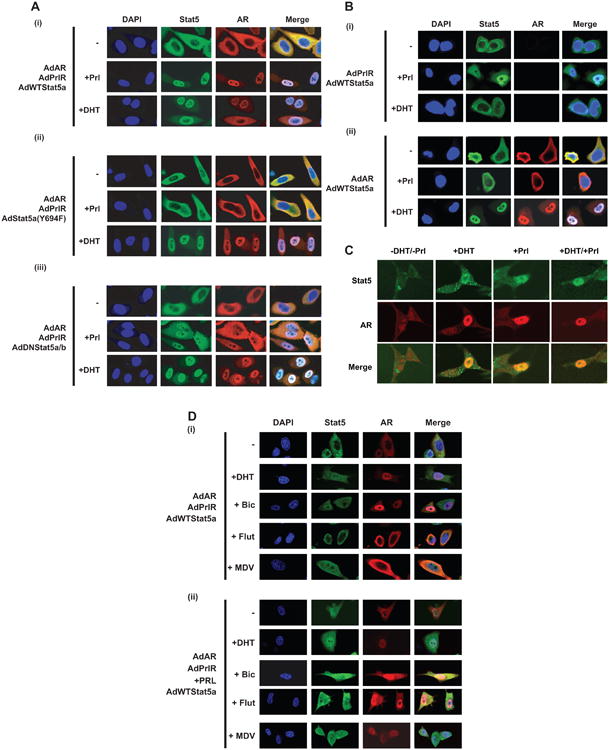
(A) (i) Prolactin (Prl) stimulation increases nuclear levels of wild-type (WT) Stat5a/b with a concurrent increase in nuclear levels of AR in PC-3 cells. Increased nuclear levels of Stat5a/b and AR are abrogated in the presence of (ii) phosphorylation-dead Stat5a(Y694F) or (iii) dominant-negative (DN) Stat5a/b. PC-3 cells were infected with AdAR, AdPrlR and AdWTStat5a, AdDNStat5a/b or AdStat5a(Y694F) (each at MOI = 4) and serum-starved (0% FBS) for 12 h before stimulation with 10 nM Prl for 30 min and/or 1 nM dihydrotestosterone (DHT) for 60 min. Cells were immunostained for 4′,6-diamidino-2-phenylindole (DAPI), Stat5a/b and AR, where indicated. Merged imaging indicates overlay of Stat5a/b and AR immunostaining. (B) Stat5a/b-induced increase in nuclear levels of AR is dependent on (i) AR expression and (ii) Stat5a/b activation via expression of upstream prolactin receptor (PrlR) in PC-3 cells. PC-3 cells were treated as described in (A). Immunostaining for DAPI, Stat5a/b, AR and merged imaging of Stat5a/b and AR is depicted. (C) Prl and/or DHT stimulation induces a concurrent increase in nuclear levels of endogenous Stat5a/b and AR in LNCaP cells. LNCaP cells were infected with AdPrlR and serum-starved for 12 h prior to stimulation with 10 nM Prl for 30 min and/or 1 nM DHT for 60 min. Endogenous Stat5a/b, AR and merged imaging of Stat5a/b and AR are shown by immunostaining. (D) Active Stat5a/b increases nuclear levels of antiandrogen-liganded AR in PC-3 cells. PC-3 cells were infected with AdAR, AdPrlR and AdWTStat5a, serum-starved for 12 h, and pretreated with 1 nM DHT, 10 μM Bicalutamide (Bic), 10 μM Flutamide (Flu) or 10 μM MDV3100 (MDV) for 1 h before stimulation with 10 nM Prl for 30 min, where indicated. Immunostaining depicts DAPI, Stat5a/b, AR and merged imaging of Stat5a/b and AR.
Quantitative real-time polymerase chain reaction (qRT-PCR)
RNA was extracted from LNCaP or LAPC-4 cells using RNeasy Mini Kit (Qiagen) and reverse-transcribed to cDNA using SuperScript III First Strand Synthesis System for RT-PCR (Life Technologies, Carlsbad, CA), according to manufacturers' protocols. Primer sequences used were as follows: PSA (F: 5′-CATCAGGAACAAAAGCGTGA-3′ and R: 5′-AGCTGTGGCTGACCTGAAAT-3′), TMPRSS2 (F: 5′GTGATGGTAATTCACGGACTGG-3′ and R: 5′-CAGCCCCATTGTTTTCTTGTA-3′), AR (F: 5′-AACAGAAGTACCTGTGCGCC-3′ and R: 5′-TTCAGATTACCAAGTTTCTTCAGC-3′), and GAPDH (F: 5′-TCAAGAAGGTGGTGAAGCAG-3′ and R: 5′-CTTACTCCTTGGAGGCAATG-3′).
Chromatin immunoprecipitation (ChIP) assay
LNCaP cells were crosslinked with 1% formaldehyde, quenched with 125 mM glycine and solubilized in lysis buffer (10mM HEPES 7.5, 10mM KCl, 1.5mM MgCl2, 0.5mM DTT, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 1 μg/ml pepstatin A and 2 μg/ml leupeptin). Nuclei were pelleted from whole cell lysates and solubilized in nuclear lysis buffer (50mM Tris pH 8.0, 10mM EDTA, 1% SDS, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 1 μg/ml pepstatin A and 2 μg/ml leupeptin). Nuclear lysates were sonicated to shear DNA, yielding fragments of approximately 500 bp. Extracts were pre-cleared using protein A sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ) and diluted 5-fold with RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40, 0.5% deoxycholate, 0.1%SDS) prior to immunoprecipitation with anti-AR pAb (1.2 μg/ml, Santa Cruz). Immunoprecipitated complexes were washed 2× with Super RIPA buffer (275 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) and diluted with 100 μl elution buffer (0.1 M NaHCO3, 1% SDS). Crosslinking was reversed by heating at 65°C overnight in 5 M NaCl. DNA was extracted using QIAquick PCR purification kit (Qiagen, Venlo, Netherlands) according to the manufacturer's protocol and subjected to PCR amplification. Primers used for PSA promoter were forward: 5′-TCTGCCTTTGTCCCCTAGAT-3′ and reverse: 5′-AACCTTCATTCCCCAGGACT-3′.
Generation of Stat5a and AR deletion constructs
Full-length Stat5a and AR were amplified by polymerase chain reaction (PCR) and subcloned to pCMV-3FLAG vector (Stratagene, La Jolla, CA) with EcoRI and SalI sites. Stat5a deletion constructs were constructed by PCR-directed mutagenesis, with pCMV-3FLAG-Stat5a (full-length Stat5a) used as a template. PCR primers for each Stat5a deletion construct contain different Stat5a truncated boundary sequences. PCR products were digested with EcoRI/SalI and ligated with pCMV-3FLAG vector to form Stat5a deletion constructs and subjected to sequencing analysis for verification. Full-length AR was amplified by PCR and subcloned to pCMV-3MYC vector (Stratagene) with BamHI and XhoI sites. AR deletion constructs were then generated and verified as described for Stat5a deletion constructs.
Coimmunoprecipitation of FLAG-Stat5a and MYC-AR deletion constructs
PC-3 cells were co-transfected with plasmid pCMV-3FLAG-Stat5a deletion constructs, pCMV-3MYC-AR (full-length AR) and pPrlR (Fig. 3D) or pCMV-3MYC-AR deletion constructs, pCMV-3FLAG-Stat5a (full-length Stat5a) and pPrlR (Fig. 3E) using FuGENE6 (Promega, Madison, WI). Cells were serum-starved, stimulated with 10 nM Prl and 1 nM DHT for 16 hours. The antibodies used for the co-immunoprecipitations were anti-AR pAb (200 μg/mL; Santa Cruz), 25 μl anti-FLAG pAb (1 μg/mL; Stratagene) or rabbit IgG polyclonal (12 mg/mL; Sigma) followed by immunoblotting with anti-FLAG mAb (1:1000; Stratagene) or anti-MYC mAb (1:000; Sigma).
Figure 3. Active Stat5a/b up-regulates expression of AR-regulated genes, increases AR occupancy of the PSA promoter and physically interacts with antiandrogen-liganded AR in prostate cancer cells.
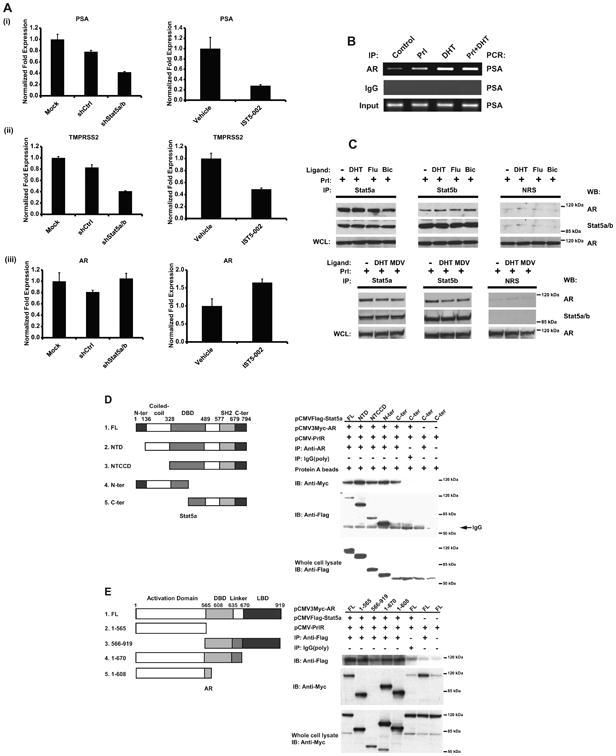
(A) Disruption of Stat5a/b activity through genetic knockdown (left panels) or IST5-002 treatment (right panels) down-regulates mRNA levels of AR target genes (i) PSA and (ii) TMPRSS2, but does not affect mRNA levels of AR (iii). LNCaP cells were transduced with lentiviral shRNA targeting Stat5a/b (shStat5a/b) or scrambled control sequence (shCtrl), or treated with 25 μM IST5-002 or vehicle for 48 h, followed by quantification of PSA, TMPRSS2 and AR mRNA levels by quantitative real-time PCR. (B) Prl and/or DHT stimulation induces AR co-occupancy of the PSA promoter. LNCaP cells were serum-starved for 20 h, stimulated by 10 nM Prl and/or 1 nM DHT, crosslinked, solubilized and nuclei were extracted. Following nuclear lysis, DNA was sheared by sonication and AR was immunoprecipitated with anti-AR pAb. DNA was amplified by PCR using primers targeting the PSA promoter. (C) Stat5a and Stat5b physically interact with AR liganded by androgens or antiandrogens. LNCaP cells were treated with or without 1 nM DHT, 10 μM Bicalutamide (Bic), 10 μM Flutamide (Flu) or 10 μM MDV3100 (MDV) for 1 h in the presence of 10 nM Prl. Stat5a and Stat5b were immunoprecipitated with polyclonal antibodies (pAbs) against Stat5a or Stat5b vs. normal rabbit serum (NRS) as control. Immunoprecipitated complexes were immunoblotted with monoclonal antibodies (mAbs) against AR and Stat5a/b. (D) The DNA-binding domain of Stat5a/b physically interacts with AR, as determined by coimmunoprecipitation with a series of Stat5a deletion constructs. PC-3 cells were cotransfected with pCMV-3FLAG-Stat5a deletion constructs, pCMV-3MYC-AR and pPrlR, serum-starved for 20 h and stimulated with 10 nM Prl and 1 nM DHT for 16 h. MYC-AR was immunoprecipitated with anti-AR pAb vs. polyclonal rabbit IgG as control and immunoblotted with anti-FLAG M2 pAb and anti-MYC mAb. (E) The N-terminal domain of AR physically interacts with Stata/b5, as determined by coimmunoprecipitation with a series of AR deletion constructs. PC-3 cells were cotransfected with pCMV-3MYC-AR deletion constructs, pCMV-3FLAG Stat5a and pPrlR, serum-starved for 20 h and stimulated with 10 nM Prl and 1 nM DHT for 16 h. FLAG-Stat5a was immunoprecipitated with anti-FLAG M2 pAb vs. polyclonal rabbit IgG as control and immunoblotted with anti-MYC mAb and anti-FLAG M2 pAb.
Generation of adenoviruses for gene delivery of wild-type (WT), dominant-negative (DN) and phosphorylation-dead (Y694F) Stat5a and WT androgen receptor (AR)
pcDNA-CMV-WT Stat5a, pcDNA-CMV-DNStat5a, pcDNA-CMV-Stat5a(Y694F) and pAR constructs were cloned to adenoviral vector using BD Adeno-X Expression System 2 (BD Biosciences Clontech, Palo Alto, CA) per the manufacturer's protocol. Purified recombinant adenoviruses were linearized by PacI digestion and transfected to QBI-293A cells (Qbiogene, Carlsbad, CA) to produce infectious adenoviruses. AdWTStat5a, AdDNStat5, AdStat5a(Y694F) and AdWTAR viral stocks were expanded in large-scale cultures, purified using double cesium chloride gradient centrifugation and titrated by a standard plaque assay method in QBI-293A cells, as per the manufacturer's instructions. AdPrlR was a gift from Dr. Hallgeir Rui (Thomas Jefferson University).
Generation of Stat5a/b antisense oligodeoxynucleotides (ASO) and transfection of prostate cancer cells
Stat5a/b ASO (5′-GGGCCTGGTCCATGTACGTG-3′; shared sequence within both human Stat5a and Stat5b transcripts; bp 2153-2173 in open reading frame) were synthesized by Isis Pharmaceuticals (Carlsbad, CA), using a phosphorothioate backbone with 2′-O-methoxyethyl modification of five terminal nucleotides (underlined) to increase stability (ISIS 130826). As control, mismatch (MM) oligodeoxynucleotides for the same chemistry were synthesized as a mixture of all four nucleotide bases. LNCaP cells were transfected using jetPEI reagent (QBiogene, Irvine, CA) with Stat5a/b ASO or MM control (each at 900 pmol) for 48 hours.
shRNA constructs and lentivirus production
The RNAi Consortium (TRC) pLKO.1 lentiviral vectors containing shRNA targeting AR (shAR), Stat5a (shStat5a), Stat5b (shStat5b) or scrambled control sequence (shCtrl) were purchased from Thermo Fisher Scientific (Waltham, MA). Second-generation VSV-G pseudotyped high-titer lentiviruses were generated by transient co-transfection of 293FT cells, kindly provided by Dr. Hallgeir Rui (Thomas Jefferson University), with a three plasmid combination as follows: one T75 cell culture flask containing 8×106 293FT cells was transfected with 9 μg pLKO.1 lentiviral vector containing shRNA of interest, 10 μg pHR'8.2ΔR packaging plasmid and 1 μg pCMV-VSV-G envelope plasmid using lipofectamine 2000 (Life Technologies, Carlsbad, CA) in Opti-MEM (Life Technologies).
Human prostate cancer xenograft studies
Castrated male athymic nude mice (Taconic, Germantown, NY), cared for according to institutional guidelines of Thomas Jefferson University, were implanted with sustained-release dihydrotestosterone (DHT) pellets (60-day release, 1 pellet/mouse, Innovative Research of America, FL) 3 days prior to inoculation of CWR22Rv1 cells. Briefly, 1.5×107 CWR22Rv1 cells were combined with 0.2 ml of Matrigel (BD Biosciences) and implanted subcutaneously (s.c.) into flanks of nude mice (one tumor/mouse) as previously described (19, 21, 31). After tumors reached approximately 100 mm3 in size (1 week), mice were randomized into four groups (n = 10 per group) and treated daily for 10 days by intraperitoneal (i.p.) injections with 0.2 ml of IST5-002 dissolved in 0.3% hydroxypropyl cellulose (HPC, Sigma-Aldrich) at 25 mg/kg or 50 mg/kg body weight, or 0.3% HPC solution alone (vehicle), or received no treatment. Tumor sizes were measured using calipers three times per week and tumor volumes were calculated using the following formula: (3.14 × length × width × depth)/6. When tumors reached 15-20 mm in diameter in the control groups, mice were sacrificed and tumor tissues were harvested.
Clinical prostate cancer sample collection, subjects and clinical information
Clinical samples, consisting of formalin-fixed, paraffin-embedded material from 132 radical prostatectomies performed as primary therapy for clinically localized prostate adenocarcinoma, were collected from the pathology archives at Turku University Central Hospital (Turku, Finland) during 1986-1997. None of the patients received adjuvant therapy before or right after surgery. Clinical post-prostatectomy follow-up data and clinical information on pre- and post-operative conditions were collected retrospectively from patient files in the hospital archives and the information concerning the cause and date of death was obtained from the National Center for Statistics (Statistics Finland). The study protocol was approved by the ethical committee of the University of Turku (Turku, Finland) and the National Data Protection Ombudsman (Helsinki, Finland) was notified about the collection of subject information. The immunohistochemical analyses of the de-identified archival tissues were granted an exemption from a full Institutional Review Board by Thomas Jefferson University (Philadelphia, PA).
The post-prostatectomy follow-up was conducted according to a clinical protocol, which included digital rectal examination and serum PSA measurements. Patient demographics and clinicopathological data of patients are presented in Supplementary Table 1. The median post-surgery follow-up time was 77 months (6.35 years) and the median age at surgery was 63 years (range = 46-74 years). The threshold for post-operative biochemical recurrence was serum PSA level of 2.0 μg/L prior to year 1995 and 1.0 μg/L thereafter.
Tissue microarray construction
Prior to preparation of the actual tissue microarray (TMA), all hematoxylin & eosin-stained slides from prostatectomy specimens and metastases were re-evaluated to identify representative areas of benign, low-grade prostatic intraepithelial neoplasia (LGPIN), high-grade PIN (HGPIN) and carcinoma lesions. When possible, multiple cores per patient (range 1-20, mean 4.55) were collected. Carcinoma cores in TMAs were graded only according to the predominant Gleason pattern because of the small amount of tissue in each transferred core. TMAs with 55 separate cores in each block were constructed with a manual tissue arrayer (Beecher Instruments, Sun Prairie, WI).
AR proteasomal degradation analysis
Cells were treated with vehicle (dimethylsulfoxide, DMSO; Sigma), 1 nM DHT, 10 μM Flutamide, Bicalutamide or MDV3100 and/or transiently transduced with lentiviral shCtrl or shStat5a/b, where indicated (Figs. 4A, 4B, 5A, 5C, 5D). Cells were treated with 10 μM MG132 (Calbiochem, Billerica, MA) 6 hours before harvest and immunoblotted as previously described. In Fig. 4Biii, cells were also treated with 10 μg/mL cyclohexamide (CHX; Calbiochem) 6 hours before harvest.
Figure 4. Stat5a/b genetic knockdown decreases AR protein levels, which is partially rescued by proteasome inhibition in prostate cancer cells.
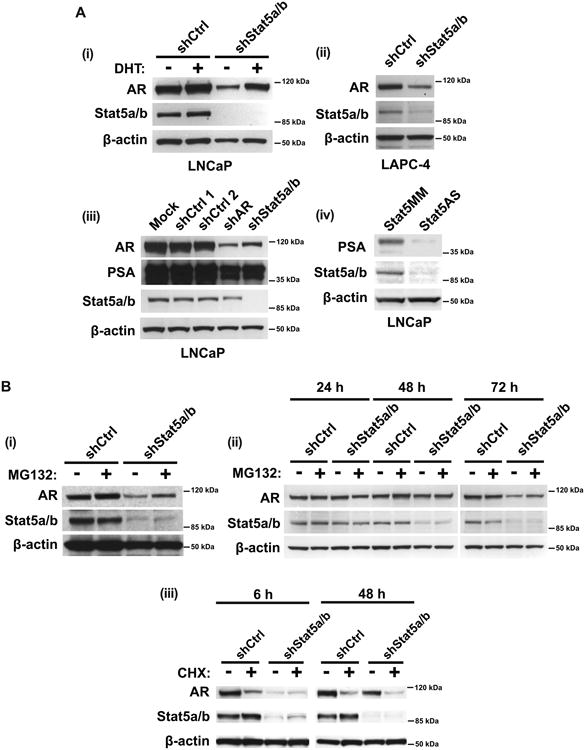
(A) Genetic knockdown of Stat5a/b decreases (i, ii) AR and (iii, iv) PSA protein levels in LNCaP and LAPC-4 cells. LNCaP and LAPC-4 cells were transduced with lentiviral shStat5a/b, shAR or shCtrl for 72 h, followed by immunoblotting of cell lysates with anti-AR mAb, anti-Stat5a/b mAb, anti-PSA pAb or anti-β-actin pAb. In panel (i), LNCaP cells were treated with 1 nM DHT 24 h prior to harvest and immunoblotting. In panel (iv), LNCaP cells were transfected with antisense oligodeoxynucleotides (ASO) targeting Stat5 or mismatched (MM) sequence as control for 72 h. (B) (i, ii) Stat5a/b knockdown-mediated decrease of AR protein levels occurs through AR proteasomal degradation and (iii) is not attributable to increased AR protein synthesis. LNCaP cells were transduced with lentiviral shStat5a/b or shCtrl for 72 h and treated with 10 μM MG132 (proteasomal inhibitor) 6 h before harvest, followed by immunoblotting of cell lysates with anti-AR mAb, anti-Stat5a/b mAb, anti-PSA pAb and anti-β-actin pAb. In panel (ii), cells were harvested at 24, 48 and 72 h after lentiviral transduction. In panel (iii), cells were treated with 10 μg/ml cyclohexamide (protein synthesis inhibitor) 6 h or 48 h before harvest.
Figure 5. Stat5a/b protects both androgen- and antiandrogen-liganded AR from proteasomal degradation in prostate cancer cells.
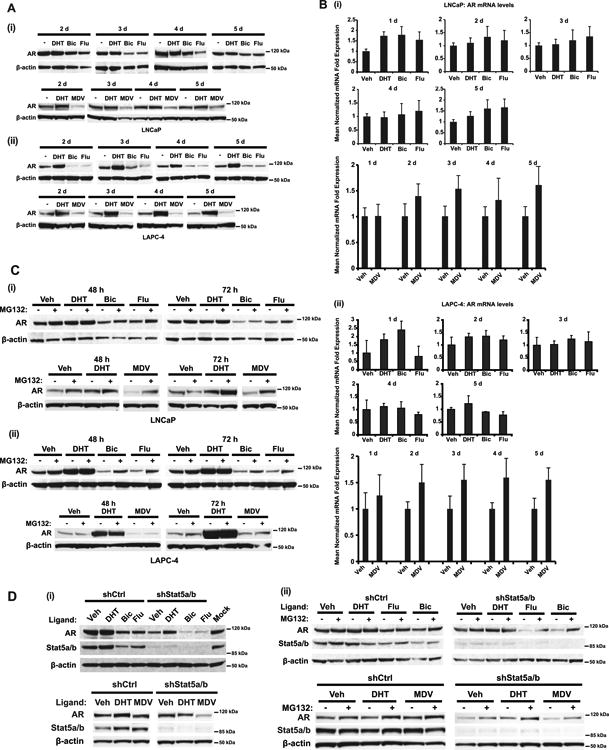
(A) Antiandrogen treatment decreases AR protein levels in (i) LNCaP and (ii) LAPC-4 cells. LNCaP and LAPC-4 cells were treated with 1 nM DHT, 10 μM Flutamide (Flu), 10 μM Bicalutamide (Bic) or 10 μM MDV3100 (MDV) for 2, 3, 4 or 5 d, followed by immunoblotting of cell lysates with anti-AR mAb and anti-β-actin pAb. (B) Antiandrogen treatment does not affect AR mRNA levels in (i) LNCaP and (ii) LAPC-4 cells. LNCaP and LAPC-4 cells were treated as described in (A) for 1, 2, 3, 4 or 5 d, followed by quantification of AR mRNA levels by qRT-PCR. (C) Antiandrogen-mediated decrease of AR protein levels occurs through proteasomal degradation in (i) LNCaP and (ii) LAPC-4 cells. LNCaP and LAPC-4 cells were treated daily with 1 nM DHT, 10 μM Flutamide, 10 μM Bicalutamide or 10 μM MDV3100 for 48 and 72 h. Before harvest (6 h), indicated samples were treated with 10 μM MG132 and the cell lysates were immunoblotted with anti-AR mAb and anti-β-actin pAb. (D) (i) Stat5a/b genetic knockdown accelerates loss of AR protein induced by antiandrogens. LNCaP cells were transduced with lentiviral shStat5a/b or shCtrl for 72 h in the presence of 1 nM DHT, 10 μM Bicalutamide, 10 μM Flutamide or 10 μM MDV3100, followed by immunoblotting of cell lysates with anti-AR mAb, anti-Stat5a/b mAb and anti-β-actin pAb. (ii) Maximal loss of AR through proteasomal degradation is achieved with combined Stat5a/b genetic knockdown and antiandrogen treatment. LNCaP cells were treated as described in (i), with indicated samples treated with 10 μM MG132 for 6 h before harvest.
Cell viability assay
Cell viability was analyzed by the CellTiter 96 Aqueous Assay Kit (Promega, Madison, WI) according to the manufacturer's protocol.
Immunohistochemical staining of paraffin-embedded tissue sections and tissue microarrays and scoring of nuclear Stat5a/b and AR levels
Immunohistochemical staining of paraffin-embedded tissue sections and tissue microarrays were performed (23, 24, 26, 33-35) and scored (33, 36) as described previously and in Supplementary Materials and Methods.
Statistical analyses
In experiments with two groups, groups were compared using two-sample t-tests with unequal variances.In experiments with more than two groups, groups were compared using ANOVA allowing for unequal variances across groups.When the overall ANOVA test of group differences was significant, pairwise comparisons were performed.For Fig. 3, p-values for pairwise comparisons were adjusted using the Bonferroni method.For Fig. 5, pairwise comparisons were made of each group with control, p-values were adjusted using the Dunnett-Hsu method.All analyses were performed using SAS 9.4 and SAS/STAT 13.2.In Fig. 6C, two-way anova with Bonferroni adjustment of the p values was used to analyze significance of IST5-002 combined with MDV3100 compared to MD3100 alone. In Fig. 2B, mixed effects regression was used to test whether AR score differed by level of Stat5a/b.Stat5a/b level was included as a fixed categorical effect and a random intercept term was included to account for correlation among samples from the same subject.
Figure 6. Stat5a/b inhibition enhances efficacy of antiandrogens in suppressing growth of prostate cancer cells.
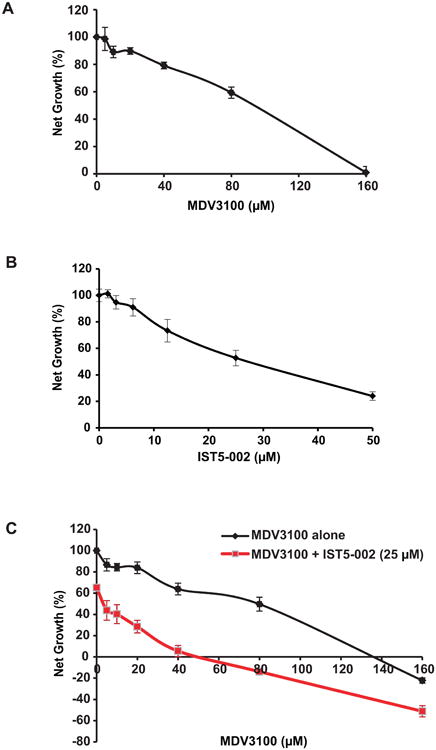
Combined inhibition of Stat5a/b and AR by IST5-002 and antiandrogens, respectively, is more effective than antiandrogens alone in suppressing growth of prostate cancer cells. LNCaP cells were treated individually with increasing doses of (A) MDV3100 or (B) IST5-002 for 72 h, followed by MTS assay to determine IC50 values for each drug. (C) LNCaP cells were treated with increasing doses of MDV3100 alone or in combination with a fixed dose of IST5-002 (25 μM, IC50 value) for 72 h, followed by MTS assay.
Figure 2. High active Stat5a/b and AR protein levels are positively associated in prostate cancer xenograft tumors and clinical prostate cancer specimens.
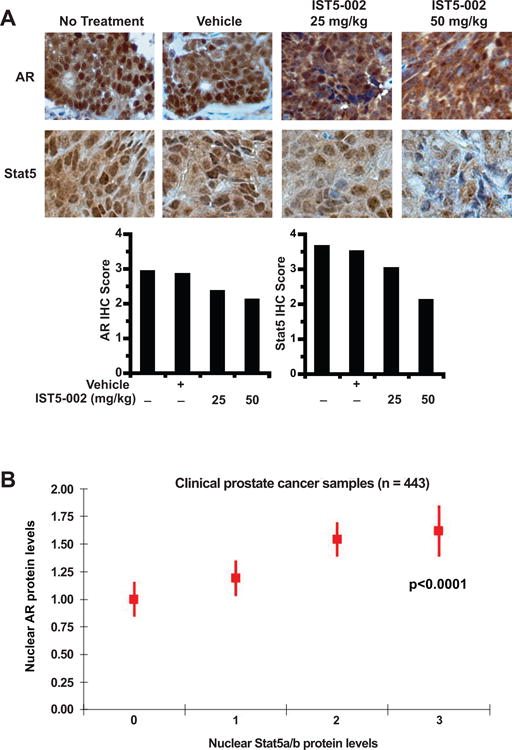
(A) Inhibition of Stat5a/b using Stat5a/b dimerization inhibitor IST5-002 decreases nuclear levels of AR and Stat5a/b in CWR22Rv1 human prostate cancer xenograft tumors in a dose-dependent manner. Castrated male athymic nude mice bearing CWR22Rv1 xenograft tumors were treated with 25 mg/kg or 50 mg/kg IST5-002, vehicle or received no treatment (n = 10 per group) daily for 10 days. On day 10, mice were sacrificed and tumor sections analyzed for nuclear AR and Stat5a/b content by immunohistochemistry (IHC) staining. Representative immunostainings from each treatment group are shown. (B) High levels of nuclear Stat5a/b were significantly associated (p<0.0001) with high levels of nuclear AR in a cohort of clinical PCs (n = 443). Association between nuclear AR and Stat5a/b is presented, along with de-identified clinical information (Supplementary Table 1).
Results
Active Stat5a/b increases nuclear levels of unliganded and antiandrogen-liganded AR in prostate cancer cells
Stat5a/b enhances transcriptional activity and nuclear localization of AR in prostate cancer cells, possibly potentiating AR signaling in prostate cancer cells during androgen deprivation (29). Since pharmacological inhibitors of Stat5a/b activation are currently in clinical development, we wanted to determine mechanistically if activation of the Stat5a/b signaling pathway is required for induction of nuclear levels of AR in the absence of androgens in prostate cancer cells. Serum-starved PC-3 cells expressing prolactin receptor and AR were infected with adenovirus expressing either wild-type Stat5a (AdWTStat5), phosphorylation-dead Stat5a (AdStat5aY694F) or dominant-negative Stat5a/b (AdDNStat5) which lacks the transactivation domain. The cells were stimulated with 10 nM human prolactin to activate Stat5a/b. As we have demonstrated previously (29), Stat5a/b activation resulted in increased levels of both Stat5a/b and AR proteins within the nuclear compartment, as detected by increased immunofluorescence of Stat5a/b and AR in the nucleus (Fig. 1A,i). In contrast, adenoviral delivery of either phosphorylation-dead Stat5a (AdStat5aY694F) or dominant-negative Stat5a/b (AdDNStat5) in place of wild-type Stat5, followed by prolactin stimulation, failed to induce comparable nuclear localization of Stat5a/b or AR, shown by equivalent distribution of Stat5a/b and AR immunofluorescence between cytosol and nucleus (Fig. 1A,ii and iii). Stimulation of PC-3 cells with 1 nM dihydrotestosterone (DHT), however, induced nuclear localization of AR with concurrent increased nuclear localization of wild-type Stat5, phosphorylation-dead Stat5a(Y694F) and dominant-negative Stat5a/b (Fig. 1A). These data indicate that transcriptionally active, phosphorylation-competent Stat5a/b is required for nuclear localization of unliganded AR in prostate cancer cells.
In a parallel set of experiments, serum-starved PC-3 cells were infected with adenovirus expressing wild-type Stat5a and prolactin receptor but not AR (Fig. 1B,i) or wild-type Stat5a and AR but not prolactin receptor (Fig. 1B,ii). While direct Stat5a/b activation via prolactin stimulation still resulted in nuclear localization of Stat5a/b, in the absence of AR protein, the nuclear localization of Stat5a/b following DHT stimulation was not detectable (Fig. 1B,i). Additionally, the absence of prolactin receptor to transduce signaling to Stat5a/b after prolactin stimulation abrogated not only nuclear localization of Stat5a/b, but of AR as well. However, DHT stimulation in the absence of prolactin receptor was still capable of increasing nuclear protein levels of not only AR, but also Stat5a/b (Fig. 1B,ii). A time course analysis of LNCaP cells, which endogenously express both AR and Stat5a/b, confirmed that active Stat5a/b induces localization of AR in the nuclei of prostate cancer cells (Fig. 1C). Peak nuclear localization of both Stat5a/b and AR concurrently occurred at 30 minutes after DHT stimulation (Supplementary Fig. 1) and at 10 minutes after prolactin stimulation (Supplementary Fig. 2). Collectively, these data indicate that active, tyrosine-phosphorylated Stat5a/b promotes nuclear localization of unliganded AR in prostate cancer cells.
To evaluate if active Stat5 is capable of inducing nuclear localization of AR liganded by antiandrogens in prostate cancer cells, PC-3 cells expressing Stat5a/b, prolactin receptor and AR were treated with DHT, Flutamide, Bicalutamide or MDV3100 followed by prolactin stimulation and immunocytochemistry of Stat5a/b and AR. DHT stimulation resulted in increased nuclear levels of both Stat5a/b and AR in the absence of prolactin (Fig. 1D). Intriguingly, prolactin-stimulated activation of Stat5a/b induced nuclear localization of not only AR liganded by DHT, but AR liganded by Flutamide, Bicalutamide or MDV3100 as well (Fig. 1D). These results suggest that active Stat5a/b can circumvent antiandrogen action in leading to increased nuclear levels of AR liganded by Flutamide, Bicalutamide or MDV3100.
To investigate if high levels of nuclear Stat5a/b and AR proteins are associated in prostate cancer in vivo, we first analyzed CWR22Rv1 xenograft tumors grown in nude mice treated with a small-molecule Stat5a/b dimerization inhibitor, IST5-002, for nuclear Stat5a/b and AR content. As expected, IST5-002 decreased nuclear Stat5a/b protein levels in a dose-dependent manner, as shown by Stat5a/b immunostaining of tumor sections accompanied by quantification (Fig. 2A). At the same time, AR immunostaining and quantification revealed that IST5-002 decreased nuclear AR protein levels in a dose-dependent manner as well (Fig. 2A). Next, we analyzed a cohort of 443 clinical prostate cancer samples (Supplementary Table 1) assembled in tissue microarray for nuclear Stat5a/b and AR content by immunostaining with quantitative scoring. A positive association was demonstrated between the levels of nuclear Stat5a/b and AR proteins, with mixed effects regression indicating that high nuclear Stat5a/b protein levels were significantly associated with high AR protein levels (p<0.0001) (Fig. 2B). In summary, the reciprocal interaction of Stat5a/b and AR characterized in prostate cell lines extends to human prostate cancer xenograft tumors grown in vivo and patient-derived clinical prostate cancer samples.
Active Stat5a/b enhances AR-regulated gene expression in prostate cancer cells and physically interacts with antiandrogen-liganded AR
Given that active Stat5a/b induced nuclear localization of antiandrogen-liganded AR in prostate cancer cells, we next investigated if active Stat5a/b modulates AR-regulated gene expression in prostate cancer. We disrupted Stat5a/b activity in LNCaP cells by genetic knockdown using lentiviral delivery of shRNA targeting Stat5a/b (shStat5a/b). Quantitative real-time PCR revealed that mRNA levels of prostate specific antigen (PSA) (p=0.044) and transmembrane protease, serine 2 (TMPRSS2) (p=0.009), two well-known representatives of AR-regulated genes (37, 38), were decreased by 50% or more after 48 hours compared to controls (Fig. 3A,i and ii). In the next set of experiments, Stat5a/b signaling was blocked by treatment of the cells with Stat5a/b dimerization inhibitor IST5-002 (25 μM). Pharmacological inhibition of Stat5a/b decreased both PSA (p=0.029) and TMPRSS2 (p=0.078) mRNA expression to the same extent as genetic knockdown of Stat5a/b (Fig. 3A,i and ii). This decrease in the expression of several AR target genes was not due to decreased AR mRNA expression, as AR mRNA levels remained unperturbed, or even slightly increased, 48 hours after Stat5a/b knockdown or IST5-002 treatment (Fig. 3A, iii). To determine if active Stat5a/b increases occupancy of AR at promoter regions within AR-regulated genes, we selected the PSA promoter for our investigation. Serum-starved LNCaP cells with endogenous Stat5a/b and AR proteins were stimulated with 10 nM prolactin and/or 1 nM DHT, followed by chromatin immunoprecipitation-PCR to identify binding of AR to a sequence within the promoter of the PSA gene. Prolactin stimulation enhanced AR occupancy of the PSA promoter in the absence of androgens (Fig. 3B). These results suggest that Stat5a/b up-regulates AR signaling in prostate cancer cells at the protein level and not by regulating AR mRNA expression.
To evaluate if Stat5a/b physically interacts with AR when liganded by the antiandrogens Flutamide, Bicalutamide and MDV3100 in prostate cancer cells, Stat5a or Stat5b were immunoprecipitated from LNCaP cells and immunoblotted with anti-AR antibody. The results demonstrate that Stat5a/b physically interacts with AR liganded not only with DHT but also with Flutamide, Bicalutamide or MDV3100 (Fig. 3C). To identify the Stat5a/b and AR domains responsible for mediating interaction of the two proteins, we generated a series of truncated FLAG-tagged Stat5a/b and MYC-tagged AR deletion constructs. Full-length MYC-AR was cotransfected into PC-3 cells along with each of the listed FLAG-Stat5a deletion constructs individually (Fig. 3D; full-length (FL); N-terminal-deleted (NTD); N-terminal and coiled-coil-deleted (NTCCD); C-terminus-deleted (N-ter); N-terminus-deleted (C-ter)). Immunoprecipitation of MYC-AR followed by immunoblotting of FLAG-Stat5a revealed that Stat5a-C-ter, corresponding to a deletion of the Stat5a N-terminal and coiled-coil domains and a partial deletion of the Stat5a DNA-binding domain, was the only construct that failed to coimmunoprecipitate with MYC-AR (Fig. 3D). Analysis of the overlap of the domains represented by Stat5a-NTCCD, -N-ter and -C-ter suggest that the AR interaction site is localized to the N-terminal portion of the Stat5a DNA-binding domain.
In the reverse scenario, full-length FLAG-Stat5a was cotransfected into PC-3 cells along with each of the listed MYC-AR deletion constructs individually (Fig. 3E; full-length (FL); 1-565 (Δ566-919); 566-919 (Δ1-565); 1-670 (Δ671-919); 1-608 (Δ609-919)). Immunoprecipitation of FLAG-Stat5a followed by immunoblotting of MYC-AR revealed that AR-566-919, corresponding to a deletion of the AR N-terminal activation domain (Δ1-565), was the only construct that failed to coimmunoprecipitate with Stat5a (Fig. 3E). Analysis of the overlap of the domains represented by the other AR deletion constructs indicate that the Stat5a interaction site is localized, albeit broadly, to the AR N-terminus between amino acids 1 and 565. To summarize, Stat5a/b physically interacts with antiandrogen-liganded AR in prostate cancer cells, and the DNA-binding domain of Stat5 and the N-terminal domain of AR contribute to physical interaction of Stat5a/b and AR.
Stat5a/b protects both androgen- and antiandrogen-liganded AR from proteasomal degradation in prostate cancer cells
Having established functional importance of the Stat5a/b-AR interaction in prostate cancer and given that genetic knockdown of Stat5a/b did not affect AR mRNA levels, we wanted to evaluate the possibility that Stat5a/b regulates AR protein levels during androgen deprivation of prostate cancer cells. We first analyzed protein levels of AR in the presence or absence of androgens following disruption of Stat5a/b function in prostate cancer cells. In both LNCaP and LAPC-4 cells, genetic knockdown of Stat5a/b (shStat5a/b) resulted in a substantial loss of AR protein after 72 hours, as visualized by immunoblotting of whole cell lysates (Fig. 4A,i and ii). Treatment with 1 nM DHT reversed this trend and partially prevented down-regulation of AR protein levels (Fig. 4A,i). Interestingly, Stat5a/b knockdown was almost as efficient as direct knockdown of AR (shAR) in decreasing AR protein levels (Fig. 4A,iii). PSA protein levels were also diminished following Stat5a/b knockdown by Stat5a/b antisense oligonucleotides during this timeframe, presumably due to loss of AR protein (Fig. 4A,iii and iv).
To assess if loss of AR protein after Stat5a/b knockdown was due to increased flux of AR through the proteasome, we initiated Stat5a/b knockdown followed by treatment with 10 μM proteasome inhibitor MG132. Proteasomal blockade partially rescued Stat5a/b knockdown-induced loss of AR, restoring AR protein levels in prostate cancer cells (Fig. 4B,i). AR loss, and subsequent AR rescue through proteasome inhibition, was time-dependent, requiring prior Stat5a/b knockdown, which generally occurred 48-72 hours after transduction of cells with lentiviral shStat5a/b (Fig. 4B,ii). AR rescue could not be attributed to generation of new AR protein, since rescue of AR protein levels occurred to a comparable extent in the presence of both MG132 and protein synthesis inhibitor cyclohexamide (Fig. 4B,iii).
Since active Stat5a/b induced nuclear localization of antiandrogen-liganded AR and physical interaction between Stat5a/b and antiandrogen-liganded AR, we next investigated if active Stat5a/b decreases proteasomal degradation of AR liganded by antiandrogens in prostate cancer cells. To evaluate the basal levels of AR in the presence of androgens vs. antiandrogens in prostate cancer cells, AR was immunoblotted from LNCaP and LAPC-4 cells treated with DHT, Flutamide, Bicalutamide or MDV3100 for 2, 3, 4 or 5 days. Unexpectedly, the data showed that antiandrogens Flutamide, Bicalutamide and MDV3100 each decreased AR protein levels in LNCaP and LAPC-4 cells (Fig. 5A). Antiandrogen effects were not exerted at the transcriptional level, since AR mRNA levels remained unaffected or were slightly increased by antiandrogen treatment during the same timeframe (LNCaP; Bicalutamide, Flutamide p=0.0004, MDV3100 p=0.037) (LAPC-4; Bicalutamide, Flutamide p=0.1213, MDV3100 p=0.011) (Fig. 5B). To investigate the mechanism of antiandrogen regulation of AR protein levels in prostate cancer cells, we treated LNCaP and LAPC-4 cells with MG132 in the presence or absence of Flutamide, Bicalutamide or MDV3100. As with Stat5a/b knockdown, the data suggest that antiandrogen treatment induced AR loss through increased proteasomal degradation in prostate cancer cells, since AR protein levels could be rescued by treatment with MG132 (Fig. 5C).
If Stat5a/b protects AR liganded by antiandrogens from proteasomal degradation in prostate cancer cells, targeting Stat5a/b in the presence of antiandrogen-liganded AR could potentially provide a strategy to accelerate proteasomal degradation of AR. To test this concept, Stat5a/b was genetically knocked down by shRNA followed by treatment with DHT, Flutamide, Bicalutamide or MDV3100 (Fig. 5D). In all instances, antiandrogen-liganded AR or AR in the absence of Stat5a/b was subjected to enhanced degradation. Indeed, the combination of Stat5a/b knockdown and antiandrogen treatment reduced AR protein levels to a greater extent than either intervention alone in LNCaP cells (Fig. 5D,i). Enhanced proteasomal degradation of AR through combined Stat5a/b knockdown and antiandrogen treatment was partially rescued by treatment with MG132, as previously seen for each intervention individually (Fig. 5D,ii). In conclusion, the data presented here indicate that Stat5a/b disruption potentiates AR proteasomal degradation induced by antiandrogens.
Combined inhibition of Stat5a/b and AR suppresses growth of prostate cancer cells to a greater extent than AR inhibition alone
To determine if pharmacological Stat5a/b inhibition potentiates the growth-inhibitory effects of antiandrogens, we evaluated the efficacy of combination therapy of antiandrogen MDV3100 and Stat5a/b dimerization inhibitor IST5-002 to MDV3100 alone in LNCaP cells. First, we determined sensitivity of LNCaP growth to MDV3100 (IC50 = 90 μM) (Fig. 6A) or IST5-002 (IC50 = 25 μM) (Fig. 6B) as single agents. Next, LNCaP cells were treated with increasing doses of MDV3100 either alone or in combination with 25 μM IST5-002 (IC50 value). At every dose of MDV3100 tested, a combination of MDV3100 and IST5-002 inhibited growth of LNCaP cells to a greater extent than MDV3100 alone (p<0.0001) (Fig. 6C). Remarkably, the addition of IST5-002 decreased the IC50 value of MDV3100 from 90 μM to approximately 5 μM in LNCaP cells, indicating substantial potentiation of antiandrogen action by Stat5a/b inhibition (p<0.0001) (Fig. 6Aiii). Collectively, these results suggest that efficacy of antiandrogens can be improved by simultaneous blockade of Stat5a/b signaling.
Discussion
In the present work, we demonstrate for the first time that Stat5a/b protects antiandrogen-liganded AR from proteasomal degradation in prostate cancer cells. Our results show that active Stat5a/b induces nuclear localization of antiandrogen-liganded AR, enhances AR occupancy of an AR-regulated promoter and increases expression of AR-regulated genes. We demonstrate that Stat5a/b physically interacts with AR through the DNA-binding and N-terminal domains of Stat5a/b and AR, respectively. We further show that antiandrogen treatment induces proteasomal degradation of AR, which can be increased by disruption of Stat5a/b expression or activity. Finally, our data indicate that combined inhibition of Stat5a/b and treatment with antiandrogens increases loss of AR through proteasomal degradation and enhances growth suppression of prostate cancer cells.
One of the key results of this work is the finding that Stat5a/b protects AR from antiandrogen-induced proteasomal degradation in prostate cancer cells. It is known that post-translational regulation of AR in prostate cancer cells occurs through ubiquitination of AR, leading to proteasomal degradation (39, 40). Specific ubiquitination of AR by E3 ligases such as E6-AP (41), CHIP (42) and Mdm2 (43) serves as the key rate-limiting step directing AR protein into the ubiquitin-proteasome pathway. While the effects of antiandrogens on AR nuclear translocation, transcriptional activity and co-activator recruitment are well-documented (16, 17, 44), the effects of antiandrogens on AR protein half-life is less clearly understood (45). Our data provide critical evidence supporting the notion that both first- and second-generation antiandrogens decrease AR protein levels in prostate cancer cells, while not reducing AR mRNA levels. Importantly, our results further show that active Stat5a/b potentiates AR signaling in the presence of antiandrogens by impeding the proteasomal degradation of antiandrogen-liganded AR in prostate cancer cells. Since AR-regulated gene expression is thought to be maintained during progression to CRPC and development of antiandrogen resistance, it is possible that Stat5a/b mediates sustained activity of AR in the presence of antiandrogens. Stat5a/b activation also induced other downstream molecular events involved in AR signaling, resulting in increased AR occupancy of the PSA promoter and expression of AR-regulated genes PSA and TMPRSS2. Future work should employ whole genome approaches to determine if Stat5a/b activation induces up-regulation of the entire AR transcriptome. While Stat5a/b disruption or antiandrogen treatment each individually increased flux of AR protein through the proteasome, a combination of these two therapeutic strategies induced maximal down-regulation of AR protein levels through proteasomal degradation in prostate cancer cells.
We show that Stat5a/b increased nuclear levels of antiandrogen-liganded AR and that transcriptionally active, phosphorylation-competent Stat5 is required for nuclear sequestration of AR. This finding is important because it suggests that pharmacological inhibitors of the Stat5a/b signaling pathway have the potential to down-regulate nuclear AR content in prostate cancer cells, resulting in decreased levels of AR available within the nucleus. We further demonstrated that active Stat5a/b and antiandrogen-liganded AR physically interact in prostate cancer cells, forming a Stat5a/b-AR complex detectable by coimmunoprecipitation (29). At present, it is unclear if the formation of a Stat5a/b-AR complex prevents recruitment of E3 ubiquitin ligases targeting AR, impedes ubiquitination of AR or interferes with some other event prior to AR being directed to the proteasome. AR ubiquitination depends on AR phosphorylation, with phosphorylated serine or tyrosine residues serving as recognition motifs for E3 ligases (phosphodegrons) (43, 46, 47). Phospho-S515 and -Y534 of AR represent the degrons for E3 ligases Mdm2 (43, 46) and CHIP (46, 47), respectively. Since our results show that Stat5a/b binds to the N-terminal region (aa 1-566) of AR, it is plausible that Stat5a/b hinders E3 ligase recruitment by masking the AR phosphodegrons. Analogous regulation of transcriptional activity and nuclear localization has been described for BRCA1 and BRCA2 via molecular masking of nuclear export signals by BARD in breast cancer (48, 49). It is also possible that Stat5a/b-induced nuclear sequestration of AR in prostate cancer cells is caused by active Stat5a/b exclusively protecting nuclear-localized AR from proteasomal degradation.
Increased Stat5a/b expression may promote transition of androgen-sensitive prostate cancer to CRPC by undermining efficacy of antiandrogen therapy. Active Stat5a/b expression is increased in 95% of clinical CRPCs (29), the Stat5a/b gene locus is amplified in 29% of distant CRPC metastases (30), and in this work, we demonstrate a significant association between high levels of nuclear Stat5a/b and AR in patient-derived clinical prostate cancers. Importantly, we show that inhibition of Stat5a/b sensitized prostate cancer cells to treatment with the second-generation antiandrogen MDV3100, providing further evidence in support of combination therapy for prostate cancer using Stat5a/b inhibitors and antiandrogens. Collectively, these findings support the concept that up-regulation of Stat5a/b signaling promotes survival of androgen-deprived prostate cancer cells. In summary, the work presented here proposes a novel means of exploiting AR proteasomal degradation and enhancing efficacy of antiandrogens through targeting of Stat5a/b. Following further clinical development, pharmacological Stat5a/b inhibitors have the potential to be deployed alongside existing antiandrogen therapies for more effective treatment of advanced prostate cancer.
Supplementary Material
Acknowledgments
This work was financially supported by a National Cancer Institute (NCI)/National Institutes of Health (NIH) Research Project Grant (2RO1CA11358-06) and an NCI/NIH Exploratory/Developmental Research Grant (1R21CA178755-01) to M.T. Nevalainen. D.T. Hoang is supported by an NCI/NIH Predoctoral Individual National Research Service Award (NRSA) Fellowship (1F31CA180626-01). Research reported in this publication utilized shared resources of Kimmel Cancer Center at Thomas Jefferson University, supported by an NCI/NIH Cancer Center Core Grant (P30CA056036).
Footnotes
All authors have no conflicts of interest to disclose.
References
- 1.Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:597–610. doi: 10.1038/nrclinonc.2011.117. [DOI] [PubMed] [Google Scholar]
- 2.Gleave ME, Goldenberg SL, Chin JL, Warner J, Saad F, Klotz LH, et al. Randomized comparative study of 3 versus 8-month neoadjuvant hormonal therapy before radical prostatectomy: biochemical and pathological effects. J Urol. 2001;166:500–6. discussion 6-7. [PubMed] [Google Scholar]
- 3.Trachtenberg J, Walsh PC. Correlation of prostatic nuclear androgen receptor content with duration of response and survival following hormonal therapy in advanced prostatic cancer. J Urol. 1982;127:466–71. doi: 10.1016/s0022-5347(17)53868-9. [DOI] [PubMed] [Google Scholar]
- 4.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature Gen. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 5.Sun C, Shi Y, Xu LL, Nageswararao C, Davis LD, Segawa T, et al. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene. 2006;25:3905–13. doi: 10.1038/sj.onc.1209424. [DOI] [PubMed] [Google Scholar]
- 6.Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–5. [PubMed] [Google Scholar]
- 7.Penning TM, Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009;1155:33–42. doi: 10.1111/j.1749-6632.2009.03700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–7. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 9.Zhu ML, Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer. 2008;15:841–9. doi: 10.1677/ERC-08-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Wang L, Lin HK, Kan PY, Xie S, Tsai MY, et al. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem Biophys Res Commun. 2003;305:462–9. doi: 10.1016/s0006-291x(03)00792-7. [DOI] [PubMed] [Google Scholar]
- 11.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sufrin G, Coffey DS. Flutamide. Mechanism of action of a new nonsteroidal antiandrogen. Invest Urol. 1976;13:429–34. [PubMed] [Google Scholar]
- 16.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem. 2002;277:26321–6. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 17.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, et al. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J Biol Chem. 2003;278:27287–92. doi: 10.1074/jbc.M304307200. [DOI] [PubMed] [Google Scholar]
- 19.Dagvadorj A, Kirken RA, Leiby B, Karras J, Nevalainen MT. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin Cancer Res. 2008;14:1317–24. doi: 10.1158/1078-0432.CCR-07-2024. [DOI] [PubMed] [Google Scholar]
- 20.Dagvadorj A, Tan SH, Liao Z, Xie J, Nurmi M, Alanen K, et al. N-terminal truncation of Stat5a/b circumvents PIAS3-mediated transcriptional inhibition of Stat5 in prostate cancer cells. Int J Biochem Cell Biol. 2011;42:2037–46. doi: 10.1016/j.biocel.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, et al. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am J Pathol. 2010;176:1959–72. doi: 10.2353/ajpath.2010.090653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu L, Vogiatzi P, Puhr M, Dagvadorj A, Lutz J, Ryder A, et al. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr Relat Cancer. 2010;17:481–93. doi: 10.1677/ERC-09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Ahonen TJ, Alanen K, Xie J, LeBaron MJ, Pretlow TG, et al. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 2004;64:4774–82. doi: 10.1158/0008-5472.CAN-03-3499. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Zhang Y, Glass A, Zellweger T, Gehan E, Bubendorf L, et al. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin Cancer Res. 2005;11:5863–8. doi: 10.1158/1078-0432.CCR-05-0562. [DOI] [PubMed] [Google Scholar]
- 25.Thomas C, Zoubeidi A, Kuruma H, Fazli L, Lamoureux F, Beraldi E, et al. Transcription factor Stat5 knockdown enhances androgen receptor degradation and delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2011;10:347–59. doi: 10.1158/1535-7163.MCT-10-0850. [DOI] [PubMed] [Google Scholar]
- 26.Mirtti T, Leiby BE, Abdulghani J, Aaltonen E, Pavela M, Mamtani A, et al. Nuclear Stat5a/b predicts early recurrence and prostate cancer-specific death in patients treated by radical prostatectomy. Hum Pathol. 2013;44:310–9. doi: 10.1016/j.humpath.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 28.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 29.Tan SH, Dagvadorj A, Shen F, Gu L, Liao Z, Abdulghani J, et al. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res. 2008;68:236–48. doi: 10.1158/0008-5472.CAN-07-2972. [DOI] [PubMed] [Google Scholar]
- 30.Haddad BR, Gu L, Mirtti T, Dagvadorj A, Vogiatzi P, Hoang DT, et al. STAT5A/B gene locus undergoes amplification during human prostate cancer progression. Am J Pathol. 2013;182:2264–75. doi: 10.1016/j.ajpath.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu L, Liao Z, Hoang DT, Dagvadorj A, Gupta S, Blackmon S, et al. Pharmacologic inhibition of Jak2-Stat5 signaling By Jak2 inhibitor AZD1480 potently suppresses growth of both primary and castrate-resistant prostate cancer. Clin Cancer Res. 2013;19:5658–74. doi: 10.1158/1078-0432.CCR-13-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 33.Ahonen TJ, Harkonen PL, Rui H, Nevalainen MT. PRL signal transduction in the epithelial compartment of rat prostate maintained as long-term organ cultures in vitro. Endocrinology. 2002;143:228–38. doi: 10.1210/endo.143.1.8576. [DOI] [PubMed] [Google Scholar]
- 34.Nevalainen MT, Xie J, Bubendorf L, Wagner KU, Rui H. Basal activation of transcription factor signal transducer and activator of transcription (Stat5) in nonpregnant mouse and human breast epithelium. Mol Endocrinol. 2002;16:1108–24. doi: 10.1210/mend.16.5.0839. [DOI] [PubMed] [Google Scholar]
- 35.Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, et al. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J Clin Oncol. 2004;22:2053–60. doi: 10.1200/JCO.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 36.Luo J, Zha S, Gage WR, Dunn TA, Hicks JL, Bennett CJ, et al. Alpha-methylacyl-CoA racemase: a new molecular marker for prostate cancer. Cancer Res. 2002;62:2220–6. [PubMed] [Google Scholar]
- 37.Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem. 1996;271:6379–88. doi: 10.1074/jbc.271.11.6379. [DOI] [PubMed] [Google Scholar]
- 38.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, et al. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999;59:4180–4. [PubMed] [Google Scholar]
- 39.Lee DK, Chang C. Endocrine mechanisms of disease: Expression and degradation of androgen receptor: mechanism and clinical implication. J Clin Endocrinol Metab. 2003;88:4043–54. doi: 10.1210/jc.2003-030261. [DOI] [PubMed] [Google Scholar]
- 40.Jaworski T. Degradation and beyond: control of androgen receptor activity by the proteasome system. Cell Mol Biol Lett. 2006;11:109–31. doi: 10.2478/s11658-006-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramamoorthy S, Nawaz Z. E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl Recept Signal. 2008;6:e006. doi: 10.1621/nrs.06006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He B, Bai S, Hnat AT, Kalman RI, Minges JT, Patterson C, et al. An androgen receptor NH2-terminal conserved motif interacts with the COOH terminus of the Hsp70-interacting protein (CHIP) J Biol Chem. 2004;279:30643–53. doi: 10.1074/jbc.M403117200. [DOI] [PubMed] [Google Scholar]
- 43.Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. Embo J. 2002;21:4037–48. doi: 10.1093/emboj/cdf406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hodgson MC, Astapova I, Hollenberg AN, Balk SP. Activity of androgen receptor antagonist bicalutamide in prostate cancer cells is independent of NCoR and SMRT corepressors. Cancer Res. 2007;67:8388–95. doi: 10.1158/0008-5472.CAN-07-0617. [DOI] [PubMed] [Google Scholar]
- 45.Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. J Biol Chem. 1992;267:968–74. [PubMed] [Google Scholar]
- 46.Chymkowitch P, Le May N, Charneau P, Compe E, Egly JM. The phosphorylation of the androgen receptor by TFIIH directs the ubiquitin/proteasome process. Embo J. 2011;30:468–79. doi: 10.1038/emboj.2010.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DaSilva J, Gioeli D, Weber MJ, Parsons SJ. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res. 2009;69:7402–11. doi: 10.1158/0008-5472.CAN-08-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henderson BR. Regulation of BRCA1, BRCA2 and BARD1 intracellular trafficking. Bioessays. 2005;27:884–93. doi: 10.1002/bies.20277. [DOI] [PubMed] [Google Scholar]
- 49.Thompson ME. BRCA1 16 years later: nuclear import and export processes. Febs J. 2010;277:3072–8. doi: 10.1111/j.1742-4658.2010.07733.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.