

Abstract
PTPN11 encodes the Shp2 non-receptor protein-tyrosine phosphatase implicated in several signaling pathways. Activating mutations in Shp2 are commonly associated with juvenile myelomonocytic leukemia (JMML) but are not as well defined in other neoplasms. Here we report that Shp2 mutations occur in human acute myeloid leukemia (AML) at a rate of 6.6% (6/91) in the ECOG E1900 dataset. We examined the role of mutated Shp2 in leukemias harboring MLL translocations which co-occur in human AML. The hyperactive Shp2E76K mutant, commonly observed in leukemia patients, significantly accelerated MLL-AF9 mediated leukemogenesis in vivo. Shp2E76K increased leukemic stem cell frequency and affords MLL-AF9 leukemic cells IL3 cytokine hypersensitivity. As Shp2 is reported to regulate anti-apoptotic genes, we investigated Bcl2, Bcl-xL and Mcl1 expression in MLL-AF9 leukemic cells with and without Shp2E76K. While the Bcl2 family of genes was upregulated in Shp2E76K cells, Mcl1 showed the highest upregulation in MLL-AF9 cells in response to Shp2E76K. Indeed, expression of Mcl1 in MLL-AF9 cells phenocopies expression of Shp2E76K suggesting Shp2 mutations cooperate through activation of anti-apoptotic genes. Finally, we show Shp2E76K mutations reduce sensitivity of AML cells to small molecule mediated Mcl1 inhibition suggesting reduced efficacy of drugs targeting MCL1 in patients with hyperactive Shp2.
Introduction
MLL rearrangements are present in ~20% of pediatric AML and can be as high as 80% of infant patients with ALL (1) and are generally associated with a poor outcome (2). Rearrangements of the MLL locus generate potent oncogenic fusion proteins that retain the N-terminus of the MLL protein but replace the C-terminus with one of >60 different partner proteins that can recruit transcriptional activation complexes (3–6). The resultant deregulated transcriptional activation mediated by MLL fusion proteins blocks hematopoietic differentiation through the sustained expression of the posterior HOXA gene cluster, namely HOXA9 (7). Interestingly, MLL leukemias display a relatively stable genome compared with other leukemic subtypes but still carry other genetic lesions at low frequency (8). Type-I mutations involving the Ras pathway are present in about 37% of MLL rearranged leukemias including mutations within NRAS, KRAS, NF1 and PTPN11 (9), consistent with the idea that pathological AML requires both type-I and type-II mutations (10). Indeed, oncogenic NRASG12V or FLT3-ITD can significantly accelerate MLL fusion protein mediated leukemogenesis in vivo (11–13). Although these mutations strongly cooperate with MLL fusion proteins to promote leukemogenesis, little is understood about the molecular mechanisms utilized by type-I mutations.
PTPN11 encodes the ubiquitously expressed SHP2 non-receptor protein tyrosine phosphatase involved in the RAS, JAK-STAT, PI3K and other pathways (14, 15). Mutations in PTPN11 are found in ~50% of patients with Noonan syndrome, as well as, ~37% of patients with hematologic malignancies such as juvenile myelomonocytic leukemia (JMML), ALL and AML (16–19). Recent genome-wide sequencing analyses have identified PTPN11 mutations in AML patients indicating this may function in a cooperative manner (20, 21). Shp2 positively regulates signal transduction pathways downstream of receptor tyrosine kinases, like Kit, where it is essential for hematopoietic stem and progenitor cells (22, 23). Hematopoietic progenitors require Shp2 for STAT5 activation and upregulation of Mcl1 and Bcl-xL (24, 25). In leukemia expression is often elevated and Shp2 can associate with FLT3-ITD leading to activation of STAT5. Shp2 co-localizes with STAT5 to activate expression of Bcl-xL protecting against cell death (26, 27). PTPN11 mutations result in amino acid changes resulting in disrupted autoinhibition and hyperactive Shp2 enzymatic activity (17, 28–30). Gain of function mutations in Shp2 result in cytokine hypersensitivity in hematopoietic progenitor cells (31). In mice, gain of function Shp2 mutations leads to a JMML-like fatal myeloproliferative disease whereas an inducible mutant Shp2 knock-in mouse model progresses to AML, as well as, B and T cell ALL with long disease latency (32–35). However, the molecular mechanisms leading to disease and the cooperative nature of hyperactive Shp2 with leukemic fusion proteins has not been explored.
To investigate whether mutations associated with PTPN11 can cooperate with oncogenic fusion proteins, we developed a mouse model of cooperative leukemogenesis with MLL-AF9 and the leukemia-associated Shp2E76K mutant that shows the highest basal phosphatase activity among all the disease-associated Shp2 mutations (17, 36). Shp2E76K strongly cooperates with MLL-AF9 to accelerate leukemogenesis in mice by altering leukemic stem cell frequency. MLL-AF9 Shp2E76K cells display cytokine hypersensitivity and activation of the Erk pathway leading to upregulation of an anti-apoptotic gene program most prominently observed with Mcl1. We find that Shp2E76K expression in both mouse and human cells reduces MLL-AF9 sensitivity to chemical inhibition of Mcl1 suggesting mutant Shp2 cooperates mechanistically with MLL fusion proteins through Mcl1 expression.
Materials and Methods
Mice
Female C57BL/6 mice at 8 to 10 weeks old were purchased from Taconic Farms. B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/J mice (TetOn mice) were purchased from Jackson laboratory. All animal studies were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Medicine.
Genetic analysis of primary patient AML samples
All primary patient samples came from pretreatment AML blood or bone marrow samples from patients treated on the ECOG E1900 clinical trial as previously described (37). All patient samples were previously evaluated for cytogenetic abnormalities and sequenced for the following genes recurrently mutated in AML: ASXL1, CEBPα, CKIT, DNMT3A, EZH2, FLT3, NPM1, HRAS, IDH1, IDH2, KRAS, NRAS, PHF6, WT1, RUNX1, TET2, and TP53 (37). For sequencing of PTPN11, all coding regions of PTPN11 were amplified using RainDance microdroplet digital PCR enrichment (RainDance, Billerica, MA) as previously described (38, 39) followed by followed by Illumina HiSeq massively parallel sequencing. Primer sequences are available upon request.
Plasmids
MSCVpuro-Flag-mMcl1 plasmid was purchased from Addgene (32982). MSCVneo-Flag-MLL-AF9 and MSCVneo-E2A-HLF plasmids have been described before (40). MIEG3 (retroviral vector utilizing the bi-cistronic MSCV backbone and expressing enhanced GFP) and MIEG3-Shp2E76K plasmids were provided by Dr. Rebecca Chan (Indiana University) (41). shRNA plasmids were generated by inserting validated shRNA sequences into the XhoI and EcoRI sites of the pTRMPV-Hygro vector (42). Mcl1 shRNAs and Renilla control shRNA sequences were described previously (43). All shRNA sequences were listed in Supplementary Table 1.
shRNA knockdown
Hematopoietic stem cells purified from TetOn mice were transformed with MLL-AF9 retrovirus. Freshly transformed (~2 weeks after transduction) cells were transduced with MIEG3 or MIEG3-Shp2E76K and sorted by GFP expression. GFP+ cells were transduced with Plat-E packaged shRNA retroviruses. Cells were selected with hygromycin for 1 week (140ug/ml for 5 days and 200μg/ml for 2 days) with fresh antibiotics added every 1 to 2 days. ShRNA containing cells were mixed with shRNA-none cells (i.e. TetOn MA9+MIEG3 or TetOn MA9+E76K) at 3:1 ratio and cultured in media containing 1μg/ml Doxcycline. 1 day after Doxcycline treatment, cells were harvested for flow cytometry to detect dsRed+ cell percentage in the mixed population. ShRNA knockdown after doxycycline treatment were verified by western blotting.
Chemical inhibition assay
All chemical inhibitors were dissolved in DMSO. Cells were cultured at 5×104 cells/ml in 12-well non-tissue culture plate in the presence of serially diluted Inhibitors. ABT-263 was added at 0.1, 0.33, 1, 2.5, 5μM. UMI-205, UMI-77, UMI-208, and UMI-212 were added at 1.25, 2.5, 5, 10, 20μM. DMSO was used as negative control. Cells were enumerated after 48 hours of culture and the growth rate k was calculated according to the exponential growth equation:
Additional methods can be found in Supplemental materials
Results
PTPN11 mutations functionally cooperate with specific oncogenes in AML
To gain insight into the mutational frequency of SHP2 in human AML, we sequenced PTPN11 by next-generation sequencing in 91 AML patients enrolled in the phase III clinical trial run by ECOG E1900 (47). Six of 91 patients were identified to contain a mutation in PTPN11 resulting in a 6.6% mutational frequency in AML; consistent with a saturation analysis of cancer genes in AML (20) suggesting PTPN11 mutations are functionally important to leukemic transformation (Table 1). All mutations were detected within exon 3 and resulted in amino acid changes previously associated with both AML and JMML (15, 17). The E76K, T31I, E76Q, D61Y, F71L and E76G mutations all reside within an auto-inhibitory region of the SHP2 protein and disrupts the N-SH2 interaction with the PTP domain leading to hyperactive phosphatase activity (16, 28). PTPN11 mutations were found to co-occur with genetic alterations in DNMT3a, NPM1, WT1, CBF and MLL suggesting mutant SHP2 may cooperate functionally in AML. As PTPN11 mutations were previously reported in MLL associated leukemias (19), we tested whether hyperactive Shp2 can functionally cooperate with MLL fusion proteins in transformation. We performed primary bone marrow transduction and colony replating assays using retroviral vectors containing the oncogenic MSCV-MLL-AF9 (MA9) fusion protein and the hyperactive Shp2 mutant MIEG3-Shp2E76K (E76K) commonly associated with hematologic malignancies. While expression of E76K alone did not lead to significant replating potential indicative of cellular transformation, the E76K mutant significantly augmented MLL-AF9 mediated colony formation as evidenced by a two-fold increase in colony formation in the second round compared to MLL-AF9 and empty MIEG3 (Figure 1A and B). Colony formation between MA9 and MA9+E76K was similar in tertiary plating that may reflect more efficient transformation or limitations in quantifying CFU potential in the absence of limiting dilutions. MA9 and MA9+E76K colonies were dense and composed of immature blast-like cells with a greater nuclear to cytoplasmic ratio compared to the diffuse colonies composed of macrophage type cells resulting from transduction with empty vectors or E76K alone (Figure 1B). Notably, while MLL-AF9 colonies were composed almost exclusively of primitive blast-like cells, MLL-AF9 and E76K co-transduced cells consistently contained sporadic differentiating macrophages (Figure 1B). To examine cooperation in other leukemic subtypes, we tested E76K expression in the presence of the unrelated leukemic oncoprotein E2A-HLF (EH) that transforms cells through an anti-apoptotic gene program, which is distinct from the HOX gene program used by MLL fusion proteins (48). Interestingly, we did not observe a difference in E2A-HLF mediated colony replating capacity in the presence of E76K (Figure 1A and B). Expression of Shp2E76K was confirmed by qPCR using Shp2 specific primer sets and RNA isolated after the first round of the colony assay (Figure 1C). Together, these data identify PTPN11 mutations as co-occurring with several common hematologic genetic alterations and functionally cooperating with MLL-AF9 mediated transformation in vitro.
Table 1.
PTPN11 mutations associated with leukemia patients from the phase III clinical trial run by ECOG E1900 (47).
| PTPN11 mutation details | Consensus Cytogenetic Risk | dfs | efs status | efs | os status | ||
|---|---|---|---|---|---|---|---|
| Genomic coordinates | Nucleotide change | AA change | |||||
| 12:112888211 | A/G | E76G | Intermediate I | 1205 | 1 | 1244 | 1 |
| 12:112888202 | C/T | T73I | Indeterminate | 352 | 1 | 385 | 1 |
| 12:112888210 | G/C | E76Q | Intermediate II | 1 | 0 | 1 | |
| 12:112888165 | G/T | D61Y | Favorable | 2114 | 0 | 2172 | 0 |
| 12:112888197 | T/G | F71L | Indeterminate | 1401 | 0 | 1429 | 0 |
| 12:112888211 | A/G | E76G | Unfavorable | 56 | 1 | 82 | 1 |
Figure 1.

Shp2E76K increases leukemia colony formation by MLL-AF9 in vitro. (A) Hematopoietic progenitor cells were transduced with empty MSCV, MIEG3 or MSCV-F-MLL-AF9 (MA9), MSCV-E2A-HLF (EH) and MIEG3-Shp2E76K (E76K) and plated in methylcellulose media for three rounds of 7 day culture. Data shows the average and standard deviation from three independent experiments with duplicated samples normalized to the highest colony formation. Data indicates cooperation between Shp2E76K and MLL-AF9 but not E2A-HLF. (* p<0.05; t-test) (B) Representative colonies after the second round of the methylcellulose colony assays were pictured using a 2x lens on an Olympus IX71 microscope (Camera: DP73; objective lens: Plan N 2x/0.06; software: cellSens Entry 1.7)(scale bar = 500μm). Images illustrate increased colony formation specifically in MLL-AF9 transduced cells with Shp2E76K. Cell morphology is obtained by staining cytospun cells following the second round of the colony assay. Cells are imaged using a 100x lens on an Olympus BX41 microscope (Camera: DP71; objective lens: 100x/1.30; software: DP Controller)(scale bars = 50μm). (C) Overexpression of Ptpn11 is confirmed from the colony assay described in (A) by real-time qPCR following RNA extraction from cells collected after the first round of the colony assay. Expression is shown relative to β-actin expression.
Hyperactive Shp2 accelerates MLL-AF9 leukemia and alters leukemic stem cell frequency
We next assessed how Shp2 mutations affected MA9 mediated leukemogenesis in vivo. Here we introduced either empty vectors, MSCV+E76K, MA9+MIEG3 or MA9+E76K into lin−c-kit+ bone marrow cells by retroviral transduction. Cells were i.v. injected into lethally irradiated syngeneic recipients and allowed to engraft. Mice receiving cells transduced with empty vectors or MSCV+E76K did not show signs of disease through 150 days suggesting overexpression of E76K alone failed to induce a lethal leukemia (Figure 2A). On the contrary, overexpression of E76K significantly accelerated MA9 mediated leukemogenesis resulting in decreased disease latency (p=0.0004, Log-Rank). The median survival of mice receiving MA9+MIEG3 cells (125 days) was more than double the median survival of mice receiving MA9+E76K (60 days) (Figure 2A). Mice were monitored for 150 days with the first moribund MA9+E76K mouse euthanized on day 38 and the first MA9+MIEG3 mouse euthanized on day 103. MA9+MIEG3 or MA9+E76K mice displayed splenomegaly (Figure 2B) and compromised organ structure in the spleen and liver (Figure 2C). Histopathology revealed infiltrating leukemic blasts in the spleen and liver while bone marrow aspirates revealed blocked myeloid differentiation in diseased MA9 and MA9+E76K mice (Figure 2C). Overexpression of Shp2E76K was confirmed by western blotting splenocytes from MA9 and MA9+E76K diseased mice (Figure 2D). It is noteworthy that MA9+E76K mice with longer disease latencies also showed the lowest expression of Shp2 by western blot (Figure 2D). A correlation between Shp2E76K expression level and disease latency was established by plotting β-Actin normalized Shp2 protein expression levels against disease latency (R2 = 0.90367) (Figure 2D). Overexpression of Ptpn11 was also confirmed by qPCR using cDNA from diseased splenocytes (Figure 2E). Remarkably, we did not detect a difference in the expression of the direct MLL-AF9 target genes Hoxa9 and Meis1 suggesting the difference in disease latency is not due to a change in the MLL-AF9 gene program induced by Shp2E76K (Figure 2E). To follow up on the difference in cell morphology observed in colony assays with MA9 or MA9+E76K (Figure 1B), we performed flow cytometry on splenocytes. Cell surface expression of Sca1 and c-kit was similar in both MA9 and MA9+E76K cells (Figure 2F). However, expression of both Cd11b and Gr1 were significantly higher in MA9+E76K cells compared to MA9 cells, consistent with a slightly more differentiated phenotype (Figure 2F). These data are consistent with a role of Shp2 in promoting hematopoietic differentiation through dephosphorylation of Runx1 (49). To understand if accelerated leukemogenesis in the presence of E76K was due to altered leukemic stem cell (LSC) frequency, we performed a limiting dilution assay with primary leukemic cells isolated from MA9 and MA9+E76K diseased mice. These were injected into secondary recipients following sublethal irradiation and monitored for 109 days for disease. While MA9 cells displayed an LSC frequency of ~1 in 285, the presence of E76K significantly increased LSC frequency to ~1 in 50 (Figure 2G). These data suggest Shp2E76K cooperates with the MLL-AF9 fusion protein in vivo to accelerate leukemogenesis by altering LSC frequency.
Figure 2.

Shp2E76K accelerates leukemia development by MLL-AF9 in vivo. (A) Survival of mice transplanted with cells transduced with MSCV+MIEG3 (n=4), MSCV+E76K (n=4), MA9+MIEG3 (n=11, median survival = 125 days), MA9+E76K (n=11, median survival = 60 days). MA9+E76K accelerates leukemogenesis compared to MA9+MIEG3 (p=0.0004, Log-Rank) (B) Splenomegaly is observed in leukemic mice injected with MA9+MIEG3 or MA9+E76K. Spleen weights for diseased mice are shown in the panel on the right. (C) Spleen, liver and bone marrow samples were collected from diseased MA9+MIEG3 and MA9+E76K mice and censored MSCV+MIEG3 and MSCV+E76K mice. Tissue sections or cytospun bone marrow was stained by H&E and imaged with an Olympus BX41 microscope (Camera: DP71; objective lens: 10x/0.3 for spleen, 100x/1.30 for liver and bone marrow; software: DP Controller) (scale bar = 500μm for the spleen and 50μm for liver and bone marrow images). Infiltrating leukemic blasts are evident in MA9 and MA9+E76K mice. (D) Top: Expression of Shp2 in mouse splenocytes was detected by western blot. Samples are numbered in chronological order of detection of disease. Bottom: β-Actin normalized Shp2 protein expression is plotted relative to disease latency. (E) Expression of Ptpn11, Hoxa9 and Meis1 detected by real-time PCR. (* p< 0.05; t-test) (F) Mean fluorescence intensity (MFI) of Sca1, c-Kit, Cd11b and Gr1 on MA9+MIEG3 and MA9+E76K splenocytes detected by flow cytometry. (* p< 0.05; t-test). In (B), (E) and (F), median value and error bar stands for interquartile range are shown with each dot representing one mouse. (G) MA9+MIEG3, MA9+E76K stem cell frequency determined by serially diluted secondary transplantation. 5 cells (n=10), 20 cells (n=5), 50 cells (n=10), 200 cells (n=10) and 1000 cells (n=5) from diseased primary mice were injected by i.v. into sublethally irradiated C57bl/6 syngeneic recipients. Figures and stem cell frequency were generated by ELDA (46).
Shp2E76K affords MLL-AF9 cells cytokine hypersensitivity
To investigate the mechanism of accelerated leukemogenesis, we generated cell lines with MA9 or MA9+E76K by retroviral transduction of lin− c-kit+ bone marrow cells. We enriched for cells transduced with empty MIEG3 or E76K by sorting for GFP, which revealed an expansion of bone marrow cells expressing E76K that was evident four days after transduction (Supplementary Figure 1). Proliferation assays demonstrated the E76K mediated bone marrow expansion was transient except in the presence of MLL-AF9 which initiated leukemic transformation (Figure 3A). Overexpression of mutant Shp2 was confirmed by western blot (Figure 3B). Previous studies had implicated hyperactive Shp2 in hematopoietic progenitor cell cytokine hypersensitivity (31, 41). We tested this in the context of MLL-AF9 leukemic cells by performing an IL3 withdrawal and proliferation assay. We observed increased tolerance for IL3 withdrawal in MA9+E76K cells compared to MA9 (Figure 3C). This was also accompanied by increased Erk phosphorylation in the absence of IL3. MA9+E76K cells displayed higher resting levels of Erk phosphorylation with limiting amounts of IL3 and showed greater Erk phosphorylation in response to IL3 compared to MA9 cells (Figure 3D).
Figure 3.

Shp2E76K induces cytokine hypersensitivity in MLL-AF9 leukemic cells. (A) Lin-c-kit+ bone marrow cells were transduced with the indicated retroviruses and selected. Cells were grown in liquid culture and counted every other day. The total number of cells is plotted. (B) Shp2 expression in MA9+MIEG3, MA9+E76K cell lines are detected by western blot following whole cell lysis. (C) MA9+MIEG3 and MA9+E76K cells were cultured in serially diluted IL3 for 3 days. The growth rate was calculated and plotted showing cytokine hypersensitivity by MA9+E76K cells. (* p<0.0001; 2-way Anova; biological duplicates). (D) Western blotting was performed for total Erk and p-Erk in MA9+MIEG3 and MA9+E76K cells. Cells were cultured in serum free OPTI-MEM media overnight before a pulse with different doses of IL3 for 10 minutes.
Several studies have linked Shp2 activity with the upregulation of anti-apoptotic genes (24, 27). Thus, we examined expression of both the Bcl2 family of genes and direct MLL fusion targets by qPCR in MA9 and MA9+E76K cells to reveal the mechanism for Shp2E76K mediated accelerated leukemogenesis and cytokine hypersensitivity. We found the expression of Hoxa9 and Meis1 not significantly changed in the presence of Shp2E76K suggesting modulation of MLL targets genes is not responsible for cytokine hypersensitivity (Figure 4A). In contrast, we observed a significant upregulation of Bcl-xL and Mcl1 in MA9+E76K cells compared to MA9 cells in the presence of low dose IL3 (0.001ng/ml) (Figure 4A). The most significant increase was observed in Mcl1 mRNA expression with a ~4 fold upregulation, which was confirmed by western blot (Figure 4B). This is consistent with genetic analyses that identified Mcl1 as a key regulator of cell survival in MLL-ENL leukemic cells compared to Bcl2 or Bcl-xL (43). Interestingly, there were only minor changes in Hoxa9, Mcl1, Bcl2 or Bcl-xL expression when Shp2E76K was over-expressed in normal lin− bone marrow suggesting a context dependent function for hyperactive Shp2 (Supplemental Figure 2). While all anti-apoptotic genes were upregulated in the presence of Shp2E76K, Mcl1 clearly emerged as the most highly expressed and upregulated in MA9+E76K cells (Figure 4A). These data suggest that the Shp2E76K mutation may accelerate leukemogenesis through the upregulation of an anti-apoptotic gene program primarily mediated by Mcl1.
Figure 4.
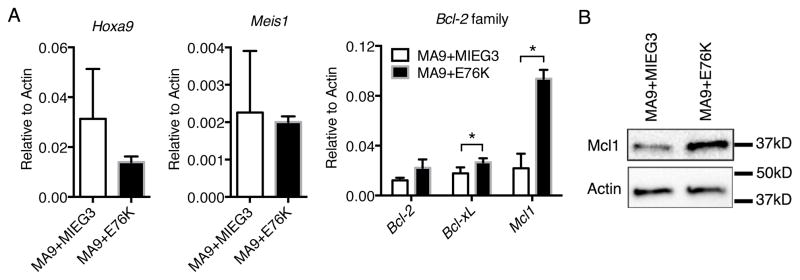
Shp2E76K up-regulates anti-apoptotic genes in MLL-AF9 cells. (A) Cells were cultured with 0.001ng/ml IL3 and Bcl2, Bcl-xL, and Mcl1 expression is shown for MA9+MIEG3 and MA9+E76K cells relative to Actin (* for Bcl2 p=0.066, for Bcl-xL p=0.0241, for Mcl1 p=0.0067). Average transcript expression and standard deviation from biological duplicate experiments is shown relative to β–actin. (B) MA9+MIEG3 and MA9+E76K cells were cultured with 10ng/ml IL3 and Mcl1 expression was detected by western blotting.
Mcl1 expression recapitulates Shp2E76K mediated cellular effects
Because Mcl1 expression was notably higher than Bcl2 and Bcl-xL, we asked whether expression of Mcl1 alone would recapitulate the effects seen by Shp2E76K in MLL-AF9 cells. To this end, we used an MSCV-puro based Mcl1 retroviral vector for co-transduction of lin− c-kit+ bone marrow cells along with MSCV-neo-MLL-AF9 and subsequent colony forming and replating assays. Similar to what was seen with co-transduction with Shp2E76K, a significant increase in colony formation was observed in the second round of the colony assay following co-transduction of MA9 and Mcl1 compared to MA9 and empty vector and was evident by visual inspection of the plates (Figure 5A). Transduction with Mcl1 alone resulted in a similar number of colonies as empty vector transduced cells and did not lead to replating capacity in the second and third round suggesting Mcl1 overexpression alone is insufficient for transformation (Figure 5A). Stable cell lines were established from lin− c-kit+ bone marrow cells following transduction with MA9+MSCVpuro or MA9+Mcl1. To test the effect of Mcl1 expression on cytokine hypersensitivity, we tested proliferation rates in serially diluted concentrations of IL3. Overexpression of Mcl1 resulted in a marked proliferative advantage in lower doses of IL3 (Figure 5B). We next examined how Mcl1 expression affected colony forming unit (CFU) frequency by performing a limiting dilution colony assay with MA9 and MA9+Mcl1. Strikingly, we observed a significantly higher frequency of CFUs per cell plated in MA9+Mcl1 (1/13.32) compared to MA9 alone (1/79.11) (Figure 5C). Over-expression of Mcl1 was confirmed by western in Figure 5D. Together, these data show expression of Mcl1 results in increased colony formation, cytokine hypersensitivity and increased CFU frequency suggesting the cellular effects mediated by Shp2E76K may be mediated by expression of Mcl1.
Figure 5.

Mcl1 partially phenocopies Shp2E76K in MLL-AF9 cells. (A) Hematopoietic progenitor cells were transduced with empty vectors, Mcl1 alone, MA9 alone or MA9+Mcl1 and plated in methylcellulose. Colonies were counted after 7 days and replated for a total of three rounds. Colonies numbers are shown relative to the highest number of colonies observed. Error bars indicate standard deviation of biological duplicate experiments. (* p<0.05; t-test). Plates were stained with INT and imaged with a ChemiDoc XRS+ Imaging System after the 3rd round. Data represents average of 3 independent experiments. (B) MA9+MSCVpuro and MA9+Mcl1 cells were cultured in serially diluted doses of IL3. Cells were counted after three days and growth rates are plotted. Data shows cytokine hypersensitivity with the overexpression of Mcl1. (* p<0.0001; 2-way Anova; biological duplicates). (C) Limiting dilution colony formation assays were performed with MA9+MIEG3 and MA9+E76K cells. The average number of colonies from biological duplicate experiments after 7 days in culture is plotted against seeding cell number with the dashed line representing a 95% confidence interval. CFU potential is determined by 1/slope. (D) Mcl1 overexpression was detected by western blotting.
AML cells with PTPN11 mutations are desensitized to Mcl1 inhibition
To further examine the role of anti-apoptotic proteins in MA9+E76K cells, we treated established mouse cell lines with chemical inhibitors with specificity towards different anti-apoptotic proteins. Here, the growth rate of MA9+MIEG3, MA9+E76K, MA9+Mcl1 and E2A-HLF cells was assessed following treatment with varying concentrations of ABT-263, UMI-212 or UMI-205. ABT-263 is a cell permeable BH3 mimetic chemical inhibitor that disrupts Bcl2 and Bcl-xL interactions with pro-death proteins like Bim (50). UMI-212 is a novel cell permeable chemical inhibitor that specifically targets Mcl1, without affecting Bcl2 or Bcl-xL, and UMI-205 is a control non-targeting compound (51, 52). As expected, no change in cell growth was observed in any cell lines following treatment of cells with UMI-205 (Figure 6A). In contrast, treatment of cells with ABT-263 resulted in decreased growth of E2A-HLF cells without affecting the growth of MA9+MIEG3, MA9+E76K or MA9+Mcl1 cells (Figure 6A). E2A-HLF cells showed an IC50 value of 1.80μM compared to >5μM for MA9+MIEG3, MA9+E76K and MA9+Mcl1 (Supplementary Table 3). These data suggest MLL fusion protein transformed cells are less sensitive to Bcl2 inhibition compared to E2A-HLF cells and are consistent with previous work implicating Bcl2 in E2A-HLF mediated leukemic transformation (48). On the contrary, MA9 cells were more sensitive to UMI-212 mediated Mcl1 inhibition compared to E2A-HLF cells (Figure 6A). The sensitivity of MA9 cells to UMI-212 was reduced with the addition of E76K such that the growth pattern was similar to E2A-HLF cells. These data are consistent with decreased cellular sensitivity to UMI-212 due to the upregulation of an anti-apoptotic program, most notably Mcl1, by E76K. In addition, direct overexpression of Mcl1 in MA9+Mcl1 cells led to less sensitivity to UMI-212 similar to expression of E76K (Figure 6A, Supplementary Table 3). MA9 cells have an IC50 of 5.197μM compared to 8.389μM, 8.711μM and 10.76μM for E2A-HLF, MA9+E76K and MA9+Mcl1 cells respectively (Supplementary Table 3). Treatment with two additional chemical inhibitors with Mcl1 specificity, MI-77 and MI-228, yielded comparable results (Supplementary Figure 3). To test if inhibition of Mcl1 led to programmed cell death, we examined cell viability by annexin V and DAPI staining following treatment with control UMI-205, ABT-263 or UMI-212. While UMI-205 did not affect cell viability, treatment with UMI-212 led to an increase in annexin V positivity indicative of programmed cell death (Figure 6B, and Supplementary Figure 4). While not statistically significant in MA9+E76K cells, a trend of decreased annexin V positivity was observed in MA9+E76K and MA9+Mcl1 cells compared to MA9 cells consistent with the above growth curves and Mcl1 expression, suggesting leukemic cells with Shp2 mutations are less responsive to UMI-212 mediated Mcl1 inhibition (Figure 6A and B, Supplementary Figure 3, and 4). These data implicate Mcl1 as a crucial mediator of MLL-AF9 cell proliferation that is significantly modulated by Shp2E76K rendering leukemic cells less sensitive to Mcl1 inhibition.
Figure 6.

Shp2E76K desensitizes MLL-AF9 cells to Mcl1 inhibition. (A) Growth rate of E2A-HLF, MA9+MIEG3, MA9+E76K, MA9+Mcl1 cells was determined after treatment with DMSO carrier or ABT-263, UMI-205, UMI-212 chemical inhibitors. Average growth rates from biological duplicate experiments are normalized to DMSO control and shown with standard deviation after 48 hours of chemical treatment and plotted against concentration with a fitted sigmoidal curve. Data indicates ABT-263 preferentially inhibits E2A-HLF while UMI-212 mediated Mcl1 inhibition preferentially inhibits MLL-AF9 cell growth, which is mitigated by Shp2E76K. (2-away Anova, * p<0.05, **p<0.01, ****p<0.0001) (B) Quantified flow cytometry data from cells co-stained with AnnexinV and DAPI. Data shows increased apoptosis with chemical inhibition of Mcl1 or Bcl2/Bcl-xL which is lessoned by Shp2E76K. Statistical analysis was performed by 2-way Anova (** p<0.01, *** p<0.001; **** p<0.0001, ns= Not Significant) (C) MA9+MIEG3, MA9+E76K cells were transduced with shRNA retroviruses targeting Mcl1 and selected with Hygromycin for 1 week before performing a competitive growth assay in the presence of Doxycycline to induce shRNA expression. Average dsRed+ population percentages with standard deviation were determined by flow cytometry after 1 day. Data is plotted relative to cells transduced with control shRNA targeting renilla (Ren sh). (D) Mcl1 knockdown was verified by western blot and the relative protein level was quantified with image lab software (BioRad).
To verify the desensitization of MA9+E76K cells to UMI-212 was mediated by Mcl1 overexpression, we engineered inducible pTRMPV retroviral vectors to express validated shRNA targeting Mcl1 or control Renilla (Ren) to allow for gene specific knockdown in leukemic cells. Freshly established TetOn MA9+MIEG3 and MA9+E76K cell lines were infected with the shRNA retroviruses and selected for 1 week. ShRNA containing cells were then mixed with parental cell lines lacking either pTRMPV vector at a 3:1 ratio and cultured with Doxycycline to induce shRNAs detectable by dsRed expression and flow cytometry. Consistent with previous data implicating Mcl1 gene expression in the survival of MLL leukemic cells (43), knockdown of Mcl1 with two separate shRNA vectors in MA9 cells led to a significantly reduced dsRed+ population in comparison to Ren controls (Figure 6C). Similarly, knockdown of Mcl1 in MA9+E76K cells lead to reduced dsRed+ population (Figure 6C). Knockdown of Mcl1 was confirmed by western 24 hours after Doxycycline treatment (Figure 6D).
We next tested how a variety of human AML cells respond to Mcl1 inhibition. To this end, we sequenced several human AML cell lines to determine the mutational status of exon 3 of PTPN11. Among the cell lines tested, only U937 cells contained a PTPN11 mutation (G60R) (Supplemental Figure 5). To determine how human leukemia cells containing a PTPN11 mutation would respond to anti-apoptotic protein inhibition, we treated the PTPN11 mutant U937 cell line and the PTPN11 wild type cell lines K562, Monomac6, THP-1 and ML2 with ABT-263, UMI-212 or UMI-205. While none of the cell lines showed a change in growth response to the control UMI-205 compound, U937 and THP1 cells were clearly more resistant to UMI-212 mediated Mcl1 inhibition compared to K562, MonoMac6 and ML2 cells (Figure 7A). U937 cells had an IC50 of 17.92μM compared to IC50 values of 9.72μM and 7.39μM for K562 and ML2 cells and 10.84μM and 15.63μM for MonoMac6 and THP1 cells (Supplementary Table 3). ML2 and THP1 cells showed slight sensitivity to ABT-263 mediated Bcl2/Bcl-xL inhibition (Figure 7A). These results were confirmed with additional Mcl1 specific inhibitors, which confirmed U937 desensitization to Mcl1 inhibition (Supplementary Figure 6). While MCL1 expression varied between cell lines, colony forming ability generally decreased in a dose dependent fashion with MCL1 inhibition (Supplementary Figure 7A, B). U937, K562 and THP1 cells displayed greater resistance to MCL1 inhibition compared to ML2 and MonoMac6 cells (Supplementary Figure 7A). The increased resistance of THP1 and K562 cells along with U937 cells in proliferation and colony assays likely reflect the utility of multiple transformation mechanisms that may compensate for loss of MCL1. Of note, THP1 cells harbor activating mutations in NRAS that may contribute to increased MCL1 expression. Despite these cell lines carrying a variety of driver mutations, including CALM-AF10, BCR-ABL, MLL-AF9 and MLL-AF6, these data suggest that a Shp2 mutation in human AML cells may result in greater resistance to MCL1 inhibition.
Figure 7.

Human leukemia cell lines with activated SHP2 mutations show resistance to Mcl1 inhibitors. (A) The growth rate of K562, U937, THP-1, Monomac6 and ML2 cells was determined following culture with UMI-205 (negative control), ABT-263 (Bcl-2/Bcl-xL inhibitor), and UMI-212 (Mcl1 inhibitor) and plotted against concentration with a fitted sigmoidal curve. Averaged data with standard deviation are shown for biological duplicate experiments normalized to DMSO treatment indicates the Shp2 mutation in U937 cells leads to greater resistance to Mcl1 inhibition. * indicates statistical significance (p<0.05) between the indicated cell line and the U937 cell line (2-way ANOVA) (B) Model of mechanistic cooperation between MLL-AF9 and Shp2E76K. A pre-leukemic clone containing an MLL-AF9 fusion protein displays blocked differentiation and increased self-renewal through upregulation of Hoxa9 and Meis1. A second genetic lesion involving a gain of function (GOF) mutation to PTPN11 leads to increased expression of Mcl1 which increases leukemic stem cell (green) frequency and cell survival.
Discussion
Cooperative leukemia
Recent deep sequencing efforts have revealed a number of mutations that together with previously identified chromosomal abnormalities give a clearer picture of the genetic landscape of acute leukemias. The data presented currently provides an experimental validation of the premise proposed by Gilliland and others that complete leukemogenesis require mutations conferring a block in differentiation and promoting cell survival (10). Although mouse models of both MLL-AF9 and Shp2E76K result in lethal leukemias (35, 53, 54), the long latencies of these diseases suggest cooperating events contribute to the disease. The presence of an MLL fusion protein, like MLL-AF9, leads to a block in hematopoietic differentiation through the sustained expression of Hoxa9 and Meis1. The addition of PTPN11 gain of function mutations lead to functional cooperation between the MLL-AF9 fusion protein and mutated Shp2 that results in significantly accelerated leukemogenesis by increasing LSC frequency through upregulation of an anti-apoptotic gene program that primarily includes Mcl1 (Figure 7B). Upregulation of the Bcl2 family, namely Mcl1, induced by activating mutations in Shp2, leads to decreased sensitivity of both murine and human leukemia cell lines to Mcl1 inhibitors, suggesting that patients harboring PTPN11 mutations (or others within the Ras pathway) may show a poorer response to Mcl1 inhibition than patients without. Our data also suggest that the signaling pathways linking Shp2E76K and Mcl1 overexpression may represent candidates for therapeutic targeting in combination with Mcl1 inhibitors.
Mcl1 is associated with transformation and modulated by cooperating mutations
Elegant genetic studies have clearly demonstrated the importance of an anti-apoptotic gene program in AML cells using both overexpression and gene depletion techniques. For example, transgenic mice over-expressing Mcl1 in hematopoietic cells leads to increased survival in several hematopoietic lineages and immortalization of myeloid cells in the presence of IL3 ex vivo (55). Further, mice expressing a modified allele of Mcl1 that encodes an abnormally stabilized form of Mcl1 showed accelerated AML induced by overexpression of c-MYC (56). These studies are important since MCL1 was shown to be the predominant Bcl2 family member overexpressed in AML patient samples. Using conditional knock out alleles, Glaser et. al. demonstrated the importance of Mcl1, Bcl2 and Bcl-xL in AML cell survival (43). Interestingly, it was shown that MLL fusion protein mediated leukemogenesis was most severely inhibited by deletion of Mcl1. However, AML cell death following loss of Mcl1 can be compensated for by expression of Bcl2. Further, cell death induced by loss of Mcl1 is augmented by treatment with ABT-737 suggesting non-overlapping and cooperative roles for Mcl1 and Bcl2 or Bcl-xL in AML cell survival (43). Together, these data are consistent with our gene expression analysis showing Mcl1 as the most highly expressed of the anti-apoptotic genes in MLL-AF9+Shp2E76K cells (Figure 4). Further, we observed differential sensitivity between MLL-AF9 and E2A-HLF AML cells following treatment with the Bcl2/Bcl-xL inhibitor ABT-263 and a Mcl1 inhibitor (Figure 6). E2A-HLF cells showed more sensitivity to Bcl2 inhibition compared to MLL-AF9 cells, however, MLL-AF9 cells are more sensitive to Mcl1 inhibition compared to E2A-HLF cells. This is consistent with the finding that E2A-HLF cells transform through the induced expression of Bcl2 while MLL-fusion cells are more dependent on Mcl1 (48) (43).
Our data suggests a strong link between Mcl1 expression, mediated by Shp2, and increased LSC frequency. Among the anti-apoptotic family of proteins, Mcl1 is indispensible for the self-renewal of normal hematopoietic stem cells (57). These data may suggest similar mechanisms governing the self-renewal of both normal hematopoietic stem cells and leukemic stem cells that may reflect an obstacle to therapeutic targeting. Indeed, upregulation of Mcl1 and Bcl-xL by Shp2 was reported in normal hematopoietic stem cells and regulation of Bcl-xL by Shp2 was reported in FLT3-ITD positive leukemic cells (24, 27). Further, differential regulation of Bcl-xL was reported by Shp2 in human leukemic HL60 cells compared to MV4;11 cells (27). Thus, Shp2 may differentially regulate anti-apoptotic genes dependent on the leukemic subtype. Further studies characterizing anti-apoptotic gene expression in various leukemic subtypes will be required to better understand this regulation.
Supplementary Material
Table 2.
| os | Response | Co-occurring genetic alterations | |||||
|---|---|---|---|---|---|---|---|
| DNMT3A (12/91) | NPM1 (11/91) | WT1 (6/91) | CBF (18/91) | MLL translocation (7/91) | Complex Karyotype | ||
| 1250 | 1 | R882H | FS | WT | WT | WT | WT |
| 552 | 1 | G543C | WT | WT | WT | WT | WT |
| 1208 | 3 | WT | WT | FS (303) | WT | WT | WT |
| 2172 | 1 | WT | WT | WT | Inv16 | WT | WT |
| 1429 | 1 | WT | WT | WT | WT | WT | WT |
| 310 | 1 | WT | WT | WT | WT | t(11;19) | Complex |
Dfs = disease free survival, efs = event free survival, os = overall survival (0=alive, 1=deceased). Response (1=complete remission, 3 = No Change/Stable, 8 = unevaluated, -1 = unknown/missing). The number of patients with a given mutation in the 91 patient cohort are shown in parentheses.
Acknowledgments
The authors thank Dr. Jay Hess for helpful discussion. This work was supported by NIH grants R01-CA149442 (Z.N-C), R00 CA158136 (A.G.M.) and an American Society of Hematology Scholar Award (A.G.M.)
Footnotes
The authors declare no conflict of interest.
The authors have no competing financial interests.
Supplementary information is available on Leukemia’s website.
References
- 1.Raimondi SC, Chang MN, Ravindranath Y, Behm FG, Gresik MV, Steuber CP, et al. Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood. 1999 Dec 1;94(11):3707–16. [PubMed] [Google Scholar]
- 2.Rubnitz JE, Link MP, Shuster JJ, Carroll AJ, Hakami N, Frankel LS, et al. Frequency and prognostic significance of HRX rearrangements in infant acute lymphoblastic leukemia: a Pediatric Oncology Group study. Blood. 1994 Jul 15;84(2):570–3. [PubMed] [Google Scholar]
- 3.Huret JL, Minor SL, Dorkeld F, Dessen P, Bernheim A. Atlas of genetics and cytogenetics in oncology and haematology, an interactive database. Nucleic Acids Res. 2000 Jan 1;28(1):349–51. doi: 10.1093/nar/28.1.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007 Jan 1;16(1):92–106. doi: 10.1093/hmg/ddl444. Epub 2006/12/01. eng. [DOI] [PubMed] [Google Scholar]
- 5.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010 Feb 12;37(3):429–37. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010 Feb 17;17(2):198–212. doi: 10.1016/j.ccr.2009.12.040. Epub 2010/02/16. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002 Jan;30(1):41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 8.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007 Apr 12;446(7137):758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 9.Balgobind BV, Zwaan CM, Pieters R, Van den Heuvel-Eibrink MM. The heterogeneity of pediatric MLL-rearranged acute myeloid leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2011 Aug;25(8):1239–48. doi: 10.1038/leu.2011.90. [DOI] [PubMed] [Google Scholar]
- 10.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002 Sep 1;100(5):1532–42. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 11.Ono R, Nakajima H, Ozaki K, Kumagai H, Kawashima T, Taki T, et al. Dimerization of MLL fusion proteins and FLT3 activation synergize to induce multiple-lineage leukemogenesis. The Journal of clinical investigation. 2005 Apr;115(4):919–29. doi: 10.1172/JCI22725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stubbs MC, Kim YM, Krivtsov AV, Wright RD, Feng Z, Agarwal J, et al. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2008 Jan;22(1):66–77. doi: 10.1038/sj.leu.2404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim WI, Matise I, Diers MD, Largaespada DA. RAS oncogene suppression induces apoptosis followed by more differentiated and less myelosuppressive disease upon relapse of acute myeloid leukemia. Blood. 2009 Jan 29;113(5):1086–96. doi: 10.1182/blood-2008-01-132316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer. Current opinion in genetics & development. 2007 Feb;17(1):23–30. doi: 10.1016/j.gde.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer metastasis reviews. 2008 Jun;27(2):179–92. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 16.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001 Dec;29(4):465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003 Jun;34(2):148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 18.Paulsson K, Horvat A, Strombeck B, Nilsson F, Heldrup J, Behrendtz M, et al. Mutations of FLT3, NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes, chromosomes & cancer. 2008 Jan;47(1):26–33. doi: 10.1002/gcc.20502. [DOI] [PubMed] [Google Scholar]
- 19.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004 Jul 15;104(2):307–13. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014 Jan 23;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012 Jul 20;150(2):264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends in biochemical sciences. 2003 Jun;28(6):284–93. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 23.Zhu HH, Ji K, Alderson N, He Z, Li S, Liu W, et al. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. 2011 May 19;117(20):5350–61. doi: 10.1182/blood-2011-01-333476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Modi H, McDonald T, Rossi J, Yee JK, Bhatia R. A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood. 2011 Aug 11;118(6):1504–15. doi: 10.1182/blood-2010-06-288910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan G, Cheung LS, Yang W, Milyavsky M, Sanders AD, Gu S, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. 2011 Apr 21;117(16):4253–61. doi: 10.1182/blood-2010-11-319517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu R, Yu Y, Zheng S, Zhao X, Dong Q, He Z, et al. Overexpression of Shp2 tyrosine phosphatase is implicated in leukemogenesis in adult human leukemia. Blood. 2005 Nov 1;106(9):3142–9. doi: 10.1182/blood-2004-10-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nabinger SC, Li XJ, Ramdas B, He Y, Zhang X, Zeng L, et al. The protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced hematopoietic progenitor hyperproliferation and malignant disease in vivo. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2013 Feb;27(2):398–408. doi: 10.1038/leu.2012.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998 Feb 20;92(4):441–50. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 29.Stein-Gerlach M, Wallasch C, Ullrich A. SHP-2, SH2-containing protein tyrosine phosphatase-2. The international journal of biochemistry & cell biology. 1998 May;30(5):559–66. doi: 10.1016/s1357-2725(98)00002-8. [DOI] [PubMed] [Google Scholar]
- 30.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998 Mar 15;6(3):249–54. doi: 10.1016/s0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 31.Schubbert S, Lieuw K, Rowe SL, Lee CM, Li X, Loh ML, et al. Functional analysis of leukemia-associated PTPN11 mutations in primary hematopoietic cells. Blood. 2005 Jul 1;106(1):311–7. doi: 10.1182/blood-2004-11-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. The Journal of experimental medicine. 2011 Sep 26;208(10):1977–88. doi: 10.1084/jem.20110450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009 Apr 30;113(18):4414–24. doi: 10.1182/blood-2008-10-182626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005 Feb;7(2):179–91. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Xu D, Wang S, Yu WM, Chan G, Araki T, Bunting KD, et al. A germline gain-of-function mutation in Ptpn11 (Shp-2) phosphatase induces myeloproliferative disease by aberrant activation of hematopoietic stem cells. Blood. 2010 Nov 4;116(18):3611–21. doi: 10.1182/blood-2010-01-265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu ZH, Xu J, Walls CD, Chen L, Zhang S, Zhang R, et al. Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J Biol Chem. 2013 Apr 12;288(15):10472–82. doi: 10.1074/jbc.M113.450023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012 Mar 22;366(12):1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ElSharawy A, Warner J, Olson J, Forster M, Schilhabel MB, Link DR, et al. Accurate variant detection across non-amplified and whole genome amplified DNA using targeted next generation sequencing. BMC genomics. 2012;13:500. doi: 10.1186/1471-2164-13-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nature biotechnology. 2009 Nov;27(11):1025–31. doi: 10.1038/nbt.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muntean AG, Chen W, Jones M, Granowicz EM, Maillard I, Hess JL. MLL fusion protein-driven AML is selectively inhibited by targeted disruption of the MLL-PAFc interaction. Blood. 2013 Jul 30; doi: 10.1182/blood-2013-02-486977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, et al. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. 2005 May 1;105(9):3737–42. doi: 10.1182/blood-2004-10-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nature biotechnology. 2011 Jan;29(1):79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012 Jan 15;26(2):120–5. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muntean AG, Giannola D, Udager AM, Hess JL. The PHD fingers of MLL block MLL fusion protein-mediated transformation. Blood. 2008 Dec 1;112(12):4690–3. doi: 10.1182/blood-2008-01-134056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, et al. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell. 2011 Nov 15;20(5):563–75. doi: 10.1016/j.ccr.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of immunological methods. 2009 Aug 15;347(1–2):70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009 Sep 24;361(13):1249–59. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inaba T, Inukai T, Yoshihara T, Seyschab H, Ashmun RA, Canman CE, et al. Reversal of apoptosis by the leukaemia-associated E2A-HLF chimaeric transcription factor. Nature. 1996 Aug 8;382(6591):541–4. doi: 10.1038/382541a0. Epub 1996/08/08. eng. [DOI] [PubMed] [Google Scholar]
- 49.Huang H, Woo AJ, Waldon Z, Schindler Y, Moran TB, Zhu HH, et al. A Src family kinase-Shp2 axis controls RUNX1 activity in megakaryocyte and T-lymphocyte differentiation. Genes Dev. 2012 Jul 15;26(14):1587–601. doi: 10.1101/gad.192054.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research. 2008 May 1;68(9):3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 51.Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, et al. A novel small-molecule inhibitor of mcl–1 blocks pancreatic cancer growth in vitro and in vivo. Molecular cancer therapeutics. 2014 Mar;13(3):565–75. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, et al. 3-Substituted-N-(4-Hydroxynaphthalen-1-yl)arylsulfonamides as a Novel Class of Selective Mcl-1 Inhibitors: Structure-Based Design, Synthesis, SAR, and Biological Evaluation. Journal of medicinal chemistry. 2014 May 22;57(10):4111–33. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobson CL, Warren AJ, Pannell R, Forster A, Lavenir I, Corral J, et al. The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. Embo J. 1999 Jul 1;18(13):3564–74. doi: 10.1093/emboj/18.13.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. Embo J. 1997 Jul 16;16(14):4226–37. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou P, Qian L, Bieszczad CK, Noelle R, Binder M, Levy NB, et al. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood. 1998 Nov 1;92(9):3226–39. [PubMed] [Google Scholar]
- 56.Okamoto T, Coultas L, Metcalf D, van Delft MF, Glaser SP, Takiguchi M, et al. Enhanced stability of Mcl1, a prosurvival Bcl2 relative, blunts stress-induced apoptosis, causes male sterility, and promotes tumorigenesis. Proc Natl Acad Sci U S A. 2014 Jan 7;111(1):261–6. doi: 10.1073/pnas.1321259110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campbell CJ, Lee JB, Levadoux-Martin M, Wynder T, Xenocostas A, Leber B, et al. The human stem cell hierarchy is defined by a functional dependence on Mcl-1 for self-renewal capacity. Blood. 2010 Sep 2;116(9):1433–42. doi: 10.1182/blood-2009-12-258095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.