

Abstract
mTOR complex 2 (mTORC2) regulates cell survival and growth through undefined mechanisms. Mcl-1, a Bcl-2 family protein, functions as an oncogenic protein. The connection between mTORC2 and Mcl-1 stability has not been established and was thus the focus of this study. Mcl-1 levels in cancer cells were decreased by mTOR kinase inhibitors (TORKinibs), which inhibit both mTORCs, by knocking down rictor and by knocking out rictor or Sin1 but not by silencing raptor. TORKinib treatment and rictor knockdown did not alter Mcl-1 mRNA levels but rather decreased its protein stability. Moreover, TORKinib-induced Mcl-1 reduction was rescued by proteasome inhibition. Consistently, TORKinib increased Mcl-1 ubiquitination. Hence, it is clear that inhibition of mTORC2 enhances Mcl-1 degradation, resulting in Mcl-1 reduction. Suppression of glycogen synthase kinase 3 (GSK3) or FBXW7 rescued Mcl-1 reduction induced by TORKinibs or rictor knockdown. Thus, mTORC2 inhibition apparently induces Mcl-1 degradation through a GSK3-dependent and SCF-FBXW7-mediated mechanism. Intriguingly, we detected a direct association between mTORC2 and SCF-FBXW7; this association could be inhibited by TORKinib treatment, suggesting that mTORC2 may directly associate with and inhibit the SCF-FBXW7 complex, resulting in delayed Mcl-1 degradation. Collectively, our findings highlight a novel mechanism by which mTORC2 regulates cell survival and growth by stabilizing Mcl-1.
INTRODUCTION
The mammalian target of rapamycin (mTOR) regulates a variety of biological functions essential for the maintenance of cancer cell survival and growth by forming two complexes through direct interaction with different partner proteins: raptor (mTOR complex 1 [mTORC1]) and rictor (mTORC2) (1, 2). mTORC1 is well known to regulate many key cellular processes, including cell growth and metabolism, primarily via regulating cap-dependent protein translation initiation. However, the biological functions of mTORC2, particularly those related to regulation of oncogenesis, and underlying mechanisms have not been fully elucidated. Nonetheless, mTOR signaling has emerged as an attractive cancer therapeutic target (3). The conventional allosteric mTOR inhibitors rapamycin and its analogues (rapalogs) have shown success in the treatment of a few types of cancer (4, 5). In addition, great efforts have also been made to develop novel mTOR kinase inhibitors (TORKinibs) that suppress both mTORC1 and mTORC2 activities. As a result, several ATP-competitive inhibitors of mTOR kinase such as INK128 and AZD8055 have been developed and are being tested in clinical trials (5, 6).
Mcl-1 is a well-known Bcl-2 family protein that negatively regulates apoptosis by binding and sequestering proapoptotic proteins such as Bax, Bak, Noxa, and Bim (7). Its expression can be controlled at various levels, including transcription, translation, and posttranslation (7). mTORC1 is known to regulate Mcl-1 translation, which contributes to mTORC1-dependent survival (8). However, it is unknown whether mTORC2 regulates Mcl-1 expression.
Mcl-1 is a short-lived protein known to undergo ubiquitination/proteasome-mediated degradation (7). One degradation mechanism involves glycogen synthase kinase 3 (GSK3), which phosphorylates Mcl-1 at Ser159, triggering Mcl-1 degradation (9, 10). Mcl-1 phosphorylation at Ser159 facilitates the association of Mcl-1 with the E3 ligase β-transducin repeats-containing protein (β-TrCP) or F-box/WD repeat-containing protein 7 (FBXW7), resulting in β-TrCP- or FBXW7-mediated ubiquitination and degradation of Mcl-1 (9, 11, 12). Therefore, GSK3 plays a critical role in the negative regulation of Mcl-1 stability.
Our recent study has revealed that GSK3 is required for TORKinibs to decrease cyclin D1 levels by enhancing its degradation and to inhibit the growth of cancer cells both in vitro and in vivo (13). Moreover, we have shown that inhibition of mTORC2 is responsible for GSK3-dependent cyclin D1 degradation induced by TORKinibs (13). In this study, we were interested in determining whether, and by which mechanisms, mTORC2 regulates Mcl-1 stability and whether inhibition of mTORC2 triggers GSK3-dependent Mcl-1 degradation. Indeed, we have demonstrated that mTORC2 stabilizes Mcl-1 by directly suppressing GSK3-dependent and FBXW7-mediated protein degradation.
MATERIALS AND METHODS
Reagents.
All TORKinibs, the GSK3 inhibitor SB216763, the proteasome inhibitor MG132, and the protein synthesis inhibitor cycloheximide (CHX) were the same as described previously (13). The GSK3 inhibitor CHIR99021 was purchased from LC Laboratories (Woburn, MA), and cis-diamminedichloroplatinum(II) (CDDP; cisplatin) was purchased from Sigma Chemical Co. (St. Louis, MO). Rabbit polyclonal Mcl-1 (sc-819), S phase kinase-associated protein 1 (Skp1; sc-7163), and p-SGK1 (S422; sc-16745R) polyclonal antibodies and mouse cullin-1 (Cul-1; sc-70895), Mcl-1 (sc-12756; used for immunoprecipitation [IP]), rictor (sc-271081; used for IP), and α-tubulin (sc-23948) monoclonal antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit GSK3α/β and SGK1 monoclonal antibodies were purchased from Upstate/EMD Millipore (Billerica, MA). rictor and raptor polyclonal antibodies were purchased from Bethyl Laboratories, Inc. (Montgomery, TX). Expression plasmids in vector pCI carrying wild-type (WT) and mutant (S159A) human Mcl-1 were provided by X. Deng (Emory University, Atlanta, GA). The myc-rictor and hemagglutinin (HA)-raptor expression plasmids used were purchased from Addgene (Cambridge MA).
Cell lines and cell culture.
The human non-small-cell lung cancer (NSCLC) cell lines used in this study, H157-scramble, H157-shRaptor, H157-shRictor, A549-pLKO.1, A549-shRaptor, and A549-shRictor stable cell lines, were described in our previous papers (14, 15). The H1299/Mcl-1 cell line was established previously (16). Immortalized WT and Rictor knockout (KO) murine embryonic fibroblasts (MEFs) were provided by M. A. Magnuson (Vanderbilt University Medical Center, Nashville, TN). rictor-KO MEFs with reexpression of myc-rictor (rictor-KO/rictor) or the matched vector (rictor-KO/vector) were provided by D. D. Sarbassov (M. D. Anderson Cancer Center, Houston, TX). WT and Sin1-KO MEFs were provided by B. Su (Yale University School of Medicine, New Haven, CT). HCT116-FBXW7-WT and HCT116-FBXW7-KO cell lines were kindly provided by B. Vogelstein (Johns Hopkins University School of Medicine, Baltimore, MD). HEK-293T cells were provided by K. Ye (Emory University, Atlanta, GA). Except for H157 and A549 cells, which were authenticated by Genetica DNA Laboratories, Inc. (Cincinnati, OH), by analyzing short tandem repeat DNA profiles, the other cell lines have not been authenticated. These cell lines were cultured in RPMI 1640, Dulbecco's modified Eagle's medium, or McCoy's medium containing 5% fetal calf serum at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Plasmid transfection was conducted primarily with HEK-293T or H1299 cells largely because of their high transfection efficiency.
Cell growth and colony formation assays.
Cells were seeded into 96-well cell culture plates and treated the next day with the agents described. Viable cell numbers were determined with a sulforhodamine B (SRB) assay as described previously (17). The combination index (CI) for drug interaction (e.g., synergy) was calculated with the CompuSyn software (ComboSyn, Inc., Paramus, NJ). The colony formation assay on plastic dishes was conducted as described previously (13).
Western blot analysis.
Preparation of whole-cell protein lysates and Western blot analysis were performed as described previously (18).
Gene knockdown by siRNA or small hairpin RNA.
The nonsilencing (control), rictor, raptor, and GSK3α/β small interfering RNAs (siRNAs) used were the same as those described previously (13). FBXW7, Cul-1, Skip1, and β-TrCP siRNAs were also described in our previous study (19). β-TrCP siRNA (sc-37178) is a pool of three different siRNA duplexes consisting of 5′-GAAGUUAAGUGGUCUGAAATT-3′, 5′-CCAGUUUGUUGUAGAAGAATT-3′, and 5′-GGAUCUCUAUUGACAAGUATT-3′ that target the 3′ untranslated region of five known β-TrCP isoforms, including isoforms 1, 2, 3, X1, and X2. Transfection of these siRNA duplexes was conducted with six-well plates and the Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA) by following the manufacturer's manual.
IP.
IP for detection of Mcl-1 ubiquitination was done as follows. H1299/Mcl-1 cells were transfected with HA-ubiquitin by using Lipofectamine 2000 transfection reagent (Life Technologies, Grand Island, NY) in accordance with the manufacturer's instructions. After 48 h, the cells were treated with INK128 or INK128 plus MG132 for an additional 4 h. Cells were then collected and lysed in 1% NP-40 buffer (50 mM Tris-Cl [pH 7.4], 150 mM NaCl, 2 mM EDTA) supplemented with protease and phosphatase inhibitors for IP with an anti-Mcl-1 antibody, followed by detection of ubiquitinated Mcl-1 by Western blot analysis with anti-HA antibody (Abgent, San Diego, CA). IP for detection of protein interaction or association was conducted as follows. Cells transfected with the plasmids indicated or treated with a TORKinib were lysed in CHAPS buffer containing 40 mM HEPES (pH 7.5), 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM glycerol phosphate, 50 mM NaF, 0.5 mM Na3VO4, and 0.3% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) and supplemented with protease and phosphatase inhibitors. Whole-cell protein lysates (500 μg) were then incubated with an appropriate antibody or antibody-conjugated beads overnight at 4°C. After being washed twice with 0.3% CHAPS lysis buffer and HEPES wash buffer (50 mM HEPES, 40 mM NaCl, 2 mM EDTA, pH 8.0), respectively, immunocomplexes were subjected to Western blotting for detection of the proteins of interest.
RT-PCR and real-time qPCR.
Total cellular RNA preparation, reverse transcription (RT)-PCR, and primers for detection of Mcl-1, FBXW7, and β-TrCP were the same as described previously (19). Quantitative PCR (qPCR) was performed with the iTaq Universal SYBR green Supermix (Bio-Rad) on a 7500 Fast real-time PCR system (Life Technologies/Applied Biosystems, Grand Island, NY) by following the manufacturer's instructions.
Lung cancer xenografts and treatments.
Lung cancer xenograft experiments were approved by the Institutional Animal Care and Use Committee of Emory University and conducted as previously described (14). Five- to 6-week-old (body weight, about 20 g) female athymic (nu/nu) mice were ordered from Harlan (Indianapolis, IN). H460 cells (5 × 106 in 100 μl of phosphate-buffered saline [PBS]) were injected subcutaneously into the right flank of each mouse. Mice (n = 6 or 7/group) were treated with the vehicle control, INK128 formulated in 5% N-methyl-2-pyrrolidone, 15% polyvinylpyrrolidone in water (0.5 mg/kg administered by oral gavage once daily) (20), CDDP in saline (2 mg/kg administered intraperitoneally once every 3 days), or a combination of INK128 and CDDP.
Statistical analysis.
The statistical significance of a difference between two groups was analyzed with a two-sided unpaired Student t test by use of InStat 3 software (GraphPad Software, San Diego, CA). Results were considered statistically significant at a P value of <0.05.
RESULTS
TORKinibs decrease Mcl-1 levels in NSCLC cells.
We first treated A549 cells with different concentrations of several representative TORKinibs, including INK128, AZD8055, and Torin 1, and detected Mcl-1 protein level alteration. As shown in Fig. 1A, these TORKinibs at concentrations ranging from 50 to 1,000 nM effectively decreased the levels of p-S6 (S235/236), p-Akt (S473), and p-SGK1 (S422), indicating their effectiveness against both mTORC1 and mTORC2 signaling. In parallel, they dose dependently decreased Mcl-1 levels. We noted that INK128 had a more potent effect than AZD8055 and Torin1 in both inhibiting mTORC signaling (i.e., suppressing the phosphorylation of S6, Akt, and SGK1) and decreasing Mcl-1 levels. The reduction of Mcl-1 occurred at 1 h posttreatment and was sustained for >12 h in both the A549 and H460 cell lines (Fig. 1B). NSCLC cell lines displayed various sensitivities to TORKinibs (e.g., INK128 and AZD8055) based on a 3-day growth inhibition assay (Fig. 1C). We found that the basal levels of Mcl-1 were, in general, higher in less sensitive cell lines (e.g., H23 and Calu-1) than in sensitive cell lines. Moreover, treatment with both INK128 and AZD8055 led to a substantial decrease in Mcl-1 levels in sensitive NSCLC cell lines (e.g., H460, A549, and H1648) but little or no decrease in relatively less sensitive cell lines (e.g., H23, Calu-1, and EKVX) (Fig. 1C). In these cell lines, our previous study has shown that both INK128 and AZD8055 effectively reduce the levels of p-Akt (S473) and p-S6 (S235/236), regardless of cell sensitivity (13). Together, these data indicate that TORKinibs effectively decrease Mcl-1 levels in NSCLC cells, particularly those sensitive to TORKinibs.
FIG 1.

TORKinibs reduce Mcl-1 levels in human NSCLC cells lines. (A) A549 cells were treated with the indicated concentrations (Conc.) of TORKinibs for 4 h. (B) The indicated cell lines were treated with 100 nM INK128 for different times as indicated. (C) The indicated NSCLC cell lines with various sensitivities to TORKinibs (lower panel) were exposed to 100 nM INK128 or AZD8055 for 4 h. After the aforementioned treatments, whole-cell protein lysates were prepared from these cell lines and subjected to Western blotting for detection of the indicated proteins. The growth curves in panel C were determined with the SRB assay after plating cells in 96-well plates and exposing them to the indicated TORKinib for 3 days. The data are means ± standard deviations of four replicate determinations. LE, long exposure; SE, short exposure; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Genetic manipulation of rictor and Sin1 but not raptor expression alters Mcl-1 levels.
To decipher the involvement of mTORCs in the positive regulation of Mcl-1 expression, we compared the effects of knockdown of rictor and raptor on Mcl-1 expression. We detected substantially reduced levels of Mcl-1 in both the A549-shRictor and H157-shRictor cell lines, in which rictor was stably silenced, but not in the A549-shRaptor and H157-shRaptor cell lines, in which raptor has been stably knocked down (Fig. 2A). In agreement, transient knockdown of rictor but not raptor by siRNA transfection also reduced Mcl-1 levels (Fig. 2B). Moreover, we detected lower levels of Mcl-1 in rictor-KO MEFs than in WT and rictor-KO/rictor MEFs (Fig. 2C), indicating that rictor deficiency reduces Mcl-1 levels and rictor reexpression can rescue Mcl-1 reduction. Consistently, KO of Sin1, an essential component of mTORC2, also decreased Mcl-1 levels (Fig. 2D). Hence, these results clearly show a rictor- or mTORC2-dependent positive regulation of Mcl-1 expression. This notion is further supported by our finding that enforced ectopic rictor but not raptor expression elevated Mcl-1 levels (Fig. 2E).
FIG 2.

Genetic inhibition of rictor or Sin1 but not raptor expression (A to D) and enforced expression of ectopic rictor but not raptor (E) alter Mcl-1 protein levels (A to E) but not its mRNA level (F). (A to E) Whole-cell protein lysates were prepared from the indicated transfectants or MEFs (A, C, and D), A549 cells 48 h after transfection with the indicated siRNAs (B), and 293T cells 48 h after transfection with plasmids harboring a given gene (E). Lysates were then subjected to Western blotting to detect the indicated proteins. SE, short exposure. (F) Total cellular RNA was prepared from the indicated MEFs and then subjected to RT-PCR for detection of Mcl-1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNAs. Ctrl, control.
Both TORKinib and rictor inhibition destabilize Mcl-1 protein by promoting its proteasomal degradation.
We next determined the underlying mechanism of Mcl-1 reduction. We found that rictor deficiency did not affect Mcl-1 mRNA levels (Fig. 2F). Consistently, TORKinibs also did not alter Mcl-1 mRNA levels, as evaluated by both RT-PCR (Fig. 3A) and qPCR (data not shown). These results suggest a posttranscriptional mechanism. Since Mcl-1 is known to be an unstable protein regulated by degradation, we compared the stabilities of Mcl-1 protein among dimethyl sulfoxide (DMSO)-, AZD8055-, and INK128-treated cells. As shown in Fig. 3B, Mcl-1 protein levels were reduced much more rapidly in AZD8055- or INK128-treated cells than in DMSO-treated cells, demonstrating that TORKinibs destabilize Mcl-1 protein. Consistently, we also detected a faster reduction of Mcl-1 protein in A549-shRictor cells than in A549-pLKO.1 and shRaptor cells (Fig. 3C). Complementarily, enforced expression of ectopic rictor but not raptor slowed Mcl-1 reduction (Fig. 3D and E). Thus, it is rictor that positively regulates Mcl-1 stability.
FIG 3.

Inhibition of mTORC2 by TORKinibs (A, B, F, and G) or rictor knockdown (C) does not alter Mcl-1 mRNA levels (A) but rather promotes Mcl-1 protein degradation (B to D), whereas enforced expression of rictor but not raptor stabilizes Mcl-1 (D and E). (A) A549 cells were treated with DMSO or the indicated TORKinibs at 100 nM for 8 h. Total cellular RNA was extracted for detection of Mcl-1 mRNA levels by RT-PCR. (B) A549 cells were treated with DMSO or 100 nM INK128 or AZD8055 for 4 h. The cells were then washed with PBS three times and refed with fresh medium containing 10 μg/ml CHX. At the indicated times, the cells were harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. Protein levels were quantified with NIH ImageJ software and normalized to tubulin. (C to E) Whole-cell protein lysates were prepared from the indicated A549 transfectants exposed to 10 μg/ml CHX for different times as indicated (C) and 293T cells 48 h after transfection with myc-rictor or HA-raptor, followed by exposure to 10 μg/ml CHX for different times as indicated (D and E). The lysates were then subjected to Western blot analysis. Protein levels were quantified with NIH ImageJ software and normalized to tubulin. (F) A549 cells were pretreated with 10 μM MG132 for 1 h and then cotreated with the indicated TORKinibs (100 nM for INK128, AZD8055, and Torin 1 and 1 μM for PP242) for 4 h. The cells were harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. (G) H1299/Mcl-1 cells were transfected with HA-ubiquitin (Ub). After 48 h, the cells were pretreated with 10 μM MG132 for 40 min and then cotreated with 100 nM INK128 for another 4 h. Whole-cell protein lysates were then prepared for IP with anti-Mcl-1 antibody, followed by Western blotting (WB) with the indicated antibodies.
We also detected Mcl-1 reduction in A549 cells exposed to different TORKinibs, including AZD8055, INK128, Torin 1, and PP242, but this effect was abolished in the presence of the proteasome inhibitor MG132 (Fig. 3F), indicating that TORKinibs induce Mcl-1 through a proteasome-mediated mechanism. Furthermore, we detected higher levels of polyubiquitinated Mcl-1 in cells exposed to INK128 plus MG132 than in cells treated with INK128 alone or MG132 alone (Fig. 3G), indicating that INK128 increases Mcl-1 polyubiquitination. Collectively, we conclude that TORKinibs or rictor inhibition induces ubiquitination/proteasome-mediated degradation of Mcl-1, resulting in reduction of Mcl-1.
TORKinibs induce GSK3-dependent Mcl-1 degradation.
Since GSK3-mediated phosphorylation of Mcl-1 at Ser159 is critical for Mcl-1 degradation (9–12), we next determined whether GSK3 is involved in Mcl-1 degradation induced by TORKinibs. All of the TORKinibs tested, including INK128, AZD8055, Torin 1, and PP242, decreased the levels of Mcl-1 in the absence of the GSK3 inhibitor SB216763 but failed to do so in its presence (Fig. 4A). Similar results were also generated with another GSK3 inhibitor, CHIR99021 (Fig. 4B). Consistently, genetic inhibition of GSK3 via siRNA-mediated gene knockdown also rescued Mcl-1 reduction induced by INK128 (Fig. 4C). Furthermore, the presence of either SB216763 or CHIR99021 blocked the reduction of Mcl-1's half-life by INK128 (Fig. 4D). These data robustly demonstrated that TORKinibs induce GSK3-dependent Mcl-1 degradation. Under the conditions tested, we found that INK128 increased p-Mcl-1 (S159/163) (Fig. 4E). In addition, INK128 or AZD8055 decreased the levels of ectopic WT Mcl-1 but not mutant Mcl-1 (S159A), in which serine 159 was changed to alanine (Fig. 4F), further supporting the notion that TORKinibs induce GSK3-dependent Mcl-1 degradation.
FIG 4.

Inhibition of GSK3 with SB216763, CHIR99021, or siRNA rescues Mcl-1 degradation induced by TORKinibs (A to D), and GSK3 phosphorylation of Mcl-1 is critical for Mcl-1 reduction by TORKinibs (E and F). (A and B) A549 cells were pretreated with 10 μM SB216763 or CHIR99021 for 30 min and then cotreated with the indicated TORKinibs (100 nM for INK128, AZD8055, and Torin 1 and 1 μM for PP242) for an additional 10 h (A) or 6 h (B). (C) A549 cells were transfected with the indicated siRNAs for 48 h and then exposed to 100 nM INK128 for an additional 4 h. (D) A549 cells were treated with DMSO, 100 nM INK128, or INK128 plus 10 μM SB216763 or CHIR99021 for 6 h. The cells were then washed with PBS three times and refed with fresh medium containing 10 μg/ml CHX. At the indicated times, the cells were harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. Protein levels were quantified with NIH ImageJ software and normalized to tubulin. (E) A549 cells were exposed to 100 nM INK128 for different times as indicated and then harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. (F) 293T cells were transfected with plasmids carrying the indicated genes, and after 48 h, they were treated for an additional 10 h with 100 nM TORKinibs as indicated. After the aforementioned treatments, whole-cell protein lysates were prepared from these cells for Western blotting to detect the indicated proteins. Ctrl, control.
TORKinibs induce Skp, cullin, F-box (SCF)-FBXW7-mediated Mcl-1 degradation.
Since both SCF E3 ligases FBXW7 and β-TrCP have been suggested to mediate GSK3-dependent Mcl-1 degradation (9, 11, 12), we then determined the possible involvement of these E3 ligases in TORKinib-induced Mcl-1 degradation. We found that disruption of the SCF complex by knocking down Skp1 and particularly by knocking down both Cul-1 and Skp1, not only elevated the baseline Mcl-1 levels but also prevented INK128-induced Mcl-1 reduction (Fig. 5A), suggesting the involvement of an SCF E3 ligase in this process. In this study, we noted that Cul-1 knockdown did not rescue Mcl-1 reduction induced by INK128, although it elevated basal Mcl-1 levels. Whether this is due to insufficient knockdown of Cul-1 needs further investigation. Subsequently, we compared the effects of knocking down FBXW7, β-TrCP, or both on INK128-induced Mcl-1 reduction and found that INK128 decreased Mcl-1 levels in control siRNA- and β-TrCP siRNA-transfected cells but not in cells transfected with FBXW7 siRNA or the FBXW7 and β-TrCP siRNAs (Fig. 5B). The knockdown efficiencies of FBXW7 and β-TrCP were confirmed by RT-PCR (Fig. 5B, lower panel). Hence, it is FBXW7, rather than β-TrCP, that is responsible for TORKinib-induced Mcl-1 degradation. In the H1299 cell line, in which transfection of FBXW7 siRNA effectively knocks down FBXW7 expression as we described previously (16), we observed that INK128, AZD8055, and Torin 1 drastically decreased Mcl-1 levels in control siRNA-transfected H1299 cells, while little or no decrease was seen in FBXW7 siRNA-transfected cells (Fig. 5C). In agreement, these TORKinibs decreased Mcl-1 levels in HCT116 parental cells but not in HCT116 cells deficient in FBXW7 (FBXW7-KO) (Fig. 5D). Similarly, rictor knockdown reduced Mcl-1 levels in HCT116 parental cells but not in FBXW7-KO HCT116 cells (Fig. 5E). These data further support our conclusion that FBXW7 mediates Mcl-1 degradation induced by TORKinibs or rictor inhibition. In our study, we found that TORKinibs did not alter FBXW7 mRNA levels, as evaluated by both RT-PCR and qPCR (data not shown).
FIG 5.

Inhibition of SCF-FBXW7 by knocking down Cul-1 and Skp1 (A) or FBXW7 (B and C) or by knocking out FBXW7 (D and E) rescues Mcl-1 reduction induced by TORKinibs (A to D) or rictor silencing (E), whereas TORKinibs decrease the levels of other known FBXW7 substrates (F). (A to C) The indicated cell lines were transfected with control (Ctrl) or other indicated siRNAs, and after 48 h, they were exposed to the indicated TORKinibs (100 nM) for an additional 4 h. (D) WT and FBXW7-KO HCT116 cells were treated with the indicated TORKinibs at 100 nM for 3 h. (E) WT and FBXW7-KO HCT116 cells were transfected with the indicated siRNAs for 48 h. After these treatments, the cells were harvested for preparation of whole-cell protein lysates and subsequent Western blotting. FBXW7 knockdown efficiency in panel B was evaluated by RT-PCR. SE, short exposure. (F) A549 cells were treated for 8 h with the different TORKinibs indicated at 100 nM and then harvested for preparation of whole-cell protein lysates and subsequent Western blotting.
Since many other proteins, such as c-Myc, c-Jun, and cyclin E, are known to undergo GSK3/SCF-FBXW7-dependent degradation (21, 22), we predicted that TORKinibs would decrease the levels of these proteins as well. Indeed, INK128, AZD8055, and Torin 1 decreased the levels of c-Myc, c-Jun, and cyclin E in addition to Mcl-1 (Fig. 5F).
mTORC2 is associated with the SCF-FBXW7 complex and inhibits its function.
It has been suggested that rictor is involved in the regulation of protein degradation (e.g., SGK1 and cyclin E1) by directly interacting with the SCF complex or FBXW7 (23, 24). To understand how mTORC2 positively regulates Mcl-1 stability or how TORKinibs induce FBXW7-mediated Mcl-1 degradation, we asked whether mTORC2 is physically associated with the SCF-FBXW7 complex. Because of the lack of a reliable FBXW7 antibody, we transfected Flag-FBXW7 into 293T cells, treated them with INK128, and then conducted IP with anti-Flag antibody and subsequent Western blotting to detect different proteins of interest in the immunoprecipitates. As shown in Fig. 6A, we detected Skp1 (a key component of SCF), rictor, mTOR, and Mcl-1, indicating that FBXW7 is associated or interacts with these proteins. Compared with cells exposed to DMSO, we detected reduced amounts of both rictor and mTOR while Skp1 remained unchanged in INK128-treated cells, suggesting that INK128 treatment promotes dissociation of mTOR and rictor from SCF-FBXW7. Using Mcl-1 antibody to pull down endogenous Mcl-1 in A549 cells, we detected Cul-1, Skp1, rictor, and mTOR in the immunoprecipitate. INK128 treatment decreased the amounts of both rictor and mTOR without altering the amounts of Cul-1 and Skp1 (Fig. 6B). In the same cell line, we pulled down endogenous rictor with a rictor antibody and detected Mcl-1, Cul-1, Skp1, and mTOR (a known partner of rictor) in the immunoprecipitate and reduced amounts of Mcl-1, Cul-1, and Skp1 in cells treated with INK128 (Fig. 6C). These results suggest that Mcl-1 is associated with Cul-1, Skp1, rictor, and mTOR and that INK128 treatment facilitates dissociation of Mcl-1 from rictor and mTOR.
FIG 6.

Detection of a possible association between mTORC2 and the SCF-FBXW7 complex (A to E) and a working model of negative regulation of SCF-FBXW7-mediated Mcl-1 degradation by mTORC2 (F). (A) 293T cells were transfected with the vector or a Flag-FBXW7 plasmid, and after 48 h, they were treated with 100 nM INK128 for 2 h. (B and C) A549 cells were exposed to DMSO or 100 nM INK128 for 3 h. (D) 293T cells were cotransfected with Flag-FBXW7 and WT or mutant Mcl-1 for 48 h. (E) 293T cells were cotransfected with myc-rictor and WT or mutant Mcl-1 for 48 h. After the aforementioned transfections or treatment, the cells were harvested for preparation of whole-cell protein lysates and subsequent IP-Western blotting (WB) to detect the indicated proteins. SE, short exposure; V, vector; *, unstripped rictor band. (F) Working model of negative regulation of SCF-FBXW7-mediated Mcl-1 degradation by mTORC2. GSK3-dependent phosphorylation of Mcl-1 at Ser159 enhances the association of Mcl-1 with both the SCF-FBXW7 complex and mTORC2; inhibition of mTORC2 (e.g., with a TORKinib) facilitates the dissociation of mTORC2 from the SCF-FBXW7 complex, allowing the SCF-FBXW7 complex to degrade Mcl-1 by the ubiquitin (Ub)-proteasome (P) pathway.
Mcl-1 phosphorylation at Ser159 is critical for Mcl-1 association with both rictor and FBXW7.
It has been shown that Mcl-1 Ser159 phosphorylation is required for FBXW7 binding to Mcl-1 (11, 12). In our study, we demonstrated the critical role of Mcl-1 Ser159 phosphorylation in modulating FBXW7 binding to Mcl-1 by cotransfection of FBXW7 and WT or mutant Mcl-1, followed by IP-Western blotting. Using an anti-Flag tag antibody to pull down FBXW7, we detected much smaller amounts of S159A mutant Mcl-1 than WT Mcl-1 (Fig. 6D). Next, we asked whether Ser159 phosphorylation impacts the association of Mcl-1 with rictor or mTORC2. To this end, we cotransfected myc-rictor and WT or mutant (S159A) Mcl-1 into 293T cells and then conducted IP with an anti-Myc tag antibody, followed by Western blotting to detect Mcl-1 and rictor. We detected smaller amounts of S159A mutant than WT Mcl-1 (Fig. 6E), indicating that Mcl-1 Ser159 phosphorylation is also important for the association of Mcl-1 with rictor.
TORKinibs enhance the cancer therapeutic efficacy of CDDP.
Despite the potent reduction of Mcl-1 by TORKinibs, as demonstrated above, TORKinibs alone at concentrations of up to 1 μM led to little or no induction of apoptosis in our tested NSCLC cell lines (data not shown). We thus wondered whether Mcl-1 downregulation might lower the thresholds at which cancer cells undergo apoptosis triggered by other stimuli (e.g., chemotherapy), leading to sensitization of cancer cells to undergo apoptosis. As a proof-of-principle study, we compared the sensitivities of WT and rictor-KO MEFs, which have differential levels of Mcl-l (Fig. 2), to CDDP, a well-known chemotherapeutic drug widely used for treatment of NSCLC and other cancers, in which Mcl-1 expression negatively impacts their responses to CDDP (25, 26). We found that rictor-KO MEFs were indeed more sensitive than WT MEFs to CDDP (Fig. 7A). This effect could be reversed by reexpression of ectopic rictor (i.e., in rictor-KO/rictor MEFs) (Fig. 7B). Furthermore, we detected larger amounts of cleaved poly(ADP-ribose) polymerase (PARP) in rictor-KO MEFs than in WT MEFs treated with CDDP (Fig. 7C), suggesting that rictor deficiency makes cells prone to CDDP treatment. These results together indicate that genetic inhibition of mTORC2 indeed sensitizes cells to CDDP treatment. Following this study, we tested the effects of CDDP in the absence or presence of a TORKinib on the growth and apoptotic death of cancer cells. In both A549 and H460 cells, in which Mcl-1 levels were substantially reduced by TORKinibs (Fig. 1C), the combination of CDDP with either INK128 or AZD8055 was much more potent than either agent alone in decreasing the survival of cancer cells in a 3-day monolayer cell culture experiment (Fig. 7D). The CIs for the combinations tested were <1, indicating synergistic effects in decreasing the survival of cancer cells. In Calu-1 and H23 cells, in which Mcl-1 levels were high and only weakly reduced by TORKinibs (Fig. 1C), the combination of CDDP with INK128 or AZD8055 also showed some degree of enhanced growth suppression (Fig. 7D). However, these effects were much weaker than those in H460 and A549 cells. In a long-term colony formation assay that allows repeated treatment, identical results were also generated. The combination of CDDP with INK128 or AZD8055 was much more effective than either agent alone in suppressing the formation and growth of both A549 and H460 colonies (Fig. 7E and data not shown). Moreover, the combination of INK128 and CDDP was also more potent than either single agent in reducing Mcl-1 levels and inducing cleavage of PARP in both A549 and H460 cells (Fig. 7F), demonstrating that the combination of INK128 and CDDP enhances induction of apoptosis.
FIG 7.

Genetic (A to C) and pharmacological (D to F) inhibition of mTORC2 enhances the effects of CDDP in suppressing cell growth (A, B, D, and E) and in inducing apoptosis (C and F). (A and B) The indicated MEFs were exposed to the indicated concentrations of CDDP for 3 days. Cell numbers were then estimated with the SRB assay. Data are mean values of four replicate determinations. Error bars show standard deviations. (C) The indicated MEFs were treated with different concentrations of CDDP for 24 h and then harvested for preparation of whole-cell protein lysates and subsequent Western blotting. (D) The indicated cell lines were plated into the wells of 96-well cell culture plates and treated the next day with the indicated concentrations of CDDP alone, 20 nM INK128 or AZD8055 alone (as indicated by arrows), or a combination of CDDP with INK128 or AZD8055 for 3 days. Cell numbers were estimated with the SRB assay. Data are mean values of four replicate determinations. Error bars show standard deviations. (E) The indicated cell lines were seeded into the wells of 12-well plates at a density of approximately 400/well. On the second day, the cells were treated with DMSO, 20 nM INK128 or AZD8055, 0.5 μM (H460), or 1 μM (A549) CDDP or CDDP plus INK128 or AZD8055. After 10 days, the plates were stained for the formation of cell colonies with crystal violet dye and photographed with a digital camera. Columns show mean values of triplicate determinations. Error bars show standard deviations. *, P < 0.001 compared with all other treatments. (F) The indicated cell lines were exposed to DMSO, 20 nM INK128, 2 μM CDDP, or CDDP plus INK128 for 24 or 48 h. The cells were harvested for preparation of whole-cell protein lysates and subsequent Western blotting to detect the indicated proteins. cPARP, cleaved PARP; LE, longer exposure.
Lastly, we validated the efficacy of the INK128 and CDDP combination on the growth of NSCLC xenografts in nude mice. While INK128 and CDDP alone at the doses tested only weakly inhibited the growth of H460 xenografts, as measured by both tumor size and weight, the combination of INK128 and CDDP significantly inhibited the growth of H460 xenografts (at least P < 0.01 compared with the vehicle control, CDDP-alone, or INK128-alone group starting from day 12) (Fig. 8). The combination did not significantly affect the body weight of mice (Fig. 8A), suggesting that the combination does not accordingly enhance toxicity. These data clearly indicate that the combination indeed displays enhanced anticancer activity without compromising safety in vivo.
FIG 8.
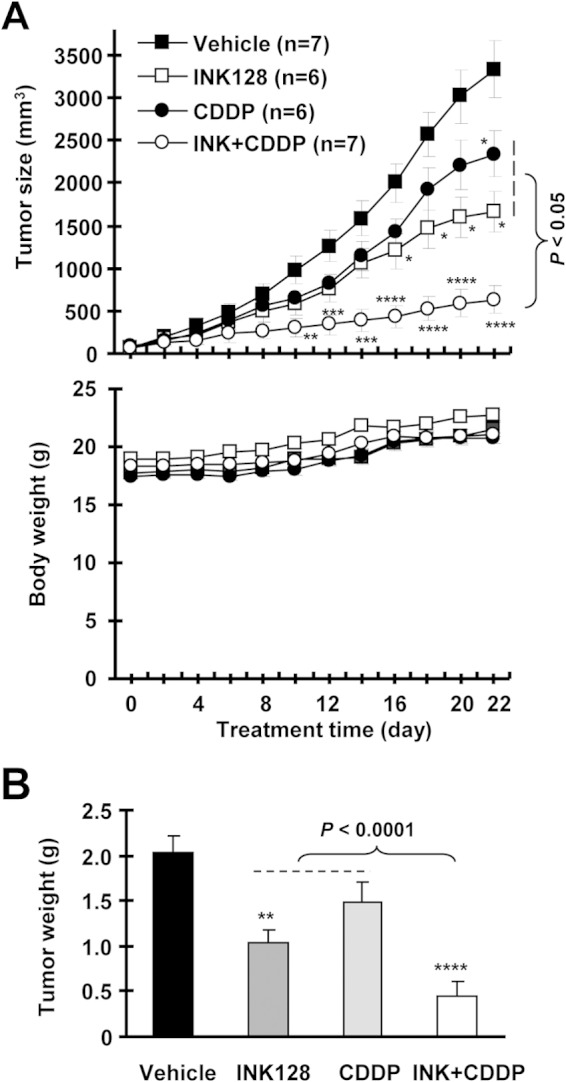
A combination of INK128 and CDDP is significantly more effective than either single agent in suppressing the growth of NSCLC xenografts (A and B) without enhancing toxicity (A) in nude mice. H460 xenografts were treated with the vehicle control, INK128 (0.5 mg/kg administered by oral gavage daily), CDDP (2 mg/kg administered intraperitoneally once every 3 days), or a combination of INK and CDDP starting on the same day after grouping. Tumor sizes (A, upper panel) and mouse body weights (A, lower panel) were measured every 2 days as indicated. The data presented are mean values ± standard errors. After 22 days, the mice were sacrificed and the tumors were removed and weighed (B). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (compared with the vehicle-treated control).
DISCUSSION
This study found that several TORKinibs, including INK128, AZD8055, and Torin1, under the conditions tested, effectively decreased Mcl-1 levels accompanied by inhibition of both mTORC1 (e.g., suppression of S6 phosphorylation) and mTORC2 (e.g., suppression of Akt and SGK1 phosphorylation) signaling in human NSCLC cell lines (Fig. 1). During the course of our studies of Mcl-1 downregulation by TORKinibs, two other groups reported that AZD8055 decreased Mcl-1 levels in rhabdomyosarcoma and colorectal cancer cells, although the underlying mechanisms were not defined (27, 28). Hence, it is clear that TORKinibs decrease Mcl-1 levels in cancer cells. We noted that INK128 was more potent than AZD8055 and Torin1 in decreasing Mcl-1 levels and inhibiting mTORC signaling, suggesting a possible association between inhibition of mTORC signaling and reduction of Mcl-1 levels.
We found that rictor deficiency and TORKinibs did not alter Mcl-1 mRNA levels (Fig. 2F and 3A). These findings are similar to the previous report that ADZ8055-induced Mcl-1 reduction is not associated with mRNA modulation (28). Thus, it is clear that TORKinibs do not downregulate Mcl-1 at the mRNA level. Rather, we have demonstrated in this study that TORKinibs reduce Mcl-1 protein levels by facilitating GSK3-dependent, SCF-FBXW7-mediated proteasomal degradation of Mcl-1 protein on the basis of the following findings: (i) both INK128 and AZD8055 enhanced the rate of Mcl-1 degradation (Fig. 3B), (ii) inhibition of the proteasome with MG132 increased basal levels of Mcl-1 and rescued Mcl-1 reduction induced by different TORKinibs (Fig. 3F), (iii) INK128 treatment increased Mcl-1 polyubiquitination (Fig. 3G), (iv) both pharmacological and genetic inhibition of GSK3 prevented Mcl-1 reduction or degradation induced by different TORKinibs (Fig. 4), and (v) SCF complex disruption or FBXW7 suppression (including knockdown and KO) blocked TORKinib-induced Mcl-1 reduction (Fig. 5). To the best of our knowledge, this is the first study to elucidate the mechanism by which TORKinibs decrease Mcl-1.
It is known that TORKinibs inhibit the activity of both mTORC1 and mTORC2. mTORC1 has been suggested to positively regulate Mcl-1 translation (8). However, it is unclear whether mTORC2 is also involved in the regulation of Mcl-1 expression and, if so, by which mechanism. In our study, we clearly show that siRNA-mediated genetic inhibition of rictor but not raptor mimics the abilities of TORKinibs to decrease Mcl-1 levels (Fig. 2A and B), to destabilize Mcl-1 protein or enhance the Mcl-1 degradation rate (Fig. 3C), and to induce FBXW7-mediated Mcl-1 degradation (Fig. 5E). In agreement, deficiency of rictor or Sin1 also reduced Mcl-1 levels; this reduction could be rescued by reintroducing exogenous rictor (Fig. 2C and D). Complementarily, enforced expression of ectopic rictor but not raptor elevated Mcl-1 levels (Fig. 2E) and stabilized Mcl-1 protein or slowed the Mcl-1 degradation rate (Fig. 3D and E). Collectively, it is apparent that both rictor and Sin1 are involved in the regulation of Mcl-1 stability. Since rictor and Sin1 are key essential components of mTORC2 and all of the TORKinibs tested induce Mcl-1 degradation, as demonstrated in this study, we conclude that mTORC2 is responsible for positive regulation of Mcl-1 stability; accordingly, inhibition of this complex, e.g., with a TORKinib, triggers Mcl-1 degradation.
Since TORKinibs induce GSK3-dependent Mcl-1 degradation, as demonstrated in this study, GSK3 is known to be a substrate of Akt (29), and mTORC2 functions as an Akt Ser473 kinase (30), it is reasonable to question whether TORKinib-induced GSK3-dependent Mcl-1 degradation is secondary to Akt inhibition. At the typical tested concentration of TORKinibs (e.g., 100 nM), we had previously shown that these TORKinibs suppress Akt Ser473 phosphorylation but do not accordingly inhibit GSK3 phosphorylation (13). Consistently, another study also reported that AZD8055 did not suppress GSK3 phosphorylation in rhabdomyosarcoma cells (27). These data indicate that TORKinibs do not activate GSK3. Moreover, in the panel of NSCLC lines with different sensitivities to TORKinibs we tested, INK128 and AZD8055 effectively reduced Mcl-1 levels in relatively sensitive cell lines but caused little or no decrease in relatively less sensitive cell lines (Fig. 1C). However, both INK128 and AZD8055 had comparable effects on inhibiting Akt Ser473 phosphorylation across these cell lines, as we demonstrated previously (13). Therefore, we believe that TORKinibs-induced GSK3-dependent Mcl-1 degradation is unlikely to be secondary to Akt inhibition.
It has previously been shown that rictor is involved in the regulation of protein degradation via interaction with FBXW7 or Cul-1, although these effects are claimed to be independent of mTOR (23, 24). This study also shows that FBXW7 is associated not only with Skp1 and Mcl-1 but also with both rictor and mTOR because we could pull down these proteins by immunoprecipitating FBXW7 (Fig. 6A). Using an anti-Mcl-1 antibody, we also pulled down both rictor and mTOR in addition to Cul-1 and Skp1 (Fig. 6B). Consistently, we could pull down Mcl-1, Cul-1, and Skp1 besides mTOR with a rictor antibody (Fig. 6C). These data together strongly suggest that mTORC2 may be physically associated with the SCF-FBXW7 complex. With INK128 treatment, we pulled down smaller amounts of rictor and mTOR by immunoprecipitation with a FBXW7 (Fig. 6A) or Mcl-1 antibody (Fig. 6B). Complementarily, we detected reduced amounts of Mcl-1, Cul-1, and Skp1 in the immunoprecipitate pulled down with a rictor antibody (Fig. 6C). These data suggest that TORKinibs induce dissociation of rictor and mTOR from Mcl-1 or the SCF complex. Furthermore, we show that Mcl-1 phosphorylation at Ser159, which is critical for FBXW7 and Mcl-1 interaction, as demonstrated previously (11, 12) and in this study (Fig. 6D), is also important for Mcl-1 and rictor association because a mutation at Ser159 substantially decreased Mcl-1 and rictor association (Fig. 6E). Hence, we propose a working model in which GSK3-dependent phosphorylation of Mcl-1 at Ser159 enhances the association of Mcl-1 with both the SCF-FBXW7 complex that mediates the degradation of Mcl-1 and mTORC2, which inhibits Mcl-1 degradation by suppressing SCF-FBXW7 activity; thus, inhibition of mTORC2 (e.g., with a TORKinib) facilitates dissociation of mTORC2 from the SCF-FBXW7 complex, allowing the SCF-FBXW7 complex to degrade Mcl-1 by the ubiquitin-proteasome pathway (Fig. 6F). Hence, our findings warrant further study in this direction and highlight a novel biological function of mTORC2 in the regulation of cell survival and growth.
Both INK128 and AZD8055 at concentrations of up to 1 μM minimally induce apoptosis in our tested cell system. In agreement, AZD8055 also minimally induces apoptosis in rhabdomyosarcoma cells (27). Hence, downregulation of Mcl-1 by TORKinibs may not be sufficient to initiate apoptosis but may lower the threshold at which cancer cells undergo apoptosis, meaning that TORKinibs may sensitize cancer cells to certain cancer therapy-induced apoptosis. Indeed, two recent studies have shown that TORKinibs such as AZD8055 enhance the effects of the Bcl-2 family inhibitors AB263 and ABT737 on the induction of apoptosis and on their cancer therapeutic efficacies (27, 28). In our study, we also show that the chemotherapeutic drug CDDP, in combination with either INK128 or AZD8055, exerts enhanced effects on decreasing Mcl-1 levels, inducing apoptosis, and suppressing the growth of cancer cells both in vitro and in vivo (Fig. 7 and 8). These findings may suggest an effective therapeutic strategy to better utilize TORKinibs in the clinic for the treatment of cancer.
In addition to its antiapoptotic function, Mcl-1 has also been suggested to play a role in supporting the high rate of proliferation of cancer cells (31). TORKinibs potently reduce Mcl-1 levels and inhibit cancer cell growth with limited apoptosis-inducing activity. We have shown that maintenance of GSK3 activity is critical for TORKinibs to inhibit cancer cell growth and, accordingly, inhibition of GSK3 antagonizes TORKinibs' growth-inhibitory effects by the blockage of cyclin D1 degradation (13). In this study, we further show that TORKinibs induce GSK3-dependent Mcl-1 degradation. Whether Mcl-1 reduction contributes to mediation of growth inhibition by TORKinibs requires further investigation.
In addition to Mcl-1, many other proteins (e.g., c-Myc, c-Jun, and cyclin E1) are known to undergo GSK3-dependent and FBXW7-mediated proteasomal degradation (21, 22). Hence, it is plausible to speculate that the degradation of these proteins will be negatively regulated by mTORC2. Our data indeed show that all of the TORKinibs tested effectively reduced the levels of not only Mcl-1 but also c-Myc, c-Jun, and cyclin E (Fig. 5F). Further investigation is thus warranted to elucidate the biological significance of the negative regulation of GSK3/FBXW7-dependent protein degradation by mTORC2 in the regulation of cancer development.
ACKNOWLEDGMENTS
We thank K. Ye, B. Vogelstein, M. A. Magnuson, B. Su, and D. D. Sarbassov for providing us with plasmids or cell lines used in this work. We are also grateful to A. Hammond in our department for editing the manuscript.
This study was supported by NIH R01 CA118450 (S.-Y. Sun) and R01 CA160522 (S.-Y. Sun). F. R. Khuri and S.-Y. Sun are Georgia Research Alliance Distinguished Cancer Scientists. S.-Y. Sun is a Halpern Research Scholar.
REFERENCES
- 1.Guertin DA, Sabatini DM. 2007. Defining the role of mTOR in cancer. Cancer Cell 12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Oh WJ, Jacinto E. 2011. mTOR complex 2 signaling and functions. Cell Cycle 10:2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Sun SY. 2009. Enhancing mTOR-targeted cancer therapy. Expert Opin Ther Targets 13:1193–1203. doi: 10.1517/14728220903225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun SY. 2013. Impact of genetic alterations on mTOR-targeted cancer therapy. Chin J Cancer 32:270–274. doi: 10.5732/cjc.013.10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun SY. 2013. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett 340:1–8. doi: 10.1016/j.canlet.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang YJ, Duan Y, Zheng XF. 2011. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today 16:325–331. doi: 10.1016/j.drudis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas LW, Lam C, Edwards SW. 2010. Mcl-1; the molecular regulation of protein function. FEBS Lett 584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 8.Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, Lowe SW, Pelletier J. 2008. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A 105:10853–10858. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. 2007. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. 2006. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O'Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC, Dixit VM. 2011. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 12.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, Xiao Y, Christie AL, Aster J, Settleman J, Gygi SP, Kung AL, Look T, Nakayama KI, DePinho RA, Wei W. 2011. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koo J, Yue P, Gal AA, Khuri FR, Sun SY. 2014. Maintaining glycogen synthase kinase-3 activity is critical for mTOR kinase inhibitors to inhibit cancer cell growth. Cancer Res 74:2555–2568. doi: 10.1158/0008-5472.CAN-13-2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Yue P, Kim YA, Fu H, Khuri FR, Sun SY. 2008. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res 68:7409–7418. doi: 10.1158/0008-5472.CAN-08-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao L, Yue P, Khuri FR, Sun SY. 2013. mTOR complex 2 is involved in regulation of Cbl-dependent c-FLIP degradation and sensitivity of TRAIL-induced apoptosis. Cancer Res 73:1946–1957. doi: 10.1158/0008-5472.CAN-12-3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren H, Zhao L, Li Y, Yue P, Deng X, Owonikoko TK, Chen M, Khuri FR, Sun SY. 2013. The PI3 kinase inhibitor NVP-BKM120 induces GSK3/FBXW7-dependent Mcl-1 degradation, contributing to induction of apoptosis and enhancement of TRAIL-induced apoptosis. Cancer Lett 338:229–238. doi: 10.1016/j.canlet.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RA, Shudo K, Hong WK, Lotan R. 1997. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res 57:4931–4939. [PubMed] [Google Scholar]
- 18.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. 2005. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 19.Ren H, Koo J, Guan B, Yue P, Deng X, Chen M, Khuri FR, Sun SY. 2013. The E3 ubiquitin ligases beta-TrCP and FBXW7 cooperatively mediates GSK3-dependent Mcl-1 degradation induced by the Akt inhibitor API-1, resulting in apoptosis. Mol Cancer 12:146. doi: 10.1186/1476-4598-12-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D. 2012. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, Wei W. 2012. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett 586:1409–1418. doi: 10.1016/j.febslet.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welcker M, Clurman BE. 2008. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 23.Gao D, Wan L, Inuzuka H, Berg AH, Tseng A, Zhai B, Shaik S, Bennett E, Tron AE, Gasser JA, Lau A, Gygi SP, Harper JW, DeCaprio JA, Toker A, Wei W. 2010. Rictor forms a complex with Cullin-1 to promote SGK1 ubiquitination and destruction. Mol Cell 39:797–808. doi: 10.1016/j.molcel.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Z, Zhou Y, Evers BM, Wang Q. 2012. Rictor regulates FBXW7-dependent c-Myc and cyclin E degradation in colorectal cancer cells. Biochem Biophys Res Commun 418:426–432. doi: 10.1016/j.bbrc.2012.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akagi H, Higuchi H, Sumimoto H, Igarashi T, Kabashima A, Mizuguchi H, Izumiya M, Sakai G, Adachi M, Funakoshi S, Nakamura S, Hamamoto Y, Kanai T, Takaishi H, Kawakami Y, Hibi T. 2013. Suppression of myeloid cell leukemia-1 (Mcl-1) enhances chemotherapy-associated apoptosis in gastric cancer cells. Gastric Cancer 16:100–110. doi: 10.1007/s10120-012-0153-6. [DOI] [PubMed] [Google Scholar]
- 26.Michels J, Obrist F, Vitale I, Lissa D, Garcia P, Behnam-Motlagh P, Kohno K, Wu GS, Brenner C, Castedo M, Kroemer G. 2014. MCL-1 dependency of cisplatin-resistant cancer cells. Biochem Pharmacol 92:55–61. doi: 10.1016/j.bcp.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 27.Preuss E, Hugle M, Reimann R, Schlecht M, Fulda S. 2013. Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J Biol Chem 288:35287–35296. doi: 10.1074/jbc.M113.495986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, Yeo AT, Edelman EJ, Song Y, Tam AT, Boisvert JL, Milano RJ, Roper J, Kodack DP, Jain RK, Corcoran RB, Rivera MN, Ramaswamy S, Hung KE, Benes CH, Engelman JA. 2014. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov 4:42–52. doi: 10.1158/2159-8290.CD-13-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen P, Frame S. 2001. The renaissance of GSK3. Nat Rev Mol Cell Biol 2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 30.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 31.Perciavalle RM, Opferman JT. 2013. Delving deeper: MCL-1's contributions to normal and cancer biology. Trends Cell Biol 23:22–29. doi: 10.1016/j.tcb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]