

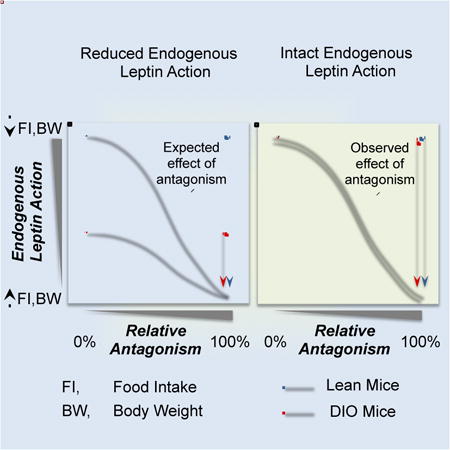
Summary
Obesity is characterized by hyperleptinemia and decreased response to exogenous leptin. This has been widely attributed to the development of leptin resistance; a state of impaired leptin signaling proposed to contribute to the development and persistence of obesity. To directly determine endogenous leptin activity in obesity, we treated lean and obese mice with a leptin receptor antagonist. The antagonist increased feeding and body weight (BW) in lean mice but not in obese models of leptin, leptin receptor or melanocortin-4 receptor deficiency. In contrast, the antagonist increased feeding and BW comparably in lean and diet-induced obese (DIO) mice, an increase associated with decreased hypothalamic expression of Socs3, a primary target of leptin. These findings demonstrate that hyperleptinemic DIO mice retain leptin-suppression of feeding comparable to lean mice, and counter the view that resistance to endogenous leptin contributes to the persistence of DIO in mice.
Graphical abstract
Introduction
Leptin is a 16kDa hormone secreted by adipocytes (Zhang et al., 1994) that plays a critical role in the control of feeding by acting on specific neurons in the Central Nervous System (CNS) (Myers et al., 2008). Individuals lacking circulating leptin are hyperphagic and obese, features that can be reversed with administration of exogenous leptin (Halaas et al., 1995). In contrast, more common forms of obesity including diet-induced obesity (DIO), exhibit hyperleptinemia proportional to the amount of body fat stores (Frederich et al., 1995). The inability of high endogenous leptin levels to reduce feeding and mitigate or reverse weight gain is referred as leptin resistance and it has been implicitly associated to the impairment of leptin action (Myers et al., 2012). Consistent with this thesis, obese hyperleptinemic animals have a blunted anorectic response to exogenously administered leptin and an associated attenuation of the leptin receptor (LEPR)-dependent intracellular signaling cascade (Enriori et al., 2007).
While the resistance of obese subjects to exogenous leptin has been widely documented, the action of endogenous leptin to control energy balance in obesity has not been rigorously tested. We hypothesized that blockade of the LEPR in mouse models of obesity would give an estimate of endogenous leptin action reflected in changes in food intake and BW. This approach was taken with genetic and diet-induced models of obese mice. Our results demonstrate that despite the presence of hyperleptinemia, wildtype DIO mice retain a similar degree of endogenous leptin action as their lean counterparts. Thus, persistance of obesity in DIO mice occurs despite ongoing endogenous leptin action.
Results
Intraperitoneal PLA increases food intake and body weight in wildtype mice but not in mice with impaired leptin signaling
We assessed the in vivo specificity of a pegylated leptin receptor antagonist (PLA) by comparing the effect of daily administration (3 mg/kg ip, daily) on energy intake and BW in leptin receptor-deficient (leprdbdb, db/db), leptin-deficient (lepobob, ob/ob) and age-matched wildtype (wt/wt) control mice over 1 wk (Fig 1). Untreated db/db and ob/ob mice had significant hyperphagia compared to untreated wt/wt controls (Fig 1a, see also Fig S1a). Consistent with a blockade of endogenous leptin action, PLA significantly increased 1-wk energy intake in wt/wt mice. In contrast, PLA failed to increase feeding in ob/ob or in db/db mice (Fig 1a). PLA significantly increased BW (Fig 1b) and BW gain (Fig 1c) in lean wt/wt control mice compared to vehicle-treated controls, but failed to affect BW in db/db mice. Interestingly, there was a small but significant reduction in BW gain in ob/ob mice treated with PLA (Fig 1c).
Figure 1. Peripheral administration of PLA (3 mg/kg, ip, once daily) in chow fed db/db, ob/ob or lean wildtype control mice.

Daily injection of PLA increases energy intake (a), BW (b) and BW change (c) in lean chow-fed control mice but not in mice voided of endogenous leptin signaling. Data shown as mean ± SEM. n=6. *=P<0.05 wt/wt Veh vs. wt/wt PLA; # P<0.05 ob/ob Veh vs. ob/ob PLA. 2-way RM ANOVA, followed by Sidak test.
Ip PLA or icv LA increases food intake and body weight in wildtype mice but not in obese melanocortin 4-receptor deficient mice
The melanocortin system plays a critical role mediating the effect of leptin on food intake and BW (Seeley et al., 1997). Mice with a homozygous deletion of MC4R (Mc4r-/- mice) are hyperphagic and develop obesity in adulthood that is associated with hyperleptinemia and reduced effectiveness of exogenous leptin (Marsh et al., 1999). To investigate the contribution of MC4R to the actions of endogenous leptin, obese Mc4r-/- mice and wildtype littermates received peripheral PLA (3 mg/kg/d, ip) for 1 wk. PLA significantly increased energy intake in wildtype (P<0.05 treatment × time) but not in obese Mc4r-/-mice (Fig 2a, see also Fig S1b). 1-wk PLA treatment did not affect total BW (Fig 2b) but promoted a significant BW change in wildtype mice relative to vehicle treated controls (Fig 2c). In contrast, leptin receptor blockade did not change BW in obese Mc4r-/- mice (Fig 2c). PLA was dosed based on BW (1.65-fold difference) but plasma leptin levels were 9.5-fold higher in obese Mc4r-/- in comparison to their wildtype controls (41.34±2.11 vs. 4.35±2.46 ng/ml, P<0.05), raising the possibility of insufficient antagonism to counteract the higher levels of endogenous leptin. To ensure maximal reduction of endogenous leptin action, obese Mc4r-/- mice and lean wildtype littermates received an infusion of non-pegylated antagonist (LA, 8 ug/d) for 1 wk directly into the lateral cerebral ventricle (icv) using osmotic minipumps. LA induced significant hyperphagia (Fig 2d, see also Fig S1c) and BW gain (Fig 2e,f) in lean wildtype but not in obese Mc4r-/- mice, despite a trend towards a BW change in the latter (P=0.059, Fig 2f). Since young, preobese Mc4r-/- mice retain responsiveness to exogenous leptin (Marsh et al., 1999), we treated a cohort of younger Mc4r-/- (28.5±1.5g) and age-matched wildtype littermates (24.1±1.0g) with PLA (3 mg/kg/d, ip) for 1 wk. PLA did not affect total BW (Fig S1d), but increased the BW gain relative to vehicle-treated controls, as well as energy intake, in Mc4r-/- mice (P<0.05 treatment × time, Fig S1e, f) and in their wildtype littermates (Fig. S1e-g).
Figure 2. Peripheral administration of PLA or central infusion of LA in chow fed obese Mc4r-/- or wildtype littermate control male mice.

Cumulative energy intake (a,d) BW (b,e) and BW change (c,f) of obese Mc4r-/- mice and wildtype controls receiving either peripheral PLA (3 nmol/kg/day, ip) (a-c) or central infusion of LA (8ug/day, icv) (d-f) for 1-wk. Data shown as mean ± SEM. n=5-8. *=P<0.05 Mc4r+/+ Veh vs. Mc4r+/+ treated mice. 2-way RM ANOVA followed by Sidak test.
Ip PLA or icv LA increase food intake and BW in lean and DIO mice
DIO mice are frequently used as a model of leptin resistance. DIO mice and age-matched, chow-fed lean controls were treated with PLA (1, 3 or 10 mg/kg, ip, once daily) for 7 d (lower doses) or 6 d (highest dose). PLA at 1 mg/kg/d significantly increased energy intake in lean mice only (Fig 3a, see also Fig S1h), and did not change BW in lean or DIO mice (Fig 3b,c). PLA at 3 mg/kg/d significantly increased food intake (Fig 3b, see also Fig S1i) and total BW (Fig 3e, P<0.05 at Day 7) in lean but not in DIO mice. However, both lean and DIO mice treated with this dose of PLA exhibited significant BW change when compared to their vehicle controls (Fig 3f), despite a 10-fold increase in circulating leptin in DIO mice compared to lean controls (29.60±2.36 vs. 2.94±0.45 ng/ml, P<0.05). PLA at 10 mg/kg/d significantly increased energy intake (Fig 3g, see also Fig S1j) and BW (Fig 3h,i) in both lean and DIO mice. To examine near-maximal effects of reducing endogenous leptin action, we infused LA (8 ug/d) icv into lean and DIO mice for 7 d using osmotic minipumps. Icv LA significantly increased food intake (Fig 3j, see also Fig.S1k) in both lean and DIO mice. LA also caused significant differences in total BW (Fig 3k) and (Fig 3l) relative BW change compared to vehicle-treated controls. When the dose-responses of PLA on energy intake and BW change were compared, the effect of endogenous leptin to restrain energy intake was comparable between lean and DIO mice, with the exception of the dose of 3 mg/kg/d (Fig 3m), whereas the BW change compared to vehicle-treated controls were similar in lean and DIO mice with all doses (Fig 3n).
Figure 3. Peripheral administration of PLA or central infusion of LA in lean and DIO mice.

(a-l) Cumulative energy intake (a,d,g,j), BW (b,e,h,k) and BW change (c,f,i,l) of lean and DIO mice receiving either peripheral PLA (1,3,10 nmol/kg/day, ip, a-i) or central infusion of LA (8ug/day, icv) (j-l). (m, n) Change in caloric intake (m) and BW (n) after 6-d of treatment with either peripheral PLA or central LA. (a-l). Data shown as mean ± SEM. n=7-8 (a-i) or n=5 (j-l). *=P<0.05 Lean Veh vs. Lean PLA; #=P<0.05 DIO Veh vs. DIO PLA. 2-way RM ANOVA followed by Sidak test (a-l) or t-Student test (m).
Effect of reduction of endogenous leptin action on Pomc, Socs3 and phosphorylated STAT3 levels in the arcuate nucleus of DIO mice
Expression of two leptin-regulated genes, proopiomelanocortin (Pomc) and Suppressor-Of-Cytokine-Signaling-3 (Socs3), was analyzed in the hypothalamic arcuate nucleus (ARC) following acute or chronic PLA. DIO mice (BW 58.8±1.2g) received a single dose of PLA (3 mg/kg, ip) 1 h prior to the onset of dark. The mice had free access to water and HFD overnight and were euthanized 1 h after the onset of light. Food intake did not differ during the experimental period (Fig S2a), but PLA-treated mice exhibited a significant BW change in comparison to vehicle-treated controls (Fig S2b), consistent with a reduction in endogenous leptin signaling. Overnight PLA treatment did not affect Pomc (Fig 4a) but significantly reduced Socs3 (Fig 4b) expression in the ARC of DIO mice.
Figure 4. Pomc and Socs3 gene expression and pSTAT3 levels in the arcuate nucleus of mice treated with leptin receptor antagonist.

Pomc (a) and Socs3 (b) expression after a single injection of PLA (3mg/kg, ip) in DIO mice 1 h before the onset of the dark phase. Pomc (c) and Socs3 (d) expression after 7-d treatment with PLA (3mg/kg, ip, once daily) on lean and DIO mice. (e,f) p-STAT3 levels relative to beta actin (e) and to total STAT3 (f) measured by inmunoblot in ARC of lean or DIO mice after 7-d infusion with icv LA (8ug/day). Data shown as mean ± SEM. n=5-7; *=P<0.05. (b) t-Student test; (c,d, e). 2-way ANOVA followed by Sidak test.
Chronic PLA treatment (3 mg/kg, ip, daily for 7 d, Fig 3d-f) significantly reduced both Pomc (Fig 4c) and Socs3 (Fig 4d) gene expression in lean and DIO mice. Pomc gene expression was similar between lean and DIO mice treated with vehicle (Fig 4c). In contrast, Socs3 expression was significantly increased in vehicle-treated DIO mice in comparison to vehicle-treated lean controls (Fig 4d).
Levels of phosphorylated Signal-Transducer-and-Activator-of-Transcription-3 (pSTAT3) were analysed by inmunoblot (Fig S3) in the ARC of lean and DIO mice receiving icv LA (8 ug/d) for 7d (Fig S2c-f). Vehicle-treated DIO mice had increased pSTAT3 in comparison to vehicle-treated lean controls when normalized to beta actin content, (Fig 4e). Icv-LA significantly reduced pSTAT3 in DIO mice (Fig 4e), and pSTAT3 normalized to total STAT3 content supported this finding (P=0.061, DIO Veh vs. DIO LA; Fig 4f).
Discussion
In this study, central or peripheral treatment with a LEPR antagonist (D23L/L39A/D40A/F41A mutant) demonstrates a significant role of endogenous leptin action regulating energy balance. More importantly, our experiments consistently show comparable contribution to the control of BW and suppression of food intake by endogenous leptin in lean and hyperleptinemic DIO mice, regardless dose and route of administration. These findings in a standard animal model of obesity often cited as leptin resistant indicate that the current view on the role of leptin action in obesity needs revision.
Consistent with our data, peripheral infusion of a different pegylated antagonist (L39A/D40A/F41A mutant) increased feeding in chow fed mice (Levi et al., 2011). In contrast, central infusion of the non-pegylated L39A/D40A/F41A antagonist failed to increase feeding in chow fed rats (Tumer et al., 2007). In addition to potential species-specific differences, this discrepancy with our results is likely accounted for by the increased potency of the antagonist used in our experiments, with greater binding to leptin receptor (60-fold) and higher antagonistic activity (14-fold) compared to the L39A/D40A/F41A mutant (Shpilman et al., 2011). This increased potency, combined with the extended duration of action provided by the addition of a polyethylene glycol moiety, provides effectiveness to PLA when admistered peripherally, results that are consistent with earlier reports (Chapnik et al., 2013; Shpilman et al., 2011; Solomon et al., 2014). Despite this increase in potency, PLA lacks orexigenic activity when given to db/db and and ob/ob mice, which confirms its selectivity in vivo. Indeed, PLA reduces BW gain in ob/ob mice, which could be the result of weak agonist activity, considering that cytokine receptors such as the LEPR, lack intrinsic activity and their signaling depends on the status of associated kinases (Ishida-Takahashi et al., 2006).
A potential factor previously suggested as contributing to leptin resistance in obesity is the impairment of the transport of leptin through the Blood Brain Barrier (BBB) into the CNS (Banks et al., 1999; Caro et al., 1996). To circumvent any role of differences in BBB permeability between lean and obese mice, we compared peripheral and icv administration of high doses of LEPR antagonist. The observation that both lean and DIO mice had comparable increases in energy intake and BW that were proportional to the doses of antagonist administered peripherally or centrally, demonstrates that both groups experienced substantial restraint of food intake by endogenous leptin, irrespective of their body weight and adiposity.
Consistent with previous reports (Levi et al., 2011; Solomon et al., 2014), blockade of LEPR signaling in lean, wildtype mice resulted in significant hyperphagia and BW gain. The lack of effect of PLA to regulate energy balance in LEPR deficient db/db mice supports the specificity of PLA for the LEPR, and a lack of “off-target” effects in vivo. The dose of PLA given to db/db mice was less than the maximally effective dose given to DIO animals, but was sufficient to induce changes in the BW of high-fat fed mice. PLA also failed to increase food intake in ob/ob mice, but did attenuate their BW gain, suggesting modest LEPR agonism of the compound in this strain, described as having increased leptin sensitivity (Harris et al., 1998).
A key neural circuit involved in the control of energy balance by leptin is the melanocortin system, including direct and/or indirect control of MC4R-expressing neurons by leptin (Ghamari-Langroudi et al., 2011). Mc4r-/- mice develop late-onset obesity (Huszar et al., 1997), with hyperleptinemia and resistance to the effect of exogenous administration of leptin (Marsh et al., 1999). The failure of PLA/LA to induce hyperphagia in obese Mc4r-/- mice supports a prominent role of the melanocortin system to convey the anorectic action of endogenous leptin in adult mice. This dramatic reduction of leptin action in obese, adult Mc4r-/- mice stands in contrast to the maintenance of leptin sensitivity found in young, non-obese mice with Mc4r deletion (Marsh et al., 1999) and with the effectiveness of PLA increasing body weight and energy intake in young Mc4r-/- mice. This discrepancy suggests an age-dependent convergence from multiple neural circuits towards the melanocortin system as the mediator of leptin effects on the homeostatic control of energy balance. Although this hypothesis remains to be corroborated experimentally, it is supported by considerable evidence suggesting age-dependent changes in leptin action (Gabriely et al., 2002; Morrison et al., 2007; Newton et al., 2013; Scarpace et al., 2000).
The reduced impact of subtracting endogenous leptin action in mouse models of obesity caused by direct (i.e. db/db or ob/ob mice) or indirect (Mc4r-/- mice) disruption of leptin signaling provides a striking contrast with the comparable induction of positive energy balance in DIO and age-matched lean mice following the peripheral administration of PLA or central infusion of LA. These findings show that DIO mice maintain intact endogenous leptin action despite hyperleptinemia and support previous observations of the susceptibility of DIO rats to weight gain when given the less potent leptin receptor antagonist (L39A/D40A/F41A) via icv (Tumer et al., 2007). In addition to hyperleptinemia, DIO mice exhibit reduced responses to exogenously administered leptin, which has been linked to impairments of the intracellular signaling cascade induced by the activated LEPR (Coppari and Bjorbaek, 2012; Myers et al., 2008). One proposed mechanism of leptin resistance involves reduced LEPR signaling as a result of increased Socs3 levels (Bjorbaek et al., 1998). Increased Socs3 prevents the phosphorylation of STAT3 by activated LEPR, providing a means of negative feedback regulation of leptin action in target cells (Myers et al., 2008). Consistent with this view, hyperleptinemic, leptin resistant DIO mice exhibit increased baseline Socs3 expression in the ARC (Enriori et al., 2007; Munzberg et al., 2004). Yet, similar to previous studies using DIO mice (Knight et al., 2010; Martin et al., 2006), we observed elevated basal p-STAT3 levels in DIO mice compared to lean controls, and a significant decrease with PLA treatment. Since leptin receptor antagonism reduced Socs3 gene expression, p-STAT3 levels, and the expression of a target gene, Pomc (Munzberg et al., 2003), our data are consistent with the hypothesis that increased Socs3 and pSTAT3 levels in the ARC of DIO mice are the direct consequence of ongoing endogenous leptin signaling in these obese animals. More importantly, this occurred at doses of antagonist sufficient to elicit similar changes in energy intake and BW in lean and DIO mice. Thus, although the increase in baseline Socs3 levels exhibited by DIO mice may attenuate the effect of exogenously administered leptin, explaining the lack of expected hypophagia or activation of LEPR signaling cascade, our results suggest that DIO mice do not experience reduced endogenous leptin action, and in fact demonstrate that it plays a critical role preventing further BW gain.
Our results suggest that DIO develops despite the sustained contribution of endogenous leptin to regulate energy balance. Thus, mechanisms opposing leptin must play a crucial role in the development or maintenance of obesity. There is evidence that some of these mechanisms may actually be LEPR-dependent, as suggested by the fact that mice over-expressing LEPR in POMC neurons are more susceptible to DIO (Gamber et al., 2012). On the other hand, transgenic mice exhibiting supraphysiological serum leptin levels remain leaner than wildtype controls on a standard low-fat diet, and reach the same BW when made DIO, suggesting that hyperleptinemia alone is not sufficient to reduce endogenous leptin action and cause obesity (Tanaka et al., 2005). Conversely, ob/ob mice supplemented with sufficient leptin to prevent obesity while fed a low-fat diet experience similar BW gain than wildtype hyperleptinemic controls once challenged with a HFD despite remaining responsive to the exogenous administration of leptin (Knight et al., 2010). Assuming the limitations due to the intrinsic differences in leptin action between ob/ob mice and wildtype mice (Bouret et al., 2004), these data suggest that factors other than leptin have a relevant role in the control of BW in conditions of energy surplus. Identifying the factors involved in counteracting the effect of leptin during the development of obesity may provide efficacious targets to prevent BW gain.
The comparable effects of PLA/LA in wildtype lean and DIO mice suggests that although hyperleptinemic mice may have close to maximal LEPR activity, suppression of steady-state of food intake by endogenous leptin remains intact, contributing to the control of energy balance. In our studies this effect is comparable to the lean control mice. This suggestion that our DIO mice have near maximal endogenous leptin action provides a caveat to the therapeutic application of leptin to treat obesity. The relatively modest effect of leptin to reduce body weight in obese humans may be due to the limited benefit of increasing leptin levels in already hyperleptinemic subjects (Heymsfield et al., 1999). In contrast, use of leptin during BW loss, when leptin levels drop and there is room for further LEPR activation, seems to be a much more effective approach (Clemmensen et al., 2013; Muller et al., 2012; Roth et al., 2008).
Overall, the findings presented here demonstrate comparable endogenous leptin activity in lean and obese hyperleptinemic diet-induced obese mice, despite different sensitivity to exogenously administered leptin. These findings challenge the general assumption of reduced leptin action in obesity, and they should be considered in the development of therapies targeting leptin signaling for the treatment of metabolic disease.
Experimental procedures
These studies were approved by the Institutional Animal Care and Use Committees at the University of Cincinnati Office in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Mice
All mice (C57/Bl6J, Lepob/ob, Leprdb/db and LoxTbMc4r) were purchased from Jackson Laboratories (Bar Harbor, ME) and were acclimated for at least one week before the study. Mice were single-housed during the study and placed in a 12-h light, 12-h dark cycle at 22 °C with free access to food and water (see also supplemental experimental procedures).
Leptin antagonists
Native (LA) and 20k-pegylated (PLA) mouse PLA (mutant D23L/L39A/D40A/F41A) were synthetized and characterized as previously described (Shpilman et al., 2011)
Intraperitoneal Injections
PLA was dissolved in PBS and administered intraperitoneally over a period of 6 d at doses of 1, 3, or 10 mg/kg daily in independent sets of mice. BW and energy intake were monitored daily.
Intracerebroventricular Infusions
Mice received a cannula in the lateral cerebral ventricle connected to a subcutanenous osmotic mini-pump (1007D; Alzet, Cupertino, CA) filled with vehicle (PBS) or LA infused at 8 ug/day for 7 d (see also supplemental experimental procedures).
Gene expression analysis
Pomc and Socs3 gene expression in the arcuate nucleus were analysed using commercially available gene-specific Taqman® probes following manufacturer instructions (see also supplemental experimental procedures) and quantified as described elsewhere (Muller et al., 2002).
Inmunoblot
levels of total STAT3 and p-STAT3 protein in the arcuate nucleus were detected by inmunoblot using commercially available antibodies, revealed using chemioluminiscense and quantified using standard imaging techinques (see also supplemental experimental procedures).
Leptin measurements
Leptin was measured using a commercially available ELISA from Alpco (Salem, NH).
Statistical analyses
Data are presented as mean ± SEM. Analyses were performed using GraphPad Prism, version 6 (GraphPad Software, Inc., San Diego, CA). T-tests were used for comparison of two groups and two-way ANOVA with or without repeated measures and Sidak multiple comparison tests were used for post hoc comparisons. P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Drs. Stephen Woods and Randy Seeley for their insightful comments on the manuscript. This work was funded by National Institutes of Health grants DK077975 to DPT and DK57900 to DDA.
Footnotes
Author contributions: NO, PM, BR and LAN performed the studies. AG provided essential research tools. All the authors analyzed the data. NO, DDA and DPT designed the experiments and wrote the manuscript.
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Banks WA, DiPalma CR, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides. 1999;20:1341–1345. doi: 10.1016/s0196-9781(99)00139-4. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Molecular cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, Lynn RB, Zhang PL, Sinha MK, Considine RV. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- Chapnik N, Solomon G, Genzer Y, Miskin R, Gertler A, Froy O. A superactive leptin antagonist alters metabolism and locomotion in high-leptin mice. The Journal of endocrinology. 2013;217:283–290. doi: 10.1530/JOE-13-0033. [DOI] [PubMed] [Google Scholar]
- Clemmensen C, Chabenne J, Finan B, Sullivan L, Fischer K, Kuchler D, Sehrer L, Ograjsek T, Hofmann S, Schriever SS, et al. GLP-1/glucagon co-agonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes. 2013 doi: 10.2337/db13-1609. [DOI] [PubMed] [Google Scholar]
- Coppari R, Bjorbaek C. Leptin revisited: its mechanism of action and potential for treating diabetes. Nature reviews. Drug discovery. 2012;11:692–708. doi: 10.1038/nrd3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nature medicine. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- Gabriely I, Ma XH, Yang XM, Rossetti L, Barzilai N. Leptin resistance during aging is independent of fat mass. Diabetes. 2002;51:1016–1021. doi: 10.2337/diabetes.51.4.1016. [DOI] [PubMed] [Google Scholar]
- Gamber KM, Huo L, Ha S, Hairston JE, Greeley S, Bjorbaek C. Over-expression of leptin receptors in hypothalamic POMC neurons increases susceptibility to diet-induced obesity. PloS one. 2012;7:e30485. doi: 10.1371/journal.pone.0030485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Srisai D, Cone RD. Multinodal regulation of the arcuate/paraventricular nucleus circuit by leptin. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:355–360. doi: 10.1073/pnas.1016785108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Harris RB, Zhou J, Redmann SM, Jr, Smagin GN, Smith SR, Rodgers E, Zachwieja JJ. A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology. 1998;139:8–19. doi: 10.1210/endo.139.1.5675. [DOI] [PubMed] [Google Scholar]
- Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA : the journal of the American Medical Association. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Ishida-Takahashi R, Rosario F, Gong Y, Kopp K, Stancheva Z, Chen X, Feener EP, Myers MG., Jr Phosphorylation of Jak2 on Ser(523) inhibits Jak2-dependent leptin receptor signaling. Molecular and cellular biology. 2006;26:4063–4073. doi: 10.1128/MCB.01589-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. PloS one. 2010;5:e11376. doi: 10.1371/journal.pone.0011376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi J, Gray SL, Speck M, Huynh FK, Babich SL, Gibson WT, Kieffer TJ. Acute disruption of leptin signaling in vivo leads to increased insulin levels and insulin resistance. Endocrinology. 2011;152:3385–3395. doi: 10.1210/en.2011-0185. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nature genetics. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. The Journal of biological chemistry. 2006;281:18933–18941. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, Zhang ZY, Gettys TW. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology. 2007;148:433–440. doi: 10.1210/en.2006-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PY, Janovjak H, Miserez AR, Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. BioTechniques. 2002;32:1372–1374. 1376, 1378–1379. [PubMed] [Google Scholar]
- Muller TD, Sullivan LM, Habegger K, Yi CX, Kabra D, Grant E, Ottaway N, Krishna R, Holland J, Hembree J, et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. Journal of peptide science : an official publication of the European Peptide Society. 2012;18:383–393. doi: 10.1002/psc.2408. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–2131. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annual review of physiology. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, Tschop MH, Yanovski JA. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012;15:150–156. doi: 10.1016/j.cmet.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AJ, Hess S, Paeger L, Vogt MC, Fleming Lascano J, Nillni EA, Bruning JC, Kloppenburg P, Xu AW. AgRP innervation onto POMC neurons increases with age and is accelerated with chronic high-fat feeding in male mice. Endocrinology. 2013;154:172–183. doi: 10.1210/en.2012-1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, Anderson CM, Parkes DG, Baron AD. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7257–7262. doi: 10.1073/pnas.0706473105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Moore RL, Tumer N. Impaired leptin responsiveness in aged rats. Diabetes. 2000;49:431–435. doi: 10.2337/diabetes.49.3.431. [DOI] [PubMed] [Google Scholar]
- Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW. Melanocortin receptors in leptin effects. Nature. 1997;390:349. doi: 10.1038/37016. [DOI] [PubMed] [Google Scholar]
- Shpilman M, Niv-Spector L, Katz M, Varol C, Solomon G, Ayalon-Soffer M, Boder E, Halpern Z, Elinav E, Gertler A. Development and characterization of high affinity leptins and leptin antagonists. The Journal of biological chemistry. 2011;286:4429–4442. doi: 10.1074/jbc.M110.196402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon G, Atkins A, Shahar R, Gertler A, Monsonego-Ornan E. Effect of peripherally administered leptin antagonist on whole body metabolism and bone microarchitecture and biomechanical properties in the mouse. American journal of physiology Endocrinology and metabolism. 2014;306:E14–27. doi: 10.1152/ajpendo.00155.2013. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Hidaka S, Masuzaki H, Yasue S, Minokoshi Y, Ebihara K, Chusho H, Ogawa Y, Toyoda T, Sato K, et al. Skeletal muscle AMP-activated protein kinase phosphorylation parallels metabolic phenotype in leptin transgenic mice under dietary modification. Diabetes. 2005;54:2365–2374. doi: 10.2337/diabetes.54.8.2365. [DOI] [PubMed] [Google Scholar]
- Tumer N, Erdos B, Matheny M, Cudykier I, Scarpace PJ. Leptin antagonist reverses hypertension caused by leptin overexpression, but fails to normalize obesity-related hypertension. Journal of hypertension. 2007;25:2471–2478. doi: 10.1097/HJH.0b013e3282e9a9fd. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.