

Abstract
Studies in vitro suggest that mitogen-activated protein kinase kinase kinase kinase 4 (Map4k4) attenuates insulin signaling, but confirmation in vivo is lacking since Map4k4 knockout is lethal during embryogenesis. We thus generated mice with floxed Map4k4 alleles and a tamoxifen-inducible Cre/ERT2 recombinase under the control of the ubiquitin C promoter to induce whole-body Map4k4 deletion after these animals reached maturity. Tamoxifen administration to these mice induced Map4k4 deletion in all tissues examined, causing decreased fasting blood glucose concentrations and enhanced insulin signaling to AKT in adipose tissue and liver but not in skeletal muscle. Surprisingly, however, mice generated with a conditional Map4k4 deletion in adiponectin-positive adipocytes or in albumin-positive hepatocytes displayed no detectable metabolic phenotypes. Instead, mice with Map4k4 deleted in Myf5-positive tissues, including all skeletal muscles tested, were protected from obesity-induced glucose intolerance and insulin resistance. Remarkably, these mice also showed increased insulin sensitivity in adipose tissue but not skeletal muscle, similar to the metabolic phenotypes observed in inducible whole-body knockout mice. Taken together, these results indicate that (i) Map4k4 controls a pathway in Myf5-positive cells that suppresses whole-body insulin sensitivity and (ii) Map4k4 is a potential therapeutic target for improving glucose tolerance and insulin sensitivity in type 2 diabetes.
INTRODUCTION
Whole-body glucose homeostasis in humans is maintained by an elaborate physiological system that involves multiorgan regulation. In the postprandial state, glucose and amino acids induce β cells in the pancreas to secrete insulin, promoting glucose uptake in skeletal muscle and adipose tissue while suppressing glucose production from the liver. These effects of insulin to maintain glucose homeostasis can be disrupted in obesity, causing an insulin-resistant state that contributes to elevated blood glucose levels and an increased incidence of type 2 diabetes (T2D) (1). While metformin (thought to mainly affect liver glucose metabolism) and insulin secretagogues are clinical mainstays for the treatment of T2D, further intervention is often required (2). Due to contraindications, treatments that address the underlying peripheral insulin resistance in T2D are now limited to the use of the thiazolidinediones (TZDs), a major drug class that alleviates insulin resistance by targeting peroxisome proliferator-activated receptor gamma (PPARγ) (3, 4). Thus, novel proteins that could be targeted with drugs to enhance peripheral insulin sensitivity would prove useful in developing therapies for T2D.
In screening the adipocyte kinome for such negative regulators of insulin signaling to glucose transport in vitro, we identified mitogen-activated protein kinase kinase kinase kinase 4 (Map4k4), a serine/threonine protein kinase with homology to Saccharomyces cerevisiae Ste20 protein kinases (5). Interestingly, single nucleotide polymorphisms (SNPs) in the Map4k4 locus appear to be associated with insulin resistance (6) and T2D (7). We therefore hypothesized that Map4k4 may be a novel therapeutic target for improving obesity-induced peripheral insulin resistance. The functions of Map4k4 in insulin signaling and energy metabolism were subsequently studied in various cell culture models. In adipocytes, Map4k4 repressed glucose transport and lipid synthesis (5, 8), while in human myotubes, Map4k4 depletion protected cells from tumor necrosis factor alpha (TNF-α)-induced insulin resistance (9). These studies suggested that systemic Map4k4 depletion might improve whole-body glucose metabolism in obesity. However, the effects of systemic Map4k4 deficiency on metabolic disease in vivo have not yet been studied due to the embryonic lethality of Map4k4-null mice (10). To circumvent this problem, we generated an inducible gene deletion mouse model to investigate the physiological effects of Map4k4 depletion that appear once mice reach maturity. We sought to address several questions employing this model. First, is Map4k4 required for viability of adult mice? Second, is Map4k4 a negative regulator of systemic insulin sensitivity and metabolic function in obese animals? Last, in what tissue or tissues might Map4k4 operate to modulate whole-body glucose homeostasis and insulin responsiveness?
Here we report that mature mice with induced systemic Map4k4 ablation (iMap4k4 knockout [iMap4k4-KO] mice) are viable and, when challenged with high-fat feeding, display lower fasting glucose levels and improved peripheral insulin sensitivity. By generating multiple tissue-specific Map4k4 knockout mice, we found that Map4k4 deficiency in cells from a Myf5 lineage, including skeletal muscles, but not selective Map4k4 deficiency in adipose cells or hepatocytes recapitulates the improved systemic insulin sensitivity observed in whole-body iMap4k4-KO mice.
MATERIALS AND METHODS
Animal studies.
All of the studies performed were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Massachusetts (UMass) Medical School. Animals were maintained under conditions of a 12-h-light/12-h-dark cycle and fed standard chow (LabDiet 5P76) unless otherwise stated. Mice with conditional Map4k4 floxed alleles were generated as described elsewhere (11) and were backcrossed to C57BL/6J mice for at least 7 generations. To inactivate Map4k4 in adult mice, homozygous Map4k4flox/flox animals were crossed to B6.Cg-Tg(UBC-cre/ERT2)1Ejb/J mice (Jackson Laboratories). Mice (both Map4k4flox/flox and Map4k4flox/flox-UBC-cre ERT2) that were 8 weeks of age were treated via intraperitoneal (i.p.) injection with 30 μg tamoxifen (dissolved in corn oil)/40 g body weight for 5 consecutive days. At 2 weeks after first tamoxifen injection, animals were fed a normal chow diet (ND) or a high-fat diet (HFD) (12492i; Harlan) for 16 weeks.
Adiponectin-cre mice [B6; FVB-Tg(Adipoq-cre)1Evdr/J], albumin-cre mice [C57BL/6-Tg(Alb-cre)21Mgn/J], and Myf-5 cre mice [B6.129S4-Myf5tm3(cre)Sor/J] were obtained from Jackson Laboratories.
Mice were fasted for 16 h for a glucose tolerance test (GTT) and a pyruvate tolerance test (PTT) or for 4 h for an insulin tolerance test (ITT). Fasted mice were i.p. injected with glucose (1 g/kg), pyruvate (1 g/kg), or insulin (1 IU/kg). Blood samples were withdrawn via the tail vein, and blood glucose levels were determined using a Breeze-2 glucose meter (Bayer).
Metabolic cage and body composition analyses were performed by personnel at the UMass Mouse Metabolic Phenotyping Center. The metabolic cages were used to measure food and water intake over a 3-day period, and average food intake/day was calculated (TSE Systems). Whole-body fat and lean mass were measured using proton magnetic resonance spectroscopy (1H-MRS) (Echo Medical System).
RNA isolation and RT-qPCR.
Total RNA was isolated from tissues using TriPure isolation reagent (Roche) following the manufacturer's protocol. Isolated RNA was DNase treated (DNA-free; Life Technologies), and cDNA was synthesized using an iScript cDNA synthesis kit (Bio-Rad). Reverse transcription-quantitative PCR (RT-qPCR) was performed using iQ Sybr green supermix, and 36B4 (Rplp0) served as the reference gene. Primer sequences were as follows: for Rplp0, 5′-TCCAGGCTTTGGGCATCA-3′ and 3′-CTTTATCAGCTGCACATCACTCAGA-5′; for Map4k4, 5′-CATCTCCAGGGAAATCCTCAGG-3′ and 3′-TTCTGTAGTCGTAAGTGGCGTCTG-5′; for Emr-1, 5′-CCCCAGTGTCCTTACAGAGTG-3′ and 3′-GTGCCCAGAGTGGATGTCT-5′; for CD68, 5′-CCATCCTTCACGATGACACCT-3′ and 3′-GGCAGGGTTATGAGTGACAGTT-5′; for Itgam, 5′-ATGGACGCTGATGGCAATACC-3′ and 3′-TCCCCATTCACGTCTCCCA-5′; for Itgax, 5′-CTGGATAGCCTTTCTTCTGCTG-3′ and 3′-GCACACTGTGTCCGAACTCA-5′; for interleukin-1 β (IL-1β), 5′-GCAACTGTTCCTGAACTCAACT-3′ and 3′-ATCTTTTGGGGTCCGTCAACT-5′; for CCL2, 5′-TTAAAAACCTGGATCGGAACCAA-3′ and 3′-GCATTAGCTTCAGATTTACGGGT-5′; for TNF-α, 5′-CAGGCGGTGCCTATGTCTC-3′ and 3′-CGATCACCCCGAAGTTCAGTAG-5′; for Pepck, 5′-CTGCATAACGGTCTGGACTTC-3′ and 3′-CAGCAACTGCCCGTACTCC-5′; for G6pc, 5′-CGACTCGCTATCTCCAAGTGA-3′ and 3′-GTTGAACCAGTCTCCGACCA-5′; for Gck, 5′-TGAGCCGGATGCAGAAGGA-3′ and 3′-GCAACATCTTTACACTGGCCT-5′; for Fasn, 5′-GGAGGTGGTGATAGCCGGTAT-3′ and 3′-TGGGTAATCCATAGAGCCCAG-5′; and for Scd-1, 5′-TTCTTGCGATACACTCTGGTGC-3′ and 3′-CGGGATTGAATGTTCTTGTCGT-5′.
Plasma analysis.
Mice were fasted overnight, and plasma was collected via cardiac puncture. Serum nonesterified fatty acid (NEFA) levels were measured using a colorimetric assay (Wako Diagnostics) according to the manufacturer's instructions. Alternatively, animals were injected with 0.5 mg/kg CL 316,243 (Santa Cruz) dissolved in phosphate-buffered saline (PBS) and blood was collected via tail vein after 1 h. Insulin measurements were performed using an insulin enzyme-linked immunosorbent assay (ELISA) kit (Millipore) according to the manufacturer's instructions.
Histology.
Tissues were isolated and fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). The UMass Morphology Core performed the embedding and sectioning.
Adipocyte isolation.
Adipose tissue was minced in digestion buffer (5% bovine serum albumin [BSA]–1 mg/ml collagenase [Sigma no. C6885]–Hanks balanced salt solution [HBSS]) and digested in a 37°C water bath for 30 to 45 min. Digested tissue was filtered through a 200-μm-pore-size nylon mesh and centrifuged at a low speed for 5 min. Floating adipocytes were washed several times in HBSS followed by centrifugation.
Hepatocyte isolation.
Hepatocytes were isolated as previously described (12). Briefly, chow-fed animals were anesthetized via i.p. ketamine-xylazine injection. Livers were perfused and then digested with 50 mg/ml collagenase (Sigma no. C2139) in HBSS that had been supplemented with 0.5 M EGTA and 1 mM CaCl2. Digested livers were collected, filtered through a 100-μm-pore-size cell strainer, and centrifuged and washed several times at a low speed.
Immunoblotting.
For in vivo insulin signaling studies, mice were fasted for 4 h and injected with PBS or insulin (1 IU/kg). At 15 min after the injection, tissues were rapidly harvested. Tissues were homogenized in lysis buffer (20 mM HEPES [pH 7.4], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 10% glycerol, 0.5% sodium deoxycholate) that had been supplemented with Halt protease and phosphatase inhibitors (Thermo Pierce). For Map4k4 expression analysis, tissues were homogenized in lysis buffer containing 150 mM NaCl, 2 mM EDTA, and 2% SDS supplemented with protease and phosphatase inhibitors. Immunoblotting was performed using standard protocols. Membranes were blotted with the following antibodies: AKT (T308, no. 2965; S473, no. 3787; total, no. 2920; Cell Signaling Technology) and Map4k4 (no. A301-503A; Bethyl Laboratories).
TG extraction.
Livers were isolated from HFD-fed animals and frozen in liquid nitrogen. Hepatic triglycerides (TGs) were extracted using the method of Folch et al. (13). Evaporated lipids with resuspended in 1% Triton X-100 dissolved in isopropanol. TG content was determined using the protocol of the manufacturer (triglyceride determination kit; Sigma).
Flow cytometry.
The stromal vascular fraction (SVF) of the visceral adipose tissue (VAT) was isolated and weighed, and subsequent flow cytometry was performed as previously described (14). Briefly, VAT was minced in digestion buffer (5% BSA–2 mg/ml collagenase–HBSS) and incubated in a 37°C water bath for 45 min. The digested tissue was passed through a 100-μm-pore-size cell strainer and centrifuged. The pelleted cells were collected as the SVF, and red blood cells were lysed by incubation with red blood cell lysis buffer. SVF cells were blocked in mouse IgG and counted. The antibodies used included F4/80 (antigen-presenting cells [APC]; AbDSerotec), CD11b (PerCP-Cy 5.5; BD), and CD11c (V450; BD).
Statistics.
Results are described as the means ± standard errors of the means (SEM). Significance was assessed using a two-tailed Student's t test.
RESULTS
iMap4k4-KO mice display low fasting blood glucose levels and improved insulin responsiveness.
To investigate the functional role of Map4k4 in whole-body glucose homeostasis, we deleted Map4k4 systemically. However, whole-body Map4k4-KO animals die at embryonic day 9.5 (E9.5) due to failure of mesodermal cells to migrate away from the primitive streak (10). To bypass the necessary role of Map4k4 during embryonic development (10), we generated Map4k4 floxed mice (bearing loxP sites around exon 7) with tamoxifen-inducible UBC-cre ERT2 recombinase to inducibly delete Map4k4 in adult mice (Fig. 1A). Tamoxifen was administered via intraperitoneal (i.p.) injection to both Map4k4flox/flox (control) and Map4k4flox/flox-UBC-cre ERT2 (iMap4k4) mice (Fig. 1B). Tamoxifen induced cre recombinase activity and Map4k4 deletion in iMap4k4 mice, resulting in animals we denote iMap4k4-KO mice. As expected, Map4k4 mRNA and protein expression was significantly reduced in all of the tissues collected from iMap4k4-KO mice 18 weeks post-tamoxifen treatment (Fig. 1C and D). Furthermore, both control and iMap4k4-KO mice continued to develop normally after tamoxifen treatment and did not display any obvious motor or behavioral defects. These results indicate that Map4k4 is not required for adult mouse viability and that this inducible Map4k4 deletion mouse model can be used to address the metabolic functions of Map4k4 in obese animals.
FIG 1.
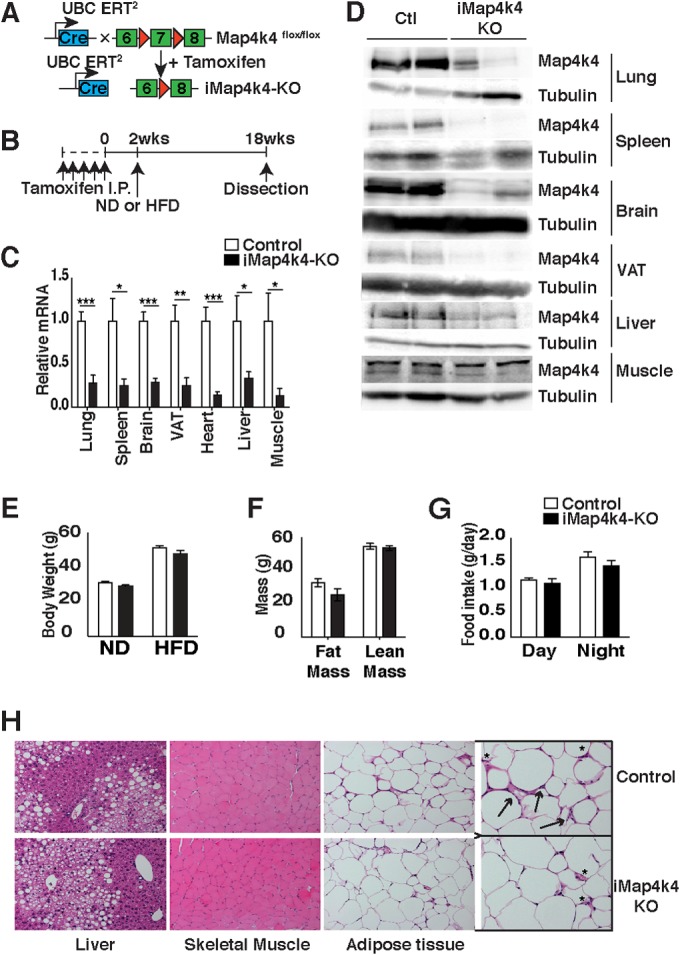
Characterization of tamoxifen-inducible whole-body iMap4k4-KO mice. (A) Schematic of alleles and transgenes used to inactivate Map4k4 systemically in adult tissues using tamoxifen. (B) Schematic of experimental design. Both control (Map4k4flox/flox) and iMap4k4 (Map4k4flox/flox-UBC-cre ERT2) mice were injected with tamoxifen as detailed in Materials and Methods to produce iMap4k4-KO mice. (C and D) Analysis was performed 18 weeks after first tamoxifen injection. (C) Map4k4 mRNA expression (n = 6 to 10). (D) Representative Map4k4 protein immunoblot with tubulin as the loading control (Ctl) (n = 6). (E to H) Control and iMap4k4-KO mice were fed a normal chow diet (ND) or a high-fat diet (HFD) for 16 weeks starting 2 weeks after the 1st tamoxifen injection. (E) Body weight (n = 16). (F) Fat and lean mass analysis in HFD-fed mice (n = 4 to 6). (G) Food intake in mice fed an HFD (n = 4 to 6). (H) Representative liver, skeletal muscle, and adipose tissue histology of HFD-fed mice stained for H&E (n = 6). Arrows indicate crown-like-structures, and asterisks sites indicate of inflammation. Data represent means ± SEM (*, P < 0.01; **, P < 0.001; ***, P < 0.0001).
To assess the role of Map4k4 in metabolic disease, animals were challenged with a lard-based high (60%)-fat diet (HFD). Animals with systemic Map4k4 deletion did not display body weight or adiposity differences from the littermate controls (Map4k4flox/flox mice treated with tamoxifen) (Fig. 1E and F). Consistent with this observation, control and iMap4k4-KO mice consumed similar quantities of food (Fig. 1G). Furthermore, although no gross histological changes were observed in metabolic tissues (Fig. 1H), the visceral adipose tissue (VAT) displayed a slight reduction in macrophage content as assessed by histology (Fig. 1H), RT-qPCR (Fig. 2A), and flow cytometry (Fig. 2B to E). Interestingly, despite similar adiposity results, HFD-fed iMap4k4-KO animals showed enhanced whole-body insulin responsiveness as assessed by an insulin tolerance test (ITT) (Fig. 3A). Strikingly, the ITT curves of HFD-fed iMap4k4-KO mice overlapped those of age-matched lean animals (Fig. 3A), suggesting that whole-body Map4k4 deficiency in obese animals improves peripheral insulin sensitivity to levels similar to those observed in lean controls. We also found that iMap4k4-KO mice had a significant (20%) reduction in fasting glucose levels (Fig. 3B), as well as lower circulating insulin levels (Fig. 3C), consistent with increased insulin responsiveness in vivo. However, no changes in the results of the glucose tolerance test (GTT) were detected (Fig. 3B).
FIG 2.
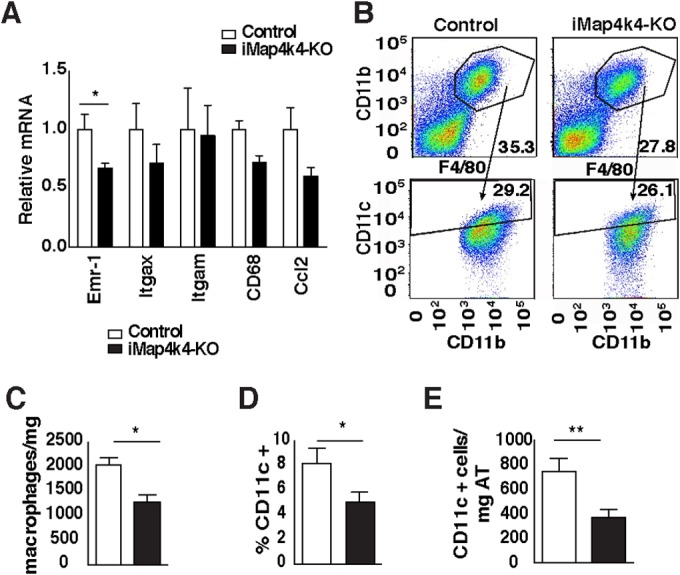
Systemic Map4k4 deletion reduces adipose tissue inflammation without changing adiposity. Animals were fed an HFD for 16 weeks, and visceral adipose tissue was isolated. (A) Relative levels of mRNA expression of various inflammatory genes as indicated (n = 7). (B) The VAT stromal vascular fraction was isolated and fluorescence-activated cell sorter (FACS) analysis performed. Representative FACS scatter plots for control mice and iMap4k4-KO mice are shown. The upper plots show F4/80- and CD11b-positive cells, while the lower plots show CD11b- and CD11c-positive cells (n = 10 to 13). (C) Number of macrophages (F4/80-positive cells) per milligram of VAT (n = 10 to 13). (D) Percentage of CD11c-positive cells (n = 10 to 13). (E) CD11c-positive cells per milligram of VAT (n = 10 to 13). Data represent average means ± SEM (*, P < 0.05; **, P < 0.005).
FIG 3.

Systemic Map4k4 deletion ameliorates obesity-induced metabolic dysfunction and enhances insulin signaling in adipose and liver. (A and B) Control and iMap4k4-KO mice were fed an ND or an HFD for 16 weeks. (A) Percentage of basal glucose remaining during insulin tolerance test (ITT) (n = 7 to 17). (B) Blood glucose levels during glucose tolerance test (GTT) (n = 10 to 17). (C) Fasting insulin levels in HFD-fed mice (n = 13). (D) Representative protein immunoblots of AKT signaling (total, thr308 [308], and ser473 [473]) in liver, adipose tissue, and skeletal muscle 15 min after PBS or insulin injection in HFD-fed animals as described in Materials and Methods. (E) Densitometry analyses of the data from the experiments described for panel D (n = 6 to 10). Results are means ± SEM (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
As an additional assessment of insulin sensitivity on a cellular level, we measured insulin signaling in adipose, liver, and muscle tissue following a bolus insulin injection into HFD-fed animals. Consistent with enhanced whole-body insulin responsiveness, iMap4k4-KO mice showed significant improvements in insulin-induced AKT phosphorylation (at Ser473 and Thr308) in the liver and in the VAT (Fig. 3D and E). These data indicate that iMap4k4-KO mice are protected from obesity-induced insulin resistance at least in part by enhancement of insulin signaling to AKT in adipose tissue and liver. Because obesity leads to glucose intolerance and insulin resistance in various organs, including adipose tissue, liver, and muscle, we aimed to identify the contribution of Map4k4 in each of these tissues to whole-body glucose homeostasis and insulin sensitivity.
Adipose Map4k4 does not influence whole-body glucose homeostasis.
Obesity-induced adipose tissue dysfunction—including adipose tissue inflammation, altered adipokine secretion, and reduced lipid-buffering capacity—contributes to the development of whole-body insulin resistance (15–19). Since Map4k4 expression is elevated in obese human adipose tissue (20) and studies performed in vitro suggest Map4k4 is a negative regulator of insulin action (5, 8), we aimed to specifically ablate Map4k4 expression in adipocytes by crossing Map4k4flox/flox mice to adiponectin-cre transgenic mice (Fig. 4A). As controls, Map4k4flox/flox littermates that do not express cre recombinase were used. The resulting adipose-specific knockout (Ad-Map4k4-KO) mice displayed disrupted Map4k4 expression in the adipocyte fractions of all the adipose tissue depots examined (visceral, subcutaneous, and brown adipose) but not in the spleen or the heart (Fig. 4B and C). Isolated adipocytes were used to confirm Map4k4 depletion in adipocytes, as the adipose tissue is composed of various cell types, including immune cells, preadipocytes, and vascular cells. To test whether the enhanced insulin sensitivity previously observed in obese iMap4k4-KO mice was mediated by the actions of Map4k4 in adipose tissue, animals were challenged with an HFD for 16 weeks. Ad-Map4k4-KO animals developed normally and gained weight and fat mass in a manner similar to that seen with control littermates fed an ND or an HFD (Fig. 4D to F). To assess whether Ad-Map4k4-KO mice displayed altered FA release, which might impact glucose metabolism, nonesterified fatty acid (NEFA) levels were measured under basal conditions and after β3-adrenergic stimulation (i.p. injection of CL 316,243). As expected, NEFA levels were increased following CL 316,243 injection; however, control and Ad-Map4k4-KO mice displayed comparable plasma NEFA levels (Fig. 4G). Metabolic functions of control and Ad-Map4k4-KO mice fed ND or HFD as assessed by glucose tolerance (GTT) and insulin sensitivity (ITT) were also similar (Fig. 4H and I). Consistent with the lack of metabolic changes, control and Ad-Map4k4-KO mice displayed similar fasting insulin levels (Fig. 4J). Furthermore, results of analysis of inflammation in both subcutaneous and visceral adipose tissue as assessed by histology (Fig. 4K) and RT-qPCR (Fig. 4L and M) were also similar between groups. Overall, these results indicate that loss of Map4k4 in adipocytes does not alter the adipose functions tested or whole-body glucose metabolism and suggest that the improved adipose insulin sensitivity observed in iMap4k4-KO mice is not mediated by the actions of Map4k4 in the adipose tissues.
FIG 4.
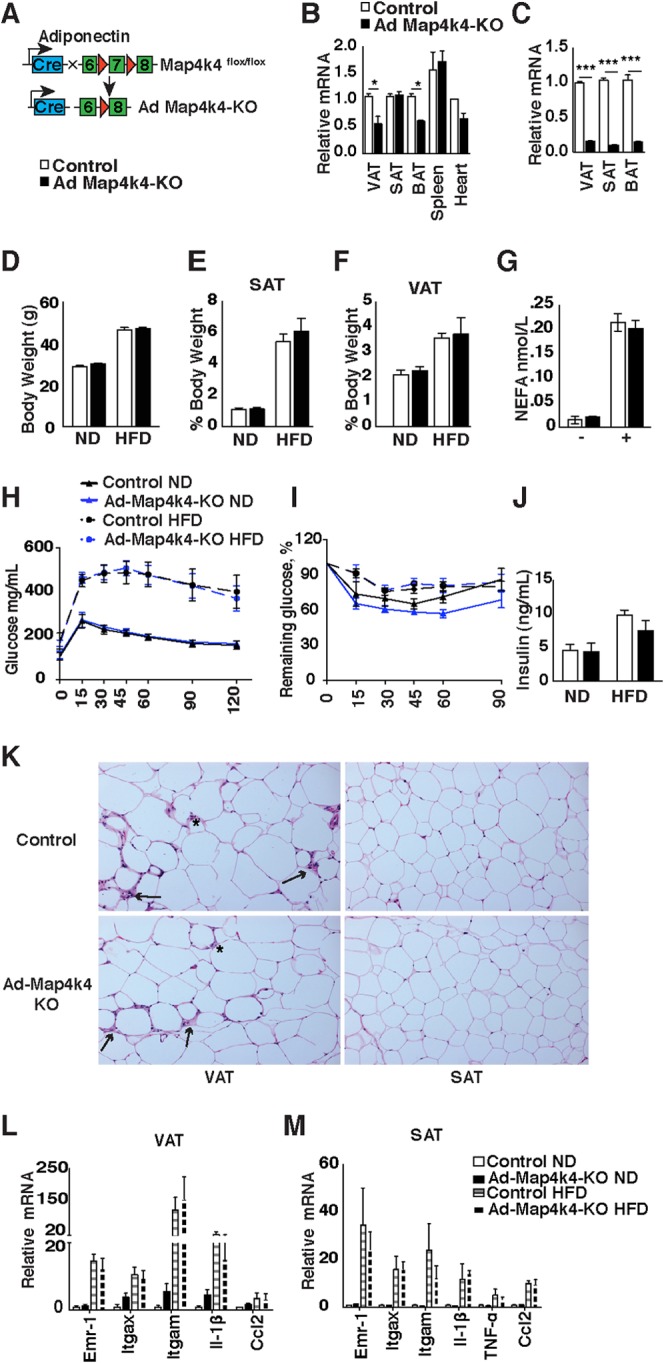
Adipose-specific Map4k4 deletion in mice does not alter systemic glucose tolerance or insulin responsiveness. (A) Schematic of alleles and transgenes used to inactivate Map4k4 in the adipose using adiponectin-cre transgene. (B) Relative levels of Map4k4 mRNA expression in adipose tissues (visceral adipose tissue [VAT], subcutaneous adipose tissue [SAT], and brown adipose tissue [BAT]), spleen, and heart (n = 4). (C) Relative levels of Map4k4 mRNA expression in isolated adipocytes from VAT, SAT, and BAT (n = 3). (D to M) Control and Ad-Map4k4-KO mice were fed an ND or an HFD for 16 weeks. (D) Body weight (n = 8 to 14). (E) SAT mass relative to body weight (n = 5). (F) VAT mass relative to body weight (n = 5). (G) NEFA levels in HFD-fed mice before and after 1 h CL 316,243 i.p. injection (n = 5). (H) GTT (n = 10 to 15). (I) ITT (n = 10 to 15). (J) Fasting insulin levels (n = 5 to 6). (K) Representative histology of VAT and SAT in HFD-fed mice. Slides were stained with H&E (n = 10). Arrows indicate crown-like-structures, and asterisks indicate sites of inflammation. (L and M) Relative mRNA levels of various inflammatory genes assessed in VAT (L) (n = 6 to 8) and SAT (M) (n = 6 to 8). Results are means ± SEM (*, P < 0.05; ***, P < 0.0001).
Hepatocyte-specific Map4k4 deletion does not improve metabolic parameters in lean or obese mice.
In the fed state, insulin suppresses glycogenolysis and gluconeogenesis while stimulating lipid synthesis in the liver, whereas, under conditions of obesity-induced insulin resistance, insulin fails to suppress hepatic glucose production, thereby contributing to elevated blood glucose levels (21). To assess whether hepatic Map4k4 could account for the improved insulin sensitivity observed in the HFD-fed iMap4k4-KO mice, Map4k4flox/flox animals were crossed to mice expressing a cre transgene driven by the hepatocyte-specific albumin promoter (Fig. 5A). Map4k4flox/flox littermates that do not express cre recombinase were used as controls. We confirmed specific deletion of Map4k4 in hepatocytes and not skeletal muscle or adipose tissue in Map4k4flox/flox-Alb-cre (Alb-Map4k4-KO) mice (Fig. 5B and C). Since the liver consists of several cell types, including Kupffer cells, we isolated hepatocytes to confirm hepatocyte-specific deletion of Map4k4 (Fig. 5B and C). To assess whether hepatic Map4k4 contributed to whole-body glucose metabolism, control and Alb-Map4k4-KO mice were challenged with an HFD for 16 weeks. Control littermates and Alb-Map4k4-KO animals gained weight at similar levels upon high-fat feeding (Fig. 5D and E), and fasting insulin and NEFA levels were unchanged between groups (Fig. 5F and G). Furthermore, no alterations in fasting glucose levels or pyruvate tolerance test (PTT), GTT, or ITT results were observed among control and Alb-Map4k4-KO animals on an ND or an HFD (Fig. 5H to K). Although liver histology of Alb-Map4k4-KO mice revealed a slight increase in hepatic lipid accumulation compared to controls (Fig. 5L), this modest increase was not statistically significant as assessed by hepatic triglyceride content (Fig. 5M). These results indicate that hepatic Map4k4 depletion does not contribute to the reduced basal glucose levels or improved insulin sensitivity observed upon inducible whole-body Map4k4 deficiency.
FIG 5.
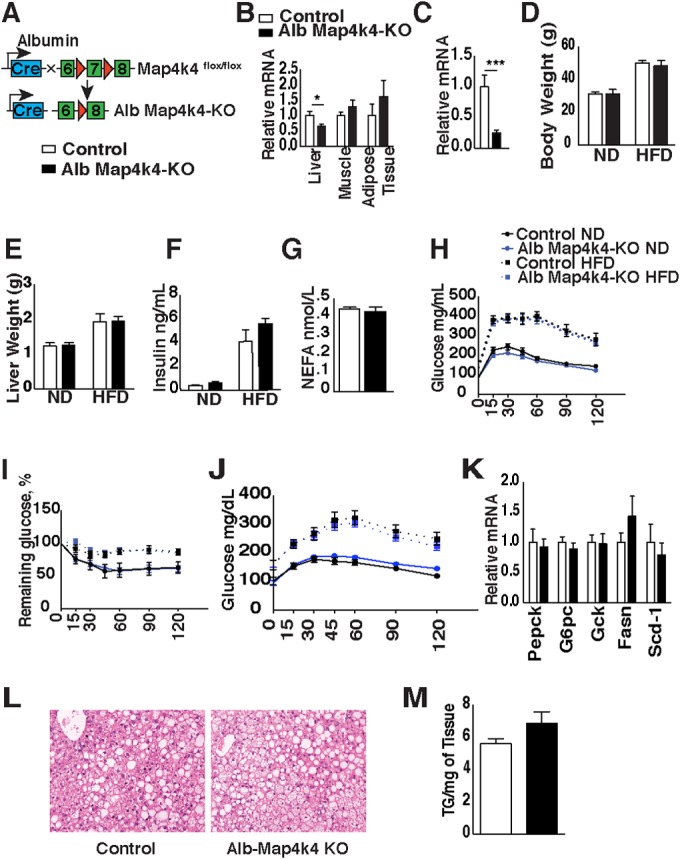
Hepatic Map4k4 does not contribute to systemic glucose tolerance or insulin responsiveness. (A) Schematic of alleles and transgenes used to inactivate Map4k4 using albumin-cre transgene. (B) Relative levels of Map4k4 mRNA expression in liver, skeletal muscle, and adipose tissue (n = 7 to 14). (C) Relative levels of Map4k4 mRNA expression in isolated hepatocytes (n = 3). (D to M) Mice were fed an ND or an HFD for 16 weeks. (D) Body weight (n = 6 to 13). (E) Liver weight (n = 6 to 17). (F) Fasting insulin levels (n = 4 to 15). (G) NEFA levels of HFD-fed mice (n = 4 to 15). (H) GTT (n = 8 to 16). (I) ITT (n = 8 to 16). (J) Blood glucose levels during pyruvate tolerance test (PTT) (n = 7 to 16). (K) Livers were isolated and RT-qPCR performed on the indicated genes of HFD-fed animals (n = 5 to 7). (L) Representative liver histology of HFD-fed mice. Slides were stained with H&E (n = 10). (M) Liver triglyceride (TG) extraction from HFD-fed mice (n = 5 to 11). Results are means ± SEM (*, P < 0.01; ***, P < 0.0001).
Map4k4 deletion in Myf5-positive cells results in improved metabolic parameters.
Skeletal muscle plays a major role in regulating whole-body glucose metabolism and is responsible for the largest fraction of insulin-mediated glucose uptake from the blood. To determine whether Map4k4 in skeletal muscle affects whole-body metabolism, we crossed Map4k4flox/flox animals to mice that express cre recombinase under the control of the Myf5 promoter (Fig. 6A). Myf5-cre was used to delete Map4k4 in muscle precursors because previous reports from our laboratory demonstrated that Map4k4 plays a key role during early stages of muscle differentiation in C2C12 cells (22). As controls, we used Map4k4flox/flox littermates, which do not express cre recombinase. As expected, Map4k4 expression was diminished in several of the skeletal muscle tissues collected (i.e., trapezius, triceps, and soleus) as well as in brown adipose tissue (BAT), as Myf5 is expressed in muscle and BAT precursors (Fig. 6B) (23–25). Consistent with previous reports showing a low (5% to 10%) prevalence of Myf5-precursor-derived adipocytes in the visceral adipose tissue (VAT), the relative levels Map4k4 mRNA expression were not altered in whole VAT (Fig. 6B) (23–25). Isolated adipocytes from VAT displayed lower Map4k4 expression levels; however, this decrease was not due to Map4k4 deletion as assessed by genomic analysis but likely reflects transcriptional downregulation of Map4k4 expression in these cells (data not shown).When challenged with an HFD, both control and Map4k4flox/flox-Myf5-cre (Myf5-Map4k4-KO) mice gained weight, although the Myf5-Map4k4-KO animals tended to be smaller (Fig. 6C). Whole-body lean and fat mass analysis, however, did not reveal any statistically significant differences between control and Myf5-Map4k4-KO mice as measured by 1H-MRS (Fig. 6D) or individual tissue weights (Fig. 6E and F). Furthermore, measurements of energy metabolism, including food intake (Fig. 6G), oxygen consumption (Fig. 6H), carbon dioxide production (Fig. 6I), energy expenditure (Fig. 6J), and physical activity levels (Fig. 6K), were not statistically different between groups. Fasting insulin and NEFA levels were also unchanged in HFD-fed Myf5-Map4k4-KO mice compared to control mice (Fig. 6L and M). Strikingly, however, despite similar adiposities, HFD-fed Myf5-Map4k4-KO animals displayed decreased fasting glucose levels and improved glucose clearance during a GTT, suggesting that Myf5-Map4k4-KO mice were more glucose tolerant than controls (Fig. 6N). We then performed an ITT, and, consistent with enhanced glucose tolerance, Myf5-Map4k4-KO mice were significantly more insulin sensitive than controls (Fig. 6O and P). These results suggest that Map4k4 deletion in Myf5-positive cells recapitulates in large part the metabolic benefits observed in HFD-fed iMap4k4-KO mice and demonstrate that Map4k4 in Myf5-positive cells promotes obesity-induced metabolic dysfunction.
FIG 6.

Map4k4 deletion in Myf5-positive tissues improves glucose tolerance and insulin responsiveness. (A) Schematic of alleles and transgenes used to inactivate Map4k4 using Myf5-cre knock-in allele. (B) Relative levels of Map4k4 mRNA expression in various muscles, adipose tissues, and liver (n = 5). Quad, quadriceps; EDL, extensor digitorum longus; TA, tibialis anterior. (C to M) Mice were fed an ND or an HFD for 8 weeks. (C) Body weight (n = 7 to 18). (D) Fat and lean mass analysis using 1H-MRS (n = 5). (E) Adipose tissues relative to body weight in ND-fed mice. Data represent visceral adipose tissue (VAT), inguinal subcutaneous adipose tissue (iWAT), axial subcutaneous adipose tissue (aWAT), perirenal adipose tissue (pWAT), and brown adipose tissue (BAT) (n = 8). (F) Muscle weight relative to body weight in ND-fed mice (n = 8). (G to M) Mice were fed an HFD for 8 weeks. (G) Food intake (n = 5). (H) Oxygen consumption rate (n = 5). (I) Carbon dioxide production rate (n = 5). (J) Energy expenditure rate (n = 5). (K) Total activity levels (n = 5). (L) Fasting insulin levels (n = 6 to 8). (M) Fasting NEFA levels (n = 5). (N) GTT (n = 7 to 17). (O) ITT of ND-fed animals (n = 11 to 19). (P) ITT of HFD-fed animals (n = 7 to 13). Results are means ± SEM (*, P < 0.05; **, P < 0.005).
To further assess peripheral insulin sensitivity in the Myf5-Map4k4-KO mice, HFD-fed control and Myf5-Map4k4-KO animals were injected with PBS or insulin followed by collection of liver, adipose tissue, and skeletal muscle samples. Myf5-Map4k4-KO mice displayed a modest increase in AKT phosphorylation in response to insulin in the soleus muscle, a tissue with near-complete Map4k4 ablation, and no changes in AKT phosphorylation in the liver (Fig. 7A and B). Surprisingly, Myf5-Map4k4-KO mice displayed a striking enhancement of AKT phosphorylation in the VAT (Fig. 7A and B), a tissue that expressed normal Map4k4 levels in these mice (Fig. 6B). No gross morphological changes were observed in either skeletal muscle or VAT that could explain improved whole-body insulin action (Fig. 7C), and the two groups of mice displayed similar adipose tissue inflammation profiles as assessed by histology and RT-qPCR of inflammatory markers (Fig. 7C and D). To assess whether muscle-secreted factors (myokines) were altered in Myf5-Map4k4-KO mice, leading to improved insulin signaling in VAT, we measured the relative levels of gene expression of known myokines in the skeletal muscle (soleus) (Fig. 7E) as well as total circulating levels of fibroblast growth factor 21 (FGF21) (Fig. 7F), interleukin-6 (IL-6) (Fig. 7G), and irisin (Fig. 7H) and found no differences between control and Myf5-Map4k4-KO mice. These results demonstrate that Map4k4 deletion in Myf5-positive tissues protects mice from obesity-induced glucose intolerance and insulin resistance and that this protection may be mediated in part via an indirect effect on the visceral adipose tissue.
FIG 7.

Map4k4 deletion in Myf-5-positive tissues improves insulin signaling in visceral adipose tissue. (A to D) Animals were fed an ND or an HFD for 8 weeks. (A) Representative protein immunoblot of AKT signaling in liver, skeletal muscle, and VAT in response to a bolus insulin injection (1 U/kg) as described in Materials and Methods. (B) Densitometry analysis of the data from the experiment described for panel A (n = 6 or 7). (C) Representative skeletal muscle (soleus) and VAT histology of HFD-fed control and Myf5-Map4k4-KO mice. Slides were stained with H&E (n = 10). (D) VAT was isolated and RT-qPCR performed on the indicated genes of HFD-fed animals (n = 6 to 12). (E to H) Animals were fed an HFD for 8 weeks. (E) Relative levels of gene expression in skeletal muscle (soleus). Data represent circulating levels of FGF21 (F), IL-6 (G), and irisin (H). Results are means ± SEM (*, P < 0.05; **, P < 0.005; ***, P < 0.0001).
Taken together, the data obtained from our four Map4k4 knockout mouse models presented here suggest that the metabolic improvements observed in iMap4k4-KO mice may be due primarily to the actions of Map4k4 in cells derived from Myf5-positive skeletal muscle cells and not to the actions of Map4k4 expressed in adipose cells or hepatocytes. Since Ad-Map4k4-KO animals (which also induce Map4k4 deletion in BAT) do not display any metabolic changes, skeletal muscle rather than BAT is more likely responsible for the metabolic improvements observed in Myf5-Map4k4-KO mice. Furthermore, these data suggest that there is physiological cross talk between Myf5-positive cells and the visceral adipose tissue that contributes to obesity-induced insulin resistance and that Map4k4 may be an important key regulator of this cross talk.
DISCUSSION
The results presented here demonstrate a role for Map4k4 in promoting obesity-induced insulin resistance, suggesting Map4k4 as a novel potential therapeutic target for the treatment of T2D. Induced systemic Map4k4 deficiency improves metabolic health in obese mice by lowering fasting glucose levels and improving insulin sensitivity (Fig. 3). While this beneficial effect of systemic Map4k4 deletion in mature mice is associated with enhanced insulin signaling to AKT in adipose tissue and liver (Fig. 3), conditional-KO mice with selective Map4k4 deletion either in adipose tissue depots (Fig. 4) or in liver (Fig. 5) display no overt metabolic phenotypes. Rather, Map4k4 deficiency in Myf5-positive cells, including all skeletal muscles, largely recapitulates the effects observed in inducible whole-body Map4k4-ablated mice, improving metabolic functions in the obese state (Fig. 6). Surprisingly, Map4k4 deletion in Myf5-positive cells causes enhancement of insulin signaling to AKT in VAT (Fig. 7), a tissue that is nearly devoid of Myf5 expression (26), indicating the presence of interesting intertissue signaling between Map4k4-deficient Myf5-positive cells and white adipocytes.
Employing iMap4k4-KO mice, we bypassed the embryonic lethality observed in constitutive Map4k4-null mice (10) and found that Map4k4 is not essential for adult viability. Indeed, the iMap4k4-KO mice whose results are presented here did not display any obvious behavioral defects and continued to gain weight throughout the study (Fig. 1E and F). Furthermore, despite similar adiposity results, inducible systemic Map4k4 deletion enhanced whole-body insulin action via improved adipose and liver insulin sensitivity (Fig. 3A, D, and E). Interestingly, iMap4k4-KO mice displayed slightly reduced adipose tissue inflammation (Fig. 2), which is consistent with previous data showing that Map4k4 silencing in macrophages in vivo dampens inflammation in lean mice (27). Future studies using conditional Map4k4 deletion in macrophages will be required to assess the role of Map4k4 in immune cells in metabolic disease.
Previous reports have shown that enhanced peripheral insulin sensitivity can be associated with lowered insulin levels, as less insulin is needed to clear glucose from the circulation (28–30). Consistent with these reports, iMap4k4-KO mice displayed reduced insulin levels compared to controls (Fig. 3C). However, although the obese iMap4k4-KO mice were more insulin sensitive and displayed lower fasting glucose levels than the controls (Fig. 3B), no changes in glucose clearance were detected (Fig. 3B). We hypothesize that the reduced insulin levels in the iMap4k4-KO mice are at least in part secondary to enhanced peripheral insulin sensitivity. However, lower insulin levels could also explain improvements in whole-body insulin action with minimal changes in glucose tolerance. Thus, these results are not inconsistent with the notion that Map4k4 plays a role in pancreatic β-cell function, potentially promoting insulin secretion in the obese state. Additional studies will be required to address a possible role for Map4k4 in pancreatic β-cell function in vivo.
Although we initially identified Map4k4 in a screen for regulators of adipocyte function and found significant effects of Map4k4 on glucose uptake and lipogenesis in cultured adipocytes (5, 8), we have not found significant metabolic effects in mice with adipose-specific Map4k4 ablation. One possibility is that the influence of Map4k4 is diminished in a more complex in vivo environment, in which these functions are regulated by multiple intertissue signals, including neuronal and endocrine signals that are absent in cultured cell models. Another possibility is that activity of related Ste20 kinases such as Traf2 and Nck interacting kinase (Tnik) and Misshapen-like kinase 1 (Mink1) compensates for the loss of Map4k4 in the adipose tissue in vivo.
Using Myf5-cre mice to deplete Map4k4 in skeletal muscle, we found that despite the equal levels of adiposity (Fig. 6C to E), Myf5-Map4k4-KO mice were resistant to obesity-induced metabolic dysfunction (Fig. 6N and P). Because previous work in our laboratory suggested that Map4k4 is highly expressed in satellite cells and may play a role during early stages of muscle differentiation (22), we selected a cre recombinase driven by a promoter that is expressed during this time frame (31–33). However, skeletal muscle weights (Fig. 6F) and histology results (Fig. 7C) appeared normal in Myf5-Map4k4-KO mice, suggesting no changes in muscle development. Although recent studies indicate that Myf5 is also expressed in certain adipose tissue depots (23–25), we hypothesize that Myf5-expressing myocytes and not brown adipose cells mediate the metabolic effects observed in Myf5-Map4k4-KO mice, as Ad-Map4k4-KO animals, which also delete in the BAT, do not display any improvements in metabolic function (Fig. 4). Interestingly, although Myf5-Map4k4-KO mice did not display any changes in Map4k4 expression in whole VAT, these mice displayed lower Map4k4 expression in isolated adipocytes from VAT; however, this decrease in Map4k4 expression was not mediated by Map4k4 deletion as indicated by analysis of genomic DNA (data not shown). Furthermore, we hypothesize that this decrease in Map4k4 expression in isolated adipocytes is not responsible for the metabolic changes observed in Myf5-Map4k4-KO mice, as adipose-specific Map4k4 deletion did not alter metabolic parameters (Fig. 4).
Map4k4 deletion in the Myf5-positive tissues largely recapitulates the improved metabolic effects observed upon systemic Map4k4 ablation, protecting mice from obesity-induced metabolic dysfunction. Although both iMap4k4-KO and Myf5-Map4k4-KO mice display enhanced whole-body insulin action and reduced fasting glucose levels (Fig. 3 and 6), glucose clearance was not altered in iMap4k4-KO mice (Fig. 3B). Since iMap4k4-KO mice deplete Map4k4 systemically, this discrepancy between models may be due to a potential role of Map4k4 in the pancreas which is observed only in iMap4k4-KO mice. Consistent with this hypothesis, iMap4k4-KO mice display reduced fasting insulin levels (Fig. 3C), whereas Myf5-Map4k4-KO mice do not (Fig. 6N). Both models, however, displayed improved peripheral insulin sensitivity in association with enhanced insulin signaling to AKT in VAT (Fig. 7A); thus, these data are consistent with previous studies that showed that silencing Map4k4 expression prevents cytokine-induced insulin resistance (9).
Because Myf5-Map4k4-KO mice displayed improved insulin sensitivity in the VAT (Fig. 7A and B), a tissue that did not show Map4k4 deletion (Fig. 6B), Map4k4 ablation in Myf5-positive cells apparently impacts Myf5-negative tissues. This observation suggests that Map4k4 could regulate production or activity of an as-yet-unidentified factor (or factors) that in turn influences adipose tissue insulin sensitivity. Several muscle-derived mediators have been identified, including FGF21, IL-6, and irisin (34). FGF21 and IL-6 are mostly secreted from other tissues, including the liver under fasting conditions (FGF21) and immune cells during an inflammatory response (IL-6) (34). We measured the mRNA expression and circulating levels of these two secreted factors and found no statistically significant differences between control and Myf5-Map4k4-KO mice (Fig. 7). Another candidate, irisin, has been reported to increase in level after exercise and to induce a white-to-brown shift in adipocytes (35), but we have not observed changes in gene expression or circulating levels of irisin and we also have not observed any white-to-brown shift in either VAT or SAT (data not shown). Thus, future studies will aim to identify the putative circulating factor(s) that improves insulin sensitivity in Myf5-Map4k4-KO mice.
ACKNOWLEDGMENTS
This work was supported by NIH Merit Award grant R37-DK030898 and by a grant from the International Research Alliance at Novo Nordisk Foundation Center for Metabolic Research. Part of this study was performed at the National Mouse Metabolic Phenotyping Center (MMPC) at UMass, funded by NIH grant U24-DK093000.
We thank members of the laboratory of M.P.C. for helpful discussions.
REFERENCES
- 1.Boucher J, Kleinridders A, Kahn CR. 1 January 2014, posting date Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meneghini LF. 2013. Intensifying insulin therapy: what options are available to patients with type 2 diabetes? Am J Med 126:S28–S37. doi: 10.1016/j.amjmed.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 3.Mansour M. 2014. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. Prog Mol Biol Transl Sci 121:217–266. doi: 10.1016/B978-0-12-800101-1.00007-7. [DOI] [PubMed] [Google Scholar]
- 4.Soccio RE, Chen ER, Lazar MA. 2014. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 20:573–591. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang X, Guilherme A, Chakladar A, Powelka AM, Konda S, Virbasius JV, Nicoloro SM, Straubhaar J, Czech MP. 2006. An RNA interference-based screen identifies MAP4K4/NIK as a negative regulator of PPARgamma, adipogenesis, and insulin-responsive hexose transport. Proc Natl Acad Sci U S A 103:2087–2092. doi: 10.1073/pnas.0507660103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sartorius T, Staiger H, Ketterer C, Heni M, Machicao F, Guilherme A, Grallert H, Schulze MB, Boeing H, Stefan N, Fritsche A, Czech MP, Haring HU. 2012. Association of common genetic variants in the MAP4K4 locus with prediabetic traits in humans. PLoS One 7:e47647. doi: 10.1371/journal.pone.0047647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elbein SC, Das SK, Hallman DM, Hanis CL, Hasstedt SJ. 2009. Genome-wide linkage and admixture mapping of type 2 diabetes in African American families from the American Diabetes Association GENNID (Genetics of NIDDM) Study Cohort. Diabetes 58:268–274. doi: 10.2337/db08-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danai LV, Guilherme A, Guntur KV, Straubhaar J, Nicoloro SM, Czech MP. 2013. Map4k4 suppresses Srebp-1 and adipocyte lipogenesis independent of JNK signaling. J Lipid Res 54:2697–2707. doi: 10.1194/jlr.M038802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Austin RL, Rune A, Bouzakri K, Zierath JR, Krook A. 2008. siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle. Diabetes 57:2066–2073. doi: 10.2337/db07-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue Y, Wang X, Li Z, Gotoh N, Chapman D, Skolnik EY. 2001. Mesodermal patterning defect in mice lacking the Ste20 NCK interacting kinase (NIK). Development 128:1559–1572. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Li S, Mana-Capelli S, Roth Flach RJ, Danai LV, Amcheslavsky A, Nie Y, Kaneko S, Yao X, Chen X, Cotton JL, Mao J, McCollum D, Jiang J, Czech MP, Xu L, Ip YT. 2014. The conserved misshapen-warts-yorkie pathway acts in enteroblasts to regulate intestinal stem cells in Drosophila. Dev Cell 31:291–304. doi: 10.1016/j.devcel.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry MN, Friend DS. 1969. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J Cell Biol 43:506–520. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folch J, Lees M, Sloane Stanley GH. 1957. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 226:497–509. [PubMed] [Google Scholar]
- 14.Roth Flach R, Matevossian A, Akie TE, Negrin KA, Paul MT, Czech MP. 2013. b3-Adrenergic receptor stimulation induces E-selectin-mediated adipose tissue inflammation. J Biol Chem 288:2882–2892. doi: 10.1074/jbc.M112.412346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sam S, Mazzone T. 2014. Adipose tissue changes in obesity and the impact on metabolic function. Transl Res 164:284–292. doi: 10.1016/j.trsl.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 16.Guilherme A, Virbasius JV, Puri V, Czech MP. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustafson B, Gogg S, Hedjazifar S, Jenndahl L, Hammarstedt A, Smith U. 2009. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am J Physiol Endocrinol Metab 297:E999–E1003. doi: 10.1152/ajpendo.00377.2009. [DOI] [PubMed] [Google Scholar]
- 18.Lafontan M. 2014. Adipose tissue and adipocyte dysregulation. Diabetes Metab 40:16–28. doi: 10.1016/j.diabet.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Blüher M. 2013. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab 27:163–177. doi: 10.1016/j.beem.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Isakson P, Hammarstedt A, Gustafson B, Smith U. 2009. Impaired preadipocyte differentiation in human abdominal obesity: role of Wnt, tumor necrosis factor-alpha, and inflammation. Diabetes 58:1550–1557. doi: 10.2337/db08-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farese RV Jr, Zechner R, Newgard CB, Walther TC. 2012. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 15:570–573. doi: 10.1016/j.cmet.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang M, Amano SU, Flach RJ, Chawla A, Aouadi M, Czech MP. 2013. Identification of Map4k4 as a novel suppressor of skeletal muscle differentiation. Mol Cell Biol 33:678–687. doi: 10.1128/MCB.00618-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atit R, Sgaier SK, Mohamed OA, Taketo MM, Dufort D, Joyner AL, Niswander L, Conlon RA. 2006. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev Biol 296:164–176. doi: 10.1016/j.ydbio.2006.04.449. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez-Gurmaches J, Hung CM, Sparks CA, Tang Y, Li H, Guertin DA. 2012. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell Metab 16:348–362. doi: 10.1016/j.cmet.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. 2008. PRDM16 controls a brown fat/skeletal muscle switch. Nature 454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Gurmaches J, Guertin DA. 2014. Adipocyte lineages: tracing back the origins of fat. Biochim Biophys Acta 1842:340–351. doi: 10.1016/j.bbadis.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostroff GR, Czech MP. 2009. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 458:1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li LO, Grevengoed TJ, Paul DS, Ilkayeva O, Koves TR, Pascual F, Newgard CB, Muoio DM, Coleman RA. 28 July 2014, posting date Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes doi: 10.2337/db13-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson JM, Seldin MM, Tan SY, Wong GW. 2014. CTRP2 overexpression improves insulin and lipid tolerance in diet-induced obese mice. PLoS One 9:e88535. doi: 10.1371/journal.pone.0088535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, Saleh M, Snowman AM, Moran TH, Mezey E, Snyder SH. 2010. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 143:897–910. doi: 10.1016/j.cell.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gensch N, Borchardt T, Schneider A, Riethmacher D, Braun T. 2008. Different autonomous myogenic cell populations revealed by ablation of Myf5-expressing cells during mouse embryogenesis. Development 135:1597–1604. doi: 10.1242/dev.019331. [DOI] [PubMed] [Google Scholar]
- 32.Tallquist MD, Weismann KE, Hellstrom M, Soriano P. 2000. Early myotome specification regulates PDGFA expression and axial skeleton development. Development 127:5059–5070. [DOI] [PubMed] [Google Scholar]
- 33.Waddell JN, Zhang P, Wen Y, Gupta SK, Yevtodiyenko A, Schmidt JV, Bidwell CA, Kumar A, Kuang S. 2010. Dlk1 is necessary for proper skeletal muscle development and regeneration. PLoS One 5:e15055. doi: 10.1371/journal.pone.0015055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eckardt K, Gorgens SW, Raschke S, Eckel J. 2014. Myokines in insulin resistance and type 2 diabetes. Diabetologia 57:1087–1099. doi: 10.1007/s00125-014-3224-x. [DOI] [PubMed] [Google Scholar]
- 35.Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Boström EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Højlund K, Gygi SP, Spiegelman BM. 2012. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]