

Abstract
Characterizing variability in the extent and nature of responses to environmental exposures is a critical aspect of human health risk assessment. Chemical toxicants act by many different mechanisms, however, and the genes involved in adverse outcome pathways (AOPs) and AOP networks are not yet characterized. Functional genomic approaches can reveal both toxicity pathways and susceptibility genes, through knockdown or knockout of all non-essential genes in a cell of interest, and identification of genes associated with a toxicity phenotype following toxicant exposure. Screening approaches in yeast and human near-haploid leukemic KBM7 cells, have identified roles for genes and pathways involved in response to many toxicants but are limited by partial homology among yeast and human genes and limited relevance to normal diploid cells. RNA interference (RNAi) suppresses mRNA expression level but is limited by off-target effects (OTEs) and incomplete knockdown. The recently developed gene editing approach called clustered regularly interspaced short palindrome repeats-associated nuclease (CRISPR)-Cas9, can precisely knock-out most regions of the genome at the DNA level with fewer OTEs than RNAi, in multiple human cell types, thus overcoming the limitations of the other approaches. It has been used to identify genes involved in the response to chemical and microbial toxicants in several human cell types and could readily be extended to the systematic screening of large numbers of environmental chemicals. CRISPR-Cas9 can also repress and activate gene expression, including that of non-coding RNA, with near-saturation, thus offering the potential to more fully characterize AOPs and AOP networks. Finally, CRISPR-Cas9 can generate complex animal models in which to conduct preclinical toxicity testing at the level of individual genotypes or haplotypes. Therefore, CRISPR-Cas9 is a powerful and flexible functional genomic screening approach that can be harnessed to provide unprecedented mechanistic insight in the field of modern toxicology.
Keywords: functional genomics, toxicity testing, yeast, haploid KBM7 cells, RNAi, CRISPR-Cas9
1. Introduction
1.1 Importance of understanding human susceptibility to chemical exposures
Humans vary in their susceptibility to toxicants. Current risk assessment approaches rely on uncertainty factors to account for inter-individual variation, which can greatly under- or over-estimate the risk for an individual or population. As genetic variation likely accounts for a significant proportion of these individual differences, an increased understanding of the genetic variability of toxicant response will enable more accurate chemical exposure risk assessment.
Currently, the main approaches to identifying gene-environment interactions in toxicant mediated disease are genome-wide association studies (GWAS) and candidate gene association studies, both of which have limitations. These methods examine associations between all variants in the human genome, or variants in sets of genes or pathways previously implicated in toxicant response, with a phenotypic outcome related to toxicant exposure. GWAS need to be conducted in large exposed and control populations in order to find significant associations among the large number of variants tested, which makes these studies expensive and often not feasible. The candidate gene association study approach, requires smaller study populations than GWAS, but is limited by incomplete understanding of modes of action, and thus relevant genes and pathways, for many toxicants. Thus, using existing techniques to identify all the chemical susceptibility genes will take many years; in the meantime, alternative approaches are needed to identify and prioritize genes for candidate gene association testing.
1.2 Identifying all toxicity pathways by functional genomics
In the contest of risk assessment, understanding mechanisms of action of toxicants in a broader systems biology context is important to understanding new information and adopting next generation risk assessment applications [1–7]. In the field of toxicology, mechanistic information is often described by simplified models called modes of action (MOAs) or adverse outcome pathways (AOPs), and by more complex models called AOP networks. The latter, called networks to indicate the interconnectedness of disease-causing AOPs, reflect the various biological pathways and mechanisms through which chemicals cause toxicity and disease. Elucidating these mechanisms and their inter-relatedness can be challenging. A genome-wide systems biology approach is needed to identify non-obvious pathways to toxicity and to understand how normal network function is altered following exposure to chemicals or stressors. Functional genomics offers just such an approach. Functional genomics uses omic data to describe gene (and protein) functions and interactions on a genome-wide scale using high-throughput methods.
1.3 The role of functional genomics in toxicity testing: evolution of the technologies
In the context of toxicology, functional genomic screening involves systematic knockdown or knockout of non-essential genes or proteins on a pathway or genome-wide basis, in a cell of interest, and measurement of a toxicity phenotype, such as lethality, viability or fitness, following exposure to various doses of a toxic compound. As it directly measures the phenotype, it informs the association of a specific gene and its corresponding loss-of-function in the cellular response to a compound [8, 9]. Thus, functional genomic screening has the potential to identify genes that confer resistance or susceptibility to toxicants and to reveal toxicity pathways, through pathway analysis. Through the application of now affordable high-throughput screening technologies, functional genomic screening provides a means to simultaneously and efficiently screen thousands of compounds in cell-based systems [10]. Thus, the approach will provide mechanistic insight across many genes, pathways and chemicals, supporting the definition of more specific in vitro toxicological endpoints and the development of targeted cell-based assays [11, 12], that ultimately will have better predictive power for adverse health effects in humans than do traditional animal toxicological studies.
Functional genomic screening has been conducted in budding and fission yeast, fruit flies, worms, and human cell lines using various techniques. In this review, we particularly discuss genomic screens using in vitro models such as yeast and haploid eukaryotes and tools such as RNA interference (RNAi) and the most recently developed clustered regularly interspaced short palindrome repeats-associated nuclease (CRISPR)-Cas9 gene editing system. This review aims to describe the main in vitro functional genomic screening approaches that have been developed and to discuss their advantages and limitations (summarized in Table 1) in the context of toxicity testing.
Table 1.
Examples of recent functional genomic screening approaches used in toxicity studies
| Methodology | Advantages | Limitations | Genome Coverage | Cell Type | Organism | Toxic Agent | Reference |
|---|---|---|---|---|---|---|---|
| PCR-based gene disruption | Yeast genome well annotated; many genes conserved between yeast and human; quantitative; high-throughput. | Not all yeast genes have human homologs; yeast tolerates higher levels of toxicants than human cells do. | All nonessential genes | Yeast | S. cervisiae | Arsenic | [33] |
| Benzene metabolites (HQ, BT, CAT) | [34] | ||||||
| Benzo[a]pyrene | [35] | ||||||
| Butanol | [37] | ||||||
| Dieldrin | [139] | ||||||
| Formaldehyde | [36, 37] | ||||||
| Toxaphene | [140] | ||||||
|
| |||||||
| Insertional mutagenesis | Extends yeast approach to human cells | Not haploid on chromosome 8; strong genomic integration bias limits true coverage; leukemic cells. | All human genes except those on chromosome 8 | KBM7 haploid CML cell line | H. sapiens | 3-bromopyruvate | [53] |
| Antimycin | [55] | ||||||
| Bacterial toxins | [46, 48] | ||||||
| Brefeldin A | [54] | ||||||
| Human Cytomegalovirus product | [52] | ||||||
| Imatinib (Gleevec) | [46, 68] | ||||||
| Influenza | [46] | ||||||
| Kaposi’s sarcoma viral product | [51] | ||||||
| Tunicamycin | [47] | ||||||
| YM155 | [56] | ||||||
| KBM7-derivative, HAP1 | Ebola | [49] | |||||
|
| |||||||
| Transposon mutagenesis | Extends yeast approach to mouse cells | Strong genomic integration bias limits true coverage | Genome wide | Mouse haploid ESC | M. musculus | 2-amino-6-mercaptopurine | [63] |
| 6-thoguanine | [64] | ||||||
| Olaparib | [59] | ||||||
| Ricin | [59] | ||||||
| RNAi | Targets RNA; applicable to many cell types; libraries targeting human genes available. | Incomplete knock down; off target effects (OTE); unsuitable for lethal phenotypes. | Genome-wide at RNA level | Human cancer cell lines | H. sapiens | Anti-cancer therapeutics | [81–83] |
| Insect | D. melanogaster | Cell cycle inhibitors | [141] | ||||
| Worm | C. elegans | Paraquat | [142] | ||||
|
| |||||||
| CRISPR-Cas9 | Precise targeting of DNA and RNA; applicable to many cell types; permanent mutations; complete knockout; libraries targeting human genes available. | Only cleave DNA at loci with a suitable proto-spacer adjacent motif (PAM) sequence; OTE | Genome wide | Cancer cell lines and ESC | H. sapiens | 6-thioguanine | [101] |
| Diphtheria toxin | [123] | ||||||
| Etoposide | [101] | ||||||
| Vemurafenib | [94] | ||||||
| 6-thioguanine | [102] | ||||||
| M. musculus | alpha-toxin | [102] | |||||
CML, chronic myeloid leukemia; HQ, hydroquinone; BT, benzenetriol; CAT, catecholamine; OTE, off-target effects; PAM, proto-spacer adjacent motif; ESC, embryonic stem cells
As discussed in detail in the following sections, each approach has its own “pros” and “cons”. For example, high-throughput yeast screening has been well established due to the availability of mutant clones for all non-essential yeast genes, but incomplete homology with mammalian genes and functions limits its relevance to human toxicity. To overcome this weakness, approaches using mammalian haploid cells such as the KBM7 human bone marrow cell line and mouse embryonic stem cells (ESCs), have been developed. However, the leukemic nature of KBM7 limits its relevance to toxicity in normal human cells. RNA interference (RNAi) and the most recently developed clustered regularly interspaced short palindrome repeats-associated nuclease (CRISPR)-Cas9 genome editing system overcome many of the limitations associated with the other previously developed approaches. They are both applicable to any transfectable cell type and organism, and CRISPR, in particular, is relatively easy to design and perform, is highly specific, efficient and well suited for high-throughput and multiplexed gene editing, and can produce precisely targeted knock-out of most regions of the genome at the DNA level [13]. Broad application of this novel CRISPR-Cas9 system has been recently proposed in many research areas, including gene-therapy, drug development, and understanding phenotype associated with genetic variations [14]. The goal of this review is to highlight the potential application of CRISPR-Cas9 application in toxicity screening of environmental toxicants and discovery of mechanism and susceptibility.
2. Functional genomics in yeast
2.1 Yeast screening process
For several reasons, the eukaryotic budding (Saccharomyces cerevisiae) and fission (Schizosaccharomyces pombe) yeasts are ideal models in which to conduct functional genomic screening studies [15]. First, yeast is a well-established and widely used eukaryotic model for molecular and cellular biology studies [16]. Second, yeast is a unicellular non-pathogenic organism with rapid and stable growth in both diploid and haploid status. Third, the genetic makeup of yeast is fully characterized and genetic manipulation is straightforward and easily implemented. In contrast to other higher eukaryotes, most of the yeast genes do not contain introns, which simplifies the process of computer-based gene identification when conducting genome-wide analyses [17]. Functional information is available for nearly every gene in yeast [15, 18]. Fourth, yeast genes are highly conserved in human cells and other higher eukaryotes. Nearly half of the human genes implicated in heritable diseases have yeast homologues [19]. Although yeast does not possess as many physiological mechanisms of response to cytotoxic compounds as do other organisms, many of the basic mechanisms in response to chemical and environmental stresses are apparently conserved between yeast and other higher eukaryotic organisms [20, 21].
Phenotype-based analysis in mutant strains is a powerful way to determine the role of genes in response to a toxicant on a gene-by-gene or genome-wide basis. Many approaches to generating mutant strains have been developed including genetic footprinting [22] random mutagenesis [23] and polymerase chain reaction (PCR) based gene disruption [24]. Genetic footprinting and random mutagenesis are both untargeted and the process of matching phenotypes to genes is slow. A novel PCR-based gene disruption strategy was developed to generate a deletion (null mutation) in each of the open reading frames (ORFs) in the yeast genome [24]. Each yeast gene can be deleted in a directed manner by replacing an individual ORF with a selectable marker (e.g. antibiotic resistance) linked to a 20-base molecular tag (or barcode) that functions as a unique strain identifier [25]. The use of molecular tags allows the mutant strains to be pooled and assayed for the phenotypes in parallel in a process called parallel deletion analysis (PDA) [26, 27].
2.2 Parallel deletion analysis (PDA)
The screening process is outlined in Figure 1 of North and Vulpe’s review [27]. Tagged (barcoded) strains are pooled and grown together in the presence (treatment) or absence (control) of a compound. After treatment, the molecular barcodes from all strains present in a pooled culture are amplified simultaneously in a single PCR reaction (using a pair of PCR primers that anneal to the common regions flanking the inserted barcodes that are present in all strains). PCR products of barcodes can be hybridized to microarrays containing probe sequences complementary to the barcodes, and the hybridization signal is proportional to the number of cells of that strain in the pooled culture. A fitness score is generated by comparing the hybridization signal of a treated strain to that of a control strain.
HTS technologies have also been used to quantify barcodes in pooled strains. One such method, Bar-seq, had improved dynamic range and throughput compared with microarray [28]. As next-generation sequencing technologies become more affordable, they will predominate. Following barcode quantification, deletion strains whose growth is significantly influenced by the compound treatment can be identified [25, 29]. The deleted genes may be direct targets, or be involved in modifications or pathways that enable the compound’s cytotoxic action [30, 31]. Application of suitable bioinformatics tools, such as Gene Ontology, network analysis and ortholog identification reveal further information about function and pathways in yeast and other organisms [27].
Four different Yeast Knockout mutant collections were created by a consortium of European and North American groups [26] containing homozygous and heterozygous diploid strains corresponding to deletions of 5,916 genes (including 1,159 essential genes) and one haploid strain of each mating type for every non-essential gene (4,757 genes). Each knockout strain is marked by two unique 20-bp barcodes, allowing quantitative and qualitative identification [32].
Screening of yeast deletion mutant collections in many published studies has revealed genes involved in response to toxicants and stressors, including toxic metal ions and pesticides, as reviewed by [15]. We and others have published data on genes involved in resistance to toxic metabolites of arsenic [33], benzene [34], and benzo[a]pyrene [35], formaldehyde [36, 37] as well as mechanisms of toxicity associated with these chemicals. In follow up studies, we validated the roles of human orthologs of some of the yeast genes associated with benzene [38–40] and arsenic [41, 42] toxicity in mammalian cell lines using RNAi-based in vitro knockdown approaches.
2.3 Limitations of screening in yeast
Although yeast functional genomic screening is a powerful tool to identify conserved cellular components required for sensitivity or tolerance to a toxicant treatment, it has certain limitations. First, yeast can tolerate higher level of toxicants than can human cells and thus is not an accurate indicator of toxic doses relevant to humans [33]. Second, information on organ or tissue-specific toxicity and cell-cell signaling is absent. Third, while many genes are conserved between yeast and human, some yeast genes have many human orthologs, making confirmatory experiments challenging. In order to address these issues, similar functional genomic screening technologies are now being developed in higher eukaryotic systems and are discussed in the following sections.
3. Functional genomics in haploid mammalian cells
Mammalian-based screening systems have the potential to generate results that are more directly relevant to toxicity and disease in humans. However, mammals are somewhat tolerant of partial loss of a gene function and inactivation of one gene copy rarely leads to severe changes in phenotype due to the fact that chromosomes are typically diploid in mammals. Therefore, utilization of haploid cells in mammalian screens is necessary. Haploid screening has been established in both human and mouse cells.
3.1 Screening in near-haploid human KBM7 cells
Near-haploid karyotypes have been reported in rare human tumors and leukemias [43] and a heterogeneous (mixed ploidy) cell line (KBM7) was established from the bone marrow of a patient with a near-haploid chronic myeloid leukemia [44]. Although around half of the cells in the initial cultures were near-haploid (apart from disomy of chromosome 8), cells with a diploid or greater DNA content tended to outgrow them with continuous passage, rendering this cell line initially unsuitable for somatic cell genetics. Two years later, this hurdle was overcome when Kotecki et al. reported the derivation of a KBM7 sub-clone (P1-55) that stably remained near-haploid for at least 12 weeks [45].
Carette et al. developed a screening method to generate null alleles for genes on all chromosomes except chromosome 8 using retrovirally-mediated insertional mutagenesis, in the karyotypically stable near-haploid KBM7 sub-clone [46]. Using this approach, they identified host factors essential for infection with influenza and genes involved in cytotoxic response to three bacterial toxins. Using the same approach, Reiling et al identified a key mediator in the response to tunicamycin, a bacterial protein that induces endoplasm reticulum (ER) stress and the unfolded protein response [47]. The original method developed by Carrette et al. was labor-intensive, and required isolation and expansion of individual clones, followed by DNA extraction, inverse-PCR and Sanger sequencing to map gene-trap insertions. More recently, they incorporated deep sequencing into their screening protocol, facilitating the analysis of millions of mutant alleles in parallel and more accurate assignment of genes to phenotypes [48]. Further, they increased improved genome-wide coverage by increasing the number of cells transfected and the transfection efficiency. Applying this improved approach, they identified 12 human genes important for intoxication by four different cytolethal distending toxins [48] and host factors required for entry of Ebola virus into human cells [49]. The latter was performed in a derivative KBM7 cell line called HAP1, which was generated through a failed attempt to induce pluripotency. HAP1 cells grow adherently, do not express hematopoietic markers and are haploid for all chromosomes including chromosome 8. Importantly, unlike KBM7 cells, HAP1 cells are susceptible to rVSV-GP-EboV, the virus used to test for Ebola-related host factors in the study. Through the inclusion of a unique DNA barcode in each gene-trap vector, Burckstrummer and colleagues subcloned individual gene trap–containing cells, creating a library of isogenic cell lines with mutations in individual genes, enabling more efficient and systematic pooled screens [50]. So far, clones covering 3,396 genes, almost one-third of the expressed genome, have been established [50].
Lehner’s group published two papers illustrating the potential of near-haploid screening to delineate the genes involved in pathogen manipulation of host immune response. First they conducted a forward genetic screen to identify genes required for the function of the Kaposi’s sarcoma herpevirus gene product K5, a ubiquitin ligase that downgreulates major histocompatibility complex-1 (MHC-1) and other immunoreceptors [51]. They identified proteolipid 2 (PLP2), a protein of unknown function, as essential for K5 activity and showed that loss of PLP2 traps the viral ligase in the ER, rendering it inactive. Comparison of the plasma proteome of K5-expressing KBM7 cells with and without PLP2, revealed novel targets of downregulation by K5. In a more recent study, Lehner’s group conducted a forward genetic screen to identify components of the ER-associated degradation (ERAD) pathway targeted by the US11 gene product of human cytomegalovirus, to promote viral immune evasion [52]. They identified TMEM129, a previously uncharacterized membrane protein, as a novel ERAD E3 ubiquitin ligase and central component of a novel ERAD complex that is essential for US11-mediated MHC-1 degradation.
Several groups have utilized near-haploid screening to identify genes and mechanisms of drug toxicity or mechanism of action. Birsoy et al. sought to identify resistance mechanisms to 3-bromopyruvate (3-BrPA), a drug candidate and glycolysis inhibitor [53]. Monocarboxylate transporter 1 (MCT1) was revealed to be a 3-BrPA transporter and key determinant of sensitivity. Further, MCT1 mRNA levels were shown to be predictive of 3-BrPA sensitivity in glycolytic cancer cells, thus representing a potential biomarker of responsive tumors. Reiling et al. performed a near-haploid screen to identify genes involved in apoptosis induced by brefeldin A (BFA), a lead chemotherapeutic compound that is toxic to ER-golgi [54]. A genome-wide haploid genetic screen led to the identification of the small G protein ADP-ribosylation factor 4 (ARF4) that protects against BFA toxicity in a signaling cascade that requires the CREB3 transcription factor. Further, the CREB3-ARF4 pathway was shown to be part of a generalized Golgi stress response. Chen et al. performed a genome-wide genetic screen in haploid human cells to identify genes that confer resistance to severe electron transport chain (ETC) dysfunction when inactivated with a mitochondrial complex III inhibitor, antimycin [55]. Loss of ATP Synthase Mitochondrial F1 Complex Assembly Factor 1 was found to strongly protect KBM7 and other cell types against antimycin-induced, and other forms of ETC dysfunction, and antimycin-induced cell death by facilitating maintenance of mitochondrial membrane potential. In a fourth study, the mechanism by which the genotoxic chemotherapeutic agent YM155 causes DNA damage toxicity was investigated [56]. A requirement for the drug-transporter solute carrier family member 35 F2 (SLC35F2) for YM-155-induced DNA intercalation was discovered and SLC35F2 expression and YM155 sensitivity were correlated across several cancer cell lines.
Though most human haploid screens have involved the selection of mutants resistant to an otherwise lethal agent, thus using cell death or survival as the outcome phenotype, KBM7 screening studies utilizing non-lethality endpoints have been published. Duncan et al. used fluorescence activated-cell sorting to identify genes involved in MHC (major histocompatibility complex) class I antigen presentation, by sorting for mutants that were defective in surface expression of MHC-1 [57]. More recently, Lee et al. used a transcriptional reporter to screen KBM7 cells for constitutive inhibitors of NF-κB and identified previously unknown inhibitors [58].
3.2 Screening in haploid mouse embryonic stem cells
Several groups [59–62] have successfully generated mouse haploid embryonic stem cells (ESC) that can stably grow after multiple passages, be efficiently subcloned, differentiate at similar kinetics as diploid ESCs, and remain in haploid karyotype through initiation of differentiation. Availability of these cells facilitates the application of functional genomics to study the effects of toxicant exposure in normal stem cells and to mimic early-life exposures.
Applying a transposon-based mutagenesis protocol with near genome-wide coverage, Elling et al. identified genes responsible for ricin toxicity [59]. Leeb and Wutz demonstrated and conducted a pilot genetic screen for mismatch repair genes involved in toxicity of 2-amino-6-mercaptopurine [63] and Pettitt et al. identified a gene that mediates olaparib toxicity [64]. These studies demonstrate the potential of functional genetic screening in mouse haploid ESCs and expand the possibility of screening in multiple cell types and developmental pathways [63]. However, mouse ESC are more challenging to culture than human KBM7 cells. Recently, haploid ESC have also been derived from monkey [65] and rat embryos [66].
3.3 Limitations of screening in haploid mammalian cells
One potential disadvantage of KBM7 is that the cells are not completely haploid, having disomy of chromosome 8. Though the KBM7-derivative HAP1, described above, lacks the second copy of chromosome 8, it retains two copies of a fragment of chromosome 15, one of which is fused to chromosome 19 [49]. Very recently, Burckstummer’s group used CRISPR/Cas9-based genome engineering to excise this chromosomal fragment and to derive a truly haploid cell line called eHAP (engineered-HAPloid) [67].
Though this is an important breakthrough, KBM7 and eHAP, as leukemic cell lines from bone marrow, might not reflect the responses of normal cells or other cell types. Carette and colleagues successfully reprogrammed KBM7 cells into induced pluripotent stem cells (iPSC) by infecting the cells with retroviruses carrying four transcription factors [68]. Though the cells retained the BCR-ABL gene fusion, they lost dependency on its signaling for survival and became resistant to imatinib. While KBM7-iPSC have potential as pluripotent cells, they retain other features of the leukemia cells from which they originate. In contrast, haploid mouse ESC are fully haploid and have a largely normal genome. The random mutagenesis approach to generate mutants for haploid screening using gene-trap retroviruses has limitations, including difficulty in reaching genome-wide coverage due to hot and cold spots, and a strong genomic integration bias of retroviruses. Further, it is difficult and time consuming. Targeted, gene-specific approaches such as RNAi and nuclease-mediated editing are required to systematically knockout or knockdown genes in mammalian cells.
4. Functional genomics by RNA interference (RNAi)
4.1 Discovery and application of RNAi
RNAi is an endogenous cellular process, conserved in most eukaryotic species, that involves targeted transcript cleavage and degradation after binding of a sequence-specific short-interfering RNA (siRNA) [69]. This natural process has been exploited as a research tool to target mRNA transcripts by introducing into cells homologous synthetic siRNAs, siRNA precursors such as short-hairpin RNAs (shRNAs), or double stranded RNA (dsRNA) [70–72]. Delivery methods include lipid-based transfection, electroporation and viral transduction with retroviruses and lentiviruses [73]. As some mammalian cell types are resistant to transfection with synthetic siRNAs, they may be alternatively transduced with viruses (e.g. lentivirus) carrying expression vectors (normally plasmid) that encode shRNAs to express gene-specific siRNAs within the targeted cells, which can achieve stable and highly effective gene suppression in a variety of mammalian cell types [74–76]. Various aspects of RNAi approaches, technical limitations and improvements have been reviewed recently [77].
RNAi has been widely used to study explore the effects of knocking out single genes in functional validation studies. As mentioned in Section 2.2., we used RNAi to knockdown and validate the functional roles of human orthologs of genes identified in yeast screens associated with benzene [38–40] and arsenic [41, 42] in mammalian cell lines. The flexibility of the approach has allowed RNAi to become one of the most powerful tools for genome-wide characterization of gene functions and it is adaptable to HTS. RNAi screens can be conducted in a pooled or arrayed manner (for illustration see Figure 1 in Willingham et al. [78]). In a pooled format, cells are transfected with a siRNA or shRNA library en masse and selected by phenotype following exposure to the agent of interest by microarray or sequencing. The phenotype may be viability or a cell marker selected by fluorescent activated cell sorting. In the arrayed format, cells are transfected with individual siRNAs or shRNAs in separate wells, exposed, and assayed in parallel by a variety of methods, including high-content microscopy or viability screening, or reporter assays [73]. In HCS, cells are labeled with multiple fluorescent markers, which can be measured in multiple channels in a highly automated manner [77].
Vector libraries, such as The RNAi Consortium lentiviral library, which contains shRNAs with unique barcodes targeting 17,200 humans genes [79], have enabled a pooled high-throughput RNAi screening approach called shRNA barcode screening. Individual shRNAs conferring cells with a specific phenotype under the conditions of toxicant treatment can be efficiently identified by PCR amplification of barcodes followed by detection by microarray or sequencing. The magnitude of the effect can be determined by measuring the abundance of identified shRNAs in treated relative to untreated cells. shRNA barcode screening is very effective at identifying genes whose knockdown confers resistance to the test conditions, and has been successfully applied to find genes whose inactivation play a role in breast cancer progression [80]. Most RNAi screens have been based on cell lines or cells with genetic modifications predisposing to certain phenotypes and they have identified genes and gene networks involved in biological processes, drug resistance and pathogen response [72, 81]. For example, in vitro screens have identified genes involved in mitotic arrest and ceramide metabolism as determinants of resistance to chemotherapeutic drugs in different cancer cell types [82] and genes involved in resistance to docetaxel in triple-negative breast cancer cells [83]. RNAi screening has also been applied to understand the functional genomics of both normal and malignant hematopoietic stem cells (HSC) and has uncovered factors regulating differentiation, targets for ex-vivo expansion, tumor suppressor genes associated with hematopoietic cancers and potential therapeutic targets [84, 85]. The GenomeRNAi database (http://www.genomernai.org) curates RNAi phenotype data from the literature for human and Drosophila [86], and contained data on 194 RNAi screens human in its 2014 release (in June, Volume 13).
4.2 Limitations of RNAi screening
Like other functional genomics tools, RNAi screening has limitations. It is limited by the incomplete target gene suppression and confounding OTEs on other mRNA that contain partial sequence homology to the reagent dsRNAs/siRNAs/shRNAs [87–91]. Bioinformatic tools, chemical modification to the seed region, and inclusion of experimental controls can reduce OTEs [72]. Reliable results may require use of more than one dsRNA/siRNA/shRNA targeting different parts of the target mRNA and determination of the silencing efficiency [92]. Functional redundancy exists in protein-coding genes and combinatorial screens targeting two or more genes or a single gene against a genetically sensitized background can be more informative. Aggregate analysis of the data, through pathway or ontology analyses, and integration of the RNAi data with complementary omics data such as transcriptomics can further leverage the power to reveal mechanistic information.
5. Functional genomics by CRISPR-Cas9
5.1 CRISPR-Cas9: Breakthrough genome editing technology
Each of the functional genomic approaches discussed above has strengths and weaknesses but they are all limited in their ability to knock out gene activity on a genome-wide basis in diploid mammalian cells. The recent development of CRISPR-Cas9 technology has provided an effective way of genome editing in a variety of cell types [93–95]. In comparison to RNAi, this RNA-guided technology can generate permanent mutations in the genome, resulting in either a loss or gain of function [95]. CRISPR-Cas9 is a cost-effective, fast, and efficient way to screen many genes. Two other genome editing nuclease technologies preceded CRISPR, zinc-finger nucleases and transcription activator-like effector nucleases (TALENs) [96, 97]. They use protein-DNA interactions for targeting and in contrast with CRISPR, they are more expensive, time-consuming and technically challenging to apply and scale up for screening [98].
CRISPR-Cas9 functions as an adaptive immune system in many bacteria and most archaea by cleaving foreign nucleic acids (e.g. viruses or plasmids) [93, 99]. There are three CRISPR-Cas systems and the simplest, type II CRISPR from Streptoccus pyogenes, utilizes a single Cas9 endonuclease to cleave the DNA and is guided by a 20 nucleotide sequence within an endogenous CRISPR transcribed RNA, and a transacting cRNA. In 2013, George Church’s group demonstrated the potential of the CRISPR-Cas9 system for targeting human cells [100] and four papers were published within a year applying pooled CRISPR screens to target multiple genes within mammalian cells [94, 101–103]. In these approaches, Cas9 is directed by a single-guide RNA (sgRNA) to induce double-strand DNA breaks (DSB) at specific genomic loci. Recognition of cleavage sites and thus target specificity is determined by sgRNA-DNA homology over 20 base pairs and an adjacent protospacer-adjacent motif (PAM), a three nucleotide NGG sequence (where N is any nucleotide) [104]. Web-based tools that facilitate the identification of potential CRISPR target sites in genes of interest are available, e.g., the ZiFiT Targeter software (http://zifit.partners.org/) [105, 106] and the CRISPR Design Tool 51 (http://crispr.mit.edu/) [107]. DSBs are repaired by either the Non-Homologous End Joining (NHEJ) DNA repair pathway or the Homology Directed Repair (HDR) pathway (Figure 1). NHEJ repair yields inserts/deletions (indels) at the DSB site leading to frame shifts and/or premature stop codons, resulting in gene knockout. The HDR pathway incorporates a repair template (donor DNA) into the DSB, introducing specific nucleotide changes into a targeted gene [13, 100, 104]. One limitation is that the NHEJ/HDR ratio is difficult to predict in a given cell type.
Figure 1.
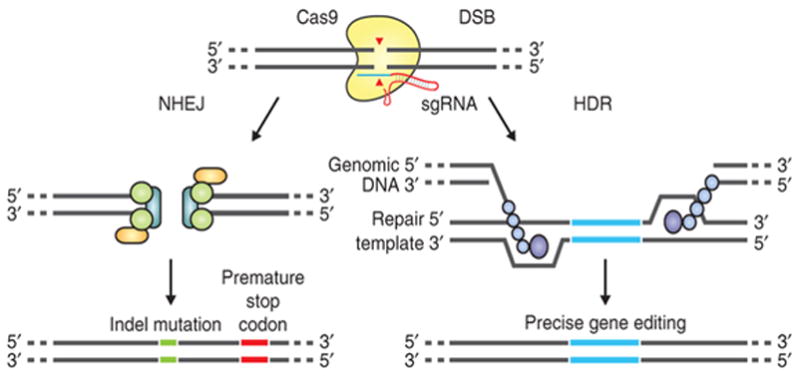
CRISPR-Cas9 DSB repair pathway and gene editing [13]. Reproduced with permission from Nature Protocols.
Use of two gRNAs coupled with Cas9 can efficiently create DNA deletions of up to 10 kb in the presence of a linear homologous repair donor [108]. Interrogation of gene function on a genome-wide scale can be facilitated by large scale oligonucleotide synthesis of guide sequences [109]. In contrast to shRNA libraries, which mediate only gene knockdown, gRNA libraries can be used with Cas9 nuclease to generate libraries of cells with knockout mutations. As reviewed in Sander et al., electroporation, nucleofection and Lipofectamine-mediated transfection have been used to transiently express Cas9 and gRNAs from plasmid DNA in cultured mammalian cells, and lentiviral vectors have been used to constitutively express Cas9 and/or gRNAs in cultured human and mouse cells [98].
Depending on the number of cells and type of cell lines used for screening, a two- or one-vector system to deliver Cas9 and sgRNA is chosen (Figure 2). In the two-vector system, cells are initially transduced with Cas9, after which clones are selected and expanded and subsequently transduced with a sgRNA library. Sabatini and Lander’s group developed a two-vector system, with a library consisting of 73,151 sgRNA plasmids that cover a total of 7,114 human genes with 100 non-targeting controls [101]. The library has been separated into distinct enriched sub libraries for gene targets of known function (e.g. kinases, cell cycle proteins, nuclear proteins, and ribosomal proteins) and genes of unknown function. Using the one-vector system, a large number of cells are transduced with both Cas9 and the sgRNA in a single vector. Such a system was developed by Zhang’s group [94], using a single lentiviral vector to deliver Cas9, a sgRNA, and a puromycin selection marker into target cells. They initially developed a genome-scale CRISPR-Cas9 knockout human library (GeCKOv1) consisting of 64,751 unique guide sequences targeting 18,080 human genes and more recently developed an improved library with 123,411 sgRNAs targeting 19,050 genes (GeCKOv2) [110]. Improvements in the latter library include the ability to target ~1000 additional genes; inclusion of a uniform number (6) of sgRNAs per gene, with three per each of two sublibraries; minimized off-target effects; and sgRNAs that inactivate miRNAs by generating mutations in pre-miRNA hairpin structures. Genome-wide mouse lentiviral sgRNA libraries consisting, of 87,897 unique sgRNA plasmids targeting 19,150 protein-coding regions was developed by Koike-Yusa et al. [102] and with 130,209 sgRNAs targeting 20,611 genes by Zhang’s group [110].
Figure 2.
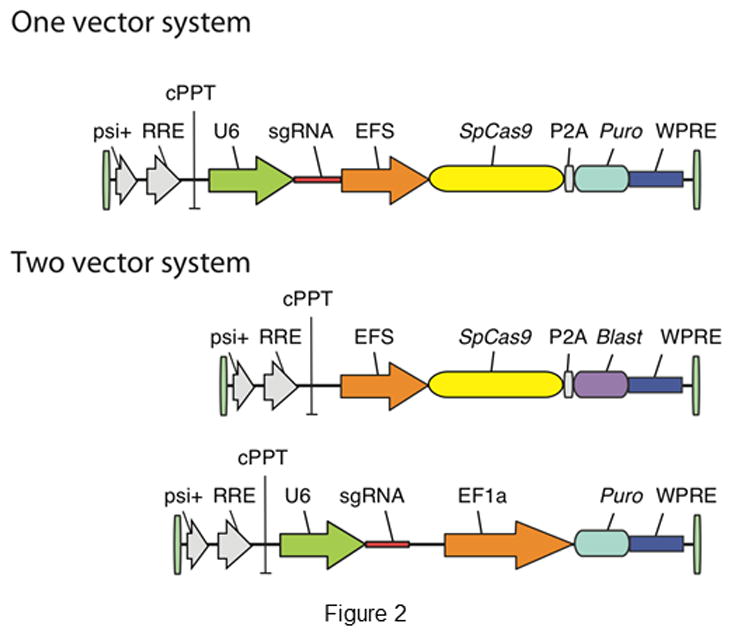
Lentiviral vector delivery system for Cas9 and sgRNA [138].
5.2 Application of CRISPR-Cas9 screening in functional genomics
Availability of these libraries has enabled genome-wide screening in various mammalian cell lines. In four CRISPR studies using pooled mutant libraries, the first three in human cells and the last in mouse cells, cell death or survival was used as the selection phenotype after exposing the cells to a toxicant and next-generation sequencing was used to identify genes responsible for resistance to each toxicant: nucleotide analog 6-thioguanine and the chemotherapeutic agent etoposide in KBM7 and HL60 cells [101], the protein kinase inhibitor vemurafenib in A375 melanoma cells and HUES62 ESC [94], anthrax and diphtheria toxin in a focused screen in HeLa cells [103], and Clostridium septicum alpha toxin and 6-thioguanine in mouse ESC [102]. Each study used multiple sgRNAs per gene and statistical tests of enrichment and discovered previously known and novel genes. Genes involved in resistance to vermurafenib correlated well with hits in a previous shRNA screen [94]. Wang and Koike-Yusa identified similar mismatch repair genes involved in 6-thioguanine toxicity in both human and mouse [101, 102].
As well as functional genomic screens, CRISPR-Cas9 is useful for generating genetic models of disease. For example, Heckl et al. used the technology to modify up to 5 genes in a single mouse HSC, leading to clonal outgrowth and myeloid malignancy [111]. Torres et al. generated human cell lines and hematopoietic mesenchymal and stem cells that included chromosomal translocations associated with acute myeloid leukaemia and Ewing’s sarcoma [112]. In another study, human iPSC were modified by a process called iCRISPR, a combination of TALEN and CRISPR-Cas, to generate biallelic knock outs with loss of function, and homozygous knock ins, that could model disease in a stage-specific, inducible manner [113]. CRISPR has also been used to create genetic models of disease in vivo in mice [114, 115]. Very recently, a Cre-dependent Cas9 knock-in mouse was established, enabling the broad application of Cas9 editing in vivo [116]. The utility of this approach was demonstrated by the simultaneous generation of mutations in KRAS, p53, and LKB1 in vivo and dynamic modeling of the resulting lung adenocarcinomas. Rats with conditional alleles in three DNA methylase transferase genes were recently generated using CRISPR-Cas9 [117]. The ability to rapidly generate complex genetic models of disease in vivo enables the examination of gene by environment (GxE) interactions in the development of disease and drug resistance, and for targeted therapy design.
Recently, the ability to repress gene expression by controlling transcription [118–120] and by targeting RNA directly [121] using CRISPR, has been described. Jonathan Weissman’s group at UCSF has developed libraries that enable reversible genome-wide repression by CRISPRi [118] or activation by CRISPRa [122] of gene expression over a wide fold-range [123]. Using a catalytically dead cas 9 protein fused to Krüppel associated box fusion to repress transcription, and a sunCas9-VP16 fusion to activate transcription, they screened for genes involved in response to a chimeric cholera diphtheria toxin (CTx-DTA) in K562 cells and identified genes and pathways involved in pathogen entry, retrotranslocation and toxicity. Previously, haploid mutagenesis revealed the diphthamide biosynthetic pathway (required to generate eEF-2-diphthamide, the target of diphtheria toxin) and the ganglioside biosynthetic pathway (required to produce GM1a, the cell-surface receptor for cholera toxin), as modulating cellular sensitivity to CTx-DTA [124]. CRISPRi provided genetic confirmation that GM1a is the relevant cell-surface receptor for CTx-DTA and revealed the role of additional components in these two pathways and complexity of their responses, enabled by the identification of both sensitizing and protective genes. Additionally, CRISPRi clarified the mechanisms by which the toxin traverses the Golgi network, through the discovery that COG and GARP complexes, which tether late endosomes to the trans-Golgi network or modulate intra-Golgi retrograde transport [125], are critical host factors for CTx-DTA. CRISPRi appeared to approach saturation, evidenced by tight clustering of many of the top hits in protein complexes and pathways. CRISPRa confirmed some of the findings of CRISPRi and revealed additional and highly complementary information, e.g. clarifying the roles of enzymes in different branches of the glycosphingolipid biosynthesis pathways in protection against toxicity. These findings highlight some of the advantages of this CRISPR approach over haploid mutagenesis.
A large part of the human transcriptome comprises non-coding RNA, including miRNAs and long non-coding RNA (lncRNA) [126]. Over 56,000 lncRNAs have been discovered to date [127] and roles for some of them in response to xenobiotic exposures have been described [128]. Methodologies to probe the full extent of lncRNA involvement are needed to fully delineate the biological response to xenobiotics. Recent studies have exemplified the power of CRISPR to target non-coding RNAs. Using CRISPRi, Gilbert et al. achieved >80% knockdown for 5 of 6 lncRNAs in K562 [123]. Ho et al. used an approach featuring HR-mediated targeting, NHEJ suppression, and a dual-guide RNA vector to generate knockouts for miRNAs and lncRNAs [129]. As mentioned above, the GeCKOv2 library developed by Feng Zhang’s group contains sgRNAs that target human miRNAs (n=1,864) [110] by directing mutations to the pre-miRNA hairpin structure [130].
5.3 Limitations of CRISPR-Cas9 screening
CRISPR-Cas9 has limitations that need to be addressed before it can be deployed, particularly in the therapeutic realm, on a large scale. Though CRISPR-Cas9 has a high validation rate, OTEs do occur partly due to the small seed region guiding specificity, albeit at a lower rate than with RNAi [131]. Several strategies to reduce OTEs have been developed. For example, a paired nickase or dual nickase strategy, in which adjacent offset nicks are generated at the target site using two gRNAs and two co-dependent Cas9 nickase monomers, has been described [98, 132–134]. Joung’s group reduced OTEs substantially by simply using sgRNAs that were truncated (tru-gRNAs) by 2–3 nucleotides at the 5′ end of their complementarity regions [135]. Another approach to minimize off-target effects is to predict them in silico, e.g. the bioinformatics tool COSMID (CRISPR Off-target Sites with Mismatches, Insertions, and Deletions) searches genomes for potential off-target sites (http://crispr.bme.gatech.edu), helping to inform the design of CRISPR-Cas systems with minimal off-target effects [136]. In a recent paper, Yang et al. used whole-genome sequencing to assess target specificity in human-induced pluripotent stem cells (hiPSC) and found that single nucleotide variants can create OTEs, with a likelihood of 1.5–8.5%, depending on the genome and site-selection method and thus has important implications for subject-specific design [127]. Joung’s group developed an approach called genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq), that relies on capture of double-stranded oligodeoxynucleotides into DSBs. The method identified many OTEs that were not detectable by existing computational methods and confirmed that tru-gRNAs exhibit reduced numbers of OTEs [137].
Another limitation is that the range of currently available target sites is restricted by the need for a PAM sequence matching the form NGG; use of alternative PAM sequences is being explored but requires further validation [98]. Use of gRNA in conjunction with Cas9 from other species may also expand target options.
Strategies to reduce OTEs, to increase the target range and specificity, and shift the balance away from NHEJ-mediated indel mutations and toward HDR-driven alterations are priorities. A final limitation is that some cell lines are more challenging to transfect/infect, and may limit the technology to less relevant cell lines.
Despite these challenges, the studies to date demonstrate the potential of the CRISPR-Cas9 system for conducting large-scale genome-wide screens in mammalian cells. This system offers several powerful features such as gene inactivation at the genomic DNA level and lower off-target effects than RNAi, which will be further reduced by ongoing improvements to the technology.
6. Potential of CRISPR-Cas9 for toxicity screening
Each of the screening methodologies described in this paper has advantages and disadvantages as discussed above. The CRISPR-Cas9 screening approach, which is evolving rapidly, overcomes many of the limitations of previous approaches. It can be used to repress and activate both coding and non-coding genes of interest, with fewer OTEs than RNAi. It is compatible with any human cell type that can be transfected and thus can identify genes that are more directly relevant to normal human cells compared with yeast and KBM7 haploid screening. Libraries are commercially available that can target a large and growing number of human genes. Therefore, CRISPR-Cas9 is a powerful functional genomic screening approach that will provide unprecedented mechanistic insight in the field of modern toxicology.
First, as described earlier, CRISPR-Cas9 has been used to identify genes involved in the response to chemical and microbial toxicants in several cell types [94, 101–103]. Thus, it could readily be extended to the systematic screening of large numbers of environmental chemicals in a variety of human cell lines. Second, as well as the knockout of gene function, CRISPRi and CRISPRa facilitate the repression and activation of gene expression with near-saturation [123]. Thus, it offers the potential to more fully characterize AOPs and AOP networks than yeast or human haploid screening. Third, as a flexible gene-editing tool, CRISPR-Cas9, as well as allowing the examination of the effects of a large number of gene knock-outs simultaneously, enables the precise targeting of individual genes and gene regions identified by CRISPR, or other screening methods, for further validation. Fourth, as well as screening genes, CRISPR-Cas9 can be used to precisely generate human cell and animal models of development and disease [111–117] that can increase the efficiency of preclinical toxicity testing and be used to discover or validate GxE interactions at the level of individual genotypes or haplotypes. Fifth, CRISPR-Cas9 can be used to target non-coding RNAs [110, 123, 129, 130], which represent a large proportion of the human transcriptome and potentially play important roles in response to toxicants.
Ongoing improvements in target range and specificity, automation, next generation sequencing, and data handling and analysis, will increase the throughput of screening techniques. High-throughput screening of chemicals of concern or unknown toxicity will elucidate common and unique mechanisms and pathways of toxicity. The time is right for the establishment of efforts to systematically evaluate chemicals of concern by CRISPR-Cas9 screening in human cells. Such an approach could be designed to address some of the major challenges of risk assessment today [1], including the identification of individual variation in susceptibility at the level of genes and toxicity pathways, as well as understanding the effect of environmental levels of exposure, co-exposures, tissue-specific effects (use of different cell types), and early-life effects (use of hESC and differentiated cells).
Acknowledgments
We acknowledge Professor Chris Vulpe for his long-term collaboration with us on screening of environmental chemicals using functional genomics in the yeast. Dr. Hua Shen is a postdoctoral trainee in the Superfund Research Program at the University of California Berkeley. This work was supported by the U.S. National Institute of Environmental Health Sciences Superfund Research Program P42 ES004705 (MTS and LZ) and by the U.S. National Institute of Health grant R01ES017452 (LZ).
Abbreviations
- 3-BrPA
3 bromopyruvate
- AOP
adverse outcome pathway
- ARF4
ADP-ribosylation factor 4
- BFA
brefeldin A
- BT
1,2,4-benzenetriol
- CML
chronic myeloid leukemia
- COSMID
CRISPR off-target sites with mismatches, insertions, and deletions
- CRISPR-Cas9
clustered regularly interspaced short palindrome repeats-associated nuclease-Cas9
- CTx-DTA
chimeric diphtheria toxin
- DSB
double-strand break
- dsRNA
double-stranded RNA
- eHAP
engineered HAPloid
- ETC
electron transport chain
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation product
- ESC
embryonic stem cell
- GeCKo
genome-scale CRISPR-Cas9 knockout
- GWAS
genome-wide association studies
- GUIDE-seq
genome-wide unbiased identification of DSBs enabled by sequencing
- HCS
high-content screening
- HDR
homolody-directed repair
- HQ
hydroquinone
- HTS
high-throughput screening
- iPSC
induced pluripotent stem cell
- lncRNA
long non-coding RNA
- MHC
major histomompatibility protein 1
- MCT1
monocarboxylate transporter 1
- MOA
mode of action
- NGS
next-generation sequencing
- NHEJ
non-homologous end-joining repair
- ORF
open reading frame
- OTE
off-target effects
- PAM
protospacer-adjacent motif
- PCR
polymerase chain reaction
- PDA
parallel deletion analysis
- PLP2
proteolipid 2
- RNAi
RNA interference
- sgRNA
single-guide RNA
- tru-gRNA
truncated sgRNA
- shRNA
short hairpin RNA
- siRNA
short-interfering RNA
- SLC35F2
solute carrier family member 35 F2
- TALEN
transcription activator-like effector nuclease
Footnotes
8. Conflict of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.US Environmental Health Protection Agency. Next Generation Risk Assessment: Incorporation of Recent Advances in Molecular, Computational, and Systems Biology (External Review Draft) Washington, DC: 2014. [Google Scholar]
- 2.Sturla SJ, Boobis AR, FitzGerald RE, Hoeng J, Kavlock RJ, Schirmer K, Whelan M, Wilks MF, Peitsch MC. Systems toxicology: from basic research to risk assessment. Chemical research in toxicology. 2014;27:314–329. doi: 10.1021/tx400410s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molinelli EJ, Korkut A, Wang W, Miller ML, Gauthier NP, Jing X, Kaushik P, He Q, Mills G, Solit DB, Pratilas CA, Weigt M, Braunstein A, Pagnani A, Zecchina R, Sander C. Perturbation biology: inferring signaling networks in cellular systems. PLoS computational biology. 2013;9:e1003290. doi: 10.1371/journal.pcbi.1003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitra S, Das S, Chakrabarti J. Systems biology of cancer biomarker detection. Cancer biomarkers : section A of Disease markers. 2013;13:201–213. doi: 10.3233/CBM-130363. [DOI] [PubMed] [Google Scholar]
- 5.Ideker T, Krogan NJ. Differential network biology. Molecular systems biology. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edwards SW, Preston RJ. Systems biology and mode of action based risk assessment. Toxicological sciences : an official journal of the Society of Toxicology. 2008;106:312–318. doi: 10.1093/toxsci/kfn190. [DOI] [PubMed] [Google Scholar]
- 7.Califano A, Butte AJ, Friend S, Ideker T, Schadt E. Leveraging models of cell regulation and GWAS data in integrative network-based association studies. Nature genetics. 2012;44:841–847. doi: 10.1038/ng.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waters MD, Fostel JM. Toxicogenomics and systems toxicology: aims and prospects. Nature reviews Genetics. 2004;5:936–948. doi: 10.1038/nrg1493. [DOI] [PubMed] [Google Scholar]
- 9.Hieter P, Boguski M. Functional genomics: it’s all how you read it. Science. 1997;278:601–602. doi: 10.1126/science.278.5338.601. [DOI] [PubMed] [Google Scholar]
- 10.Mahadevan B, Snyder RD, Waters MD, Benz RD, Kemper RA, Tice RR, Richard AM. Genetic toxicology in the 21st century: reflections and future directions. Environmental and molecular mutagenesis. 2011;52:339–354. doi: 10.1002/em.20653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen ME, Krewski D. Toxicity testing in the 21st century: bringing the vision to life. Toxicological sciences : an official journal of the Society of Toxicology. 2009;107:324–330. doi: 10.1093/toxsci/kfn255. [DOI] [PubMed] [Google Scholar]
- 12.Krewski D, Andersen ME, Mantus E, Zeise L. Toxicity testing in the 21st century: implications for human health risk assessment. Risk analysis : an official publication of the Society for Risk Analysis. 2009;29:474–479. doi: 10.1111/j.1539-6924.2008.01150.x. [DOI] [PubMed] [Google Scholar]
- 13.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dos Santos SC, Teixeira MC, Cabrito TR, Sa-Correia I. Yeast toxicogenomics: genome-wide responses to chemical stresses with impact in environmental health, pharmacology, and biotechnology. Frontiers in genetics. 2012;3:63. doi: 10.3389/fgene.2012.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Botstein D, Fink GR. Yeast: an experimental organism for 21st Century biology. Genetics. 2011;189:695–704. doi: 10.1534/genetics.111.130765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costanzo MC, Hogan JD, Cusick ME, Davis BP, Fancher AM, Hodges PE, Kondu P, Lengieza C, Lew-Smith JE, Lingner C, Roberg-Perez KJ, Tillberg M, Brooks JE, Garrels JI. The yeast proteome database (YPD) and Caenorhabditis elegans proteome database (WormPD): comprehensive resources for the organization and comparison of model organism protein information. Nucleic Acids Res. 2000;28:73–76. doi: 10.1093/nar/28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinmetz LM, Scharfe C, Deutschbauer AM, Mokranjac D, Herman ZS, Jones T, Chu AM, Giaever G, Prokisch H, Oefner PJ, Davis RW. Systematic screen for human disease genes in yeast. Nat Genet. 2002;31:400–404. doi: 10.1038/ng929. [DOI] [PubMed] [Google Scholar]
- 19.Bassett DE, Jr, Boguski MS, Hieter P. Yeast genes and human disease. Nature. 1996;379:589–590. doi: 10.1038/379589a0. [DOI] [PubMed] [Google Scholar]
- 20.Parsons AB, Geyer R, Hughes TR, Boone C. Yeast genomics and proteomics in drug discovery and target validation. Prog Cell Cycle Res. 2003;5:159–166. [PubMed] [Google Scholar]
- 21.Foury F. Human genetic diseases: a cross-talk between man and yeast. Gene. 1997;195:1–10. doi: 10.1016/s0378-1119(97)00140-6. [DOI] [PubMed] [Google Scholar]
- 22.Smith V, Chou KN, Lashkari D, Botstein D, Brown PO. Functional analysis of the genes of yeast chromosome V by genetic footprinting. Science. 1996;274:2069–2074. doi: 10.1126/science.274.5295.2069. [DOI] [PubMed] [Google Scholar]
- 23.Ross-Macdonald P, Sheehan A, Roeder GS, Snyder M. A multipurpose transposon system for analyzing protein production, localization, and function in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:190–195. doi: 10.1073/pnas.94.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz MC, Muir RS, Lim E, McElver J, Weber SC, Heitman J. Gene disruption with PCR products in Saccharomyces cerevisiae. Gene. 1995;158:113–117. doi: 10.1016/0378-1119(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 25.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet. 1996;14:450–456. doi: 10.1038/ng1296-450. [DOI] [PubMed] [Google Scholar]
- 26.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 27.North M, Vulpe CD. Functional toxicogenomics: mechanism-centered toxicology. International journal of molecular sciences. 2010;11:4796–4813. doi: 10.3390/ijms11124796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith AM, Heisler LE, Mellor J, Kaper F, Thompson MJ, Chee M, Roth FP, Giaever G, Nislow C. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009;19:1836–1842. doi: 10.1101/gr.093955.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21:278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- 30.Pierce SE, Davis RW, Nislow C, Giaever G. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat Protoc. 2007;2:2958–2974. doi: 10.1038/nprot.2007.427. [DOI] [PubMed] [Google Scholar]
- 31.Pierce SE, Davis RW, Nislow C, Giaever G. Chemogenomic approaches to elucidation of gene function and genetic pathways. Methods Mol Biol. 2009;548:115–143. doi: 10.1007/978-1-59745-540-4_7. [DOI] [PubMed] [Google Scholar]
- 32.Scherens B, Goffeau A. The uses of genome-wide yeast mutant collections. Genome Biol. 2004;5:229. doi: 10.1186/gb-2004-5-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jo WJ, Loguinov A, Wintz H, Chang M, Smith AH, Kalman D, Zhang L, Smith MT, Vulpe CD. Comparative functional genomic analysis identifies distinct and overlapping sets of genes required for resistance to monomethylarsonous acid (MMAIII) and arsenite (AsIII) in yeast. Toxicological sciences : an official journal of the Society of Toxicology. 2009;111:424–436. doi: 10.1093/toxsci/kfp162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.North M, Tandon VJ, Thomas R, Loguinov A, Gerlovina I, Hubbard AE, Zhang L, Smith MT, Vulpe CD. Genome-wide functional profiling reveals genes required for tolerance to benzene metabolites in yeast. PloS one. 2011;6:e24205. doi: 10.1371/journal.pone.0024205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connor ST, Lan J, North M, Loguinov A, Zhang L, Smith MT, Gu AZ, Vulpe C. Genome-Wide Functional and Stress Response Profiling Reveals Toxic Mechanism and Genes Required for Tolerance to Benzo[a]pyrene in S. cerevisiae. Frontiers in genetics. 2012;3:316. doi: 10.3389/fgene.2012.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Graaf B, Clore A, McCullough AK. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst) 2009;8:1207–1214. doi: 10.1016/j.dnarep.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.North M, Romero C, Loguinov A, Smith MT, Zhang L, Vulpe CD. Identification of novel biomarkers of formaldehyde toxicity in humans using functional genomics in yeast. The Toxicologist. 2011;120:548. [Google Scholar]
- 38.Galvan N, Lim S, Zmugg S, Smith MT, Zhang L. Depletion of WRN enhances DNA damage in HeLa cells exposed to the benzene metabolite, hydroquinone. Mutation research. 2008;649:54–61. doi: 10.1016/j.mrgentox.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren X, Lim S, Ji Z, Yuh J, Peng V, Smith MT, Zhang L. Comparison of proliferation and genomic instability responses to WRN silencing in hematopoietic HL60 and TK6 cells. PloS one. 2011;6:e14546. doi: 10.1371/journal.pone.0014546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren X, Lim S, Smith MT, Zhang L. Werner syndrome protein, WRN, protects cells from DNA damage induced by the benzene metabolite hydroquinone. Toxicological sciences : an official journal of the Society of Toxicology. 2009;107:367–375. doi: 10.1093/toxsci/kfn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jo WJ, Ren X, Chu F, Aleshin M, Wintz H, Burlingame A, Smith MT, Vulpe CD, Zhang L. Acetylated H4K16 by MYST1 protects UROtsa cells from arsenic toxicity and is decreased following chronic arsenic exposure. Toxicology and applied pharmacology. 2009;241:294–302. doi: 10.1016/j.taap.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren X, Aleshin M, Jo WJ, Dills R, Kalman DA, Vulpe CD, Smith MT, Zhang L. Involvement of N-6 adenine-specific DNA methyltransferase 1 (N6AMT1) in arsenic biomethylation and its role in arsenic-induced toxicity. Environmental health perspectives. 2011;119:771–777. doi: 10.1289/ehp.1002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandberg AA, Wake N, Kohno S. Chromosomes and causation of human cancer and leukemia. XLVII. severe hypodiploidy and chromosome conglomerations in ALL. Cancer genetics and cytogenetics. 1982;5:293–307. doi: 10.1016/0165-4608(82)90095-4. [DOI] [PubMed] [Google Scholar]
- 44.Andersson BS, Beran M, Pathak S, Goodacre A, Barlogie B, McCredie KB. Ph-positive chronic myeloid leukemia with near-haploid conversion in vivo and establishment of a continuously growing cell line with similar cytogenetic pattern. Cancer genetics and cytogenetics. 1987;24:335–343. doi: 10.1016/0165-4608(87)90116-6. [DOI] [PubMed] [Google Scholar]
- 45.Kotecki M, Reddy PS, Cochran BH. Isolation and characterization of a near-haploid human cell line. Experimental cell research. 1999;252:273–280. doi: 10.1006/excr.1999.4656. [DOI] [PubMed] [Google Scholar]
- 46.Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, Brummelkamp TR. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 47.Reiling JH, Clish CB, Carette JE, Varadarajan M, Brummelkamp TR, Sabatini DM. A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:11756–11765. doi: 10.1073/pnas.1018098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, Ploegh HL, Brummelkamp TR. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nature biotechnology. 2011;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burckstummer T, Banning C, Hainzl P, Schobesberger R, Kerzendorfer C, Pauler FM, Chen D, Them N, Schischlik F, Rebsamen M, Smida M, Fece de la Cruz F, Lapao A, Liszt M, Eizinger B, Guenzl PM, Blomen VA, Konopka T, Gapp B, Parapatics K, Maier B, Stockl J, Fischl W, Salic S, Taba Casari MR, Knapp S, Bennett KL, Bock C, Colinge J, Kralovics R, Ammerer G, Casari G, Brummelkamp TR, Superti-Furga G, Nijman SM. A reversible gene trap collection empowers haploid genetics in human cells. Nature methods. 2013;10:965–971. doi: 10.1038/nmeth.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Timms RT, Duncan LM, Tchasovnikarova IA, Antrobus R, Smith DL, Dougan G, Weekes MP, Lehner PJ. Haploid genetic screens identify an essential role for PLP2 in the downregulation of novel plasma membrane targets by viral E3 ubiquitin ligases. PLoS pathogens. 2013;9:e1003772. doi: 10.1371/journal.ppat.1003772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van den Boomen DJ, Timms RT, Grice GL, Stagg HR, Skodt K, Dougan G, Nathan JA, Lehner PJ. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11425–11430. doi: 10.1073/pnas.1409099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Birsoy K, Wang T, Possemato R, Yilmaz OH, Koch CE, Chen WW, Hutchins AW, Gultekin Y, Peterson TR, Carette JE, Brummelkamp TR, Clish CB, Sabatini DM. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nature genetics. 2013;45:104–108. doi: 10.1038/ng.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiling JH, Olive AJ, Sanyal S, Carette JE, Brummelkamp TR, Ploegh HL, Starnbach MN, Sabatini DM. A CREB3-ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nature cell biology. 2013;15:1473–1485. doi: 10.1038/ncb2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen WW, Birsoy K, Mihaylova MM, Snitkin H, Stasinski I, Yucel B, Bayraktar EC, Carette JE, Clish CB, Brummelkamp TR, Sabatini DD, Sabatini DM. Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell reports. 2014;7:27–34. doi: 10.1016/j.celrep.2014.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winter GE, Radic B, Mayor-Ruiz C, Blomen VA, Trefzer C, Kandasamy RK, Huber KV, Gridling M, Chen D, Klampfl T, Kralovics R, Kubicek S, Fernandez-Capetillo O, Brummelkamp TR, Superti-Furga G. The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nature chemical biology. 2014;10:768–773. doi: 10.1038/nchembio.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duncan LM, Timms RT, Zavodszky E, Cano F, Dougan G, Randow F, Lehner PJ. Fluorescence-based phenotypic selection allows forward genetic screens in haploid human cells. PloS one. 2012;7:e39651. doi: 10.1371/journal.pone.0039651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee CC, Carette JE, Brummelkamp TR, Ploegh HL. A reporter screen in a human haploid cell line identifies CYLD as a constitutive inhibitor of NF-kappaB. PloS one. 2013;8:e70339. doi: 10.1371/journal.pone.0070339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elling U, Taubenschmid J, Wirnsberger G, O’Malley R, Demers SP, Vanhaelen Q, Shukalyuk AI, Schmauss G, Schramek D, Schnuetgen F, von Melchner H, Ecker JR, Stanford WL, Zuber J, Stark A, Penninger JM. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell stem cell. 2011;9:563–574. doi: 10.1016/j.stem.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leeb M, Wutz A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011;479:131–134. doi: 10.1038/nature10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang H, Shi L, Wang BA, Liang D, Zhong C, Liu W, Nie Y, Liu J, Zhao J, Gao X, Li D, Xu GL, Li J. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell. 2012;149:605–617. doi: 10.1016/j.cell.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 62.Li W, Shuai L, Wan H, Dong M, Wang M, Sang L, Feng C, Luo GZ, Li T, Li X, Wang L, Zheng QY, Sheng C, Wu HJ, Liu Z, Liu L, Wang L, Wang XJ, Zhao XY, Zhou Q. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature. 2012;490:407–411. doi: 10.1038/nature11435. [DOI] [PubMed] [Google Scholar]
- 63.Leeb M, Dietmann S, Paramor M, Niwa H, Smith A. Genetic exploration of the exit from self-renewal using haploid embryonic stem cells. Cell stem cell. 2014;14:385–393. doi: 10.1016/j.stem.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pettitt SJ, Rehman FL, Bajrami I, Brough R, Wallberg F, Kozarewa I, Fenwick K, Assiotis I, Chen L, Campbell J, Lord CJ, Ashworth A. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PloS one. 2013;8:e61520. doi: 10.1371/journal.pone.0061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang H, Liu Z, Ma Y, Zhong C, Yin Q, Zhou C, Shi L, Cai Y, Zhao H, Wang H, Tang F, Wang Y, Zhang C, Liu XY, Lai D, Jin Y, Sun Q, Li J. Generation of haploid embryonic stem cells from Macaca fascicularis monkey parthenotes. Cell research. 2013;23:1187–1200. doi: 10.1038/cr.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li W, Li X, Li T, Jiang MG, Wan H, Luo GZ, Feng C, Cui X, Teng F, Yuan Y, Zhou Q, Gu Q, Shuai L, Sha J, Xiao Y, Wang L, Liu Z, Wang XJ, Zhao XY, Zhou Q. Genetic modification and screening in rat using haploid embryonic stem cells. Cell stem cell. 2014;14:404–414. doi: 10.1016/j.stem.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 67.Essletzbichler P, Konopka T, Santoro F, Chen D, Gapp BV, Kralovics R, Brummelkamp TR, Nijman SM, Burckstummer T. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome research. 2014;24:2059–2065. doi: 10.1101/gr.177220.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, Camargo FD, Wernig M, Jaenisch R, Brummelkamp TR. Generation of iPSCs from cultured human malignant cells. Blood. 2010;115:4039–4042. doi: 10.1182/blood-2009-07-231845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nature reviews Molecular cell biology. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 70.Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. Journal of cell science. 2001;114:4557–4565. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]
- 71.Maeda I, Kohara Y, Yamamoto M, Sugimoto A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Current biology : CB. 2001;11:171–176. doi: 10.1016/s0960-9822(01)00052-5. [DOI] [PubMed] [Google Scholar]
- 72.Mohr SE, Smith JA, Shamu CE, Neumuller RA, Perrimon N. RNAi screening comes of age: improved techniques and complementary approaches. Nature reviews Molecular cell biology. 2014;15:591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wolters NM, MacKeigan JP. From sequence to function: using RNAi to elucidate mechanisms of human disease. Cell death and differentiation. 2008;15:809–819. doi: 10.1038/sj.cdd.4402311. [DOI] [PubMed] [Google Scholar]
- 74.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O’Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, McCombie WR, Elledge SJ, Hannon GJ. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 76.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 77.Fennell M, Xiang Q, Hwang A, Chen C, Huang CH, Chen CC, Pelossof R, Garippa RJ. Impact of RNA-Guided Technologies for Target Identification and Deconvolution. Journal of biomolecular screening. 2014 doi: 10.1177/1087057114548414. pii: 1087057114548414. [DOI] [PubMed] [Google Scholar]
- 78.Willingham AT, Deveraux QL, Hampton GM, Aza-Blanc P. RNAi and HTS: exploring cancer by systematic loss-of-function. Oncogene. 2004;23:8392–8400. doi: 10.1038/sj.onc.1208217. [DOI] [PubMed] [Google Scholar]
- 79.Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nature methods. 2006;3:715–719. doi: 10.1038/nmeth924. [DOI] [PubMed] [Google Scholar]
- 80.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 81.Berns K, Bernards R. Understanding resistance to targeted cancer drugs through loss of function genetic screens. Drug Resist Updat. 2012;15:268–275. doi: 10.1016/j.drup.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 82.Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, Brenton JD, Downward J, Nicke B. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer cell. 2007;11:498–512. doi: 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 83.Singel SM, Cornelius C, Batten K, Fasciani G, Wright WE, Lum L, Shay JW. A targeted RNAi screen of the breast cancer genome identifies KIF14 and TLN1 as genes that modulate docetaxel chemosensitivity in triple-negative breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:2061–2070. doi: 10.1158/1078-0432.CCR-13-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ali N, Karlsson C, Aspling M, Hu G, Hacohen N, Scadden DT, Larsson J. Forward RNAi screens in primary human hematopoietic stem/progenitor cells. Blood. 2009;113:3690–3695. doi: 10.1182/blood-2008-10-176396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Karlsson C, Rak J, Larsson J. RNA interference screening to detect targetable molecules in hematopoietic stem cells. Current opinion in hematology. 2014;21:283–288. doi: 10.1097/MOH.0000000000000053. [DOI] [PubMed] [Google Scholar]
- 86.Schmidt EE, Pelz O, Buhlmann S, Kerr G, Horn T, Boutros M. GenomeRNAi: a database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic acids research. 2013;41:D1021–1026. doi: 10.1093/nar/gks1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N, Heidelberg Fly Array C. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- 88.Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 89.Kaelin WG., Jr Molecular biology. Use and abuse of RNAi to study mammalian gene function. Science. 2012;337:421–422. doi: 10.1126/science.1225787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Booker M, Samsonova AA, Kwon Y, Flockhart I, Mohr SE, Perrimon N. False negative rates in Drosophila cell-based RNAi screens: a case study. BMC genomics. 2011;12:50. doi: 10.1186/1471-2164-12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Niwa R, Slack FJ. Interpreting Fluorescence Microscopy Images and Measurements. In: Zuk D, editor. Evaluating Techniques in Biochemical Research. Cambridge, MA: Cell Press; 2007. [Google Scholar]
- 93.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 94.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ. An improved zinc-finger nuclease architecture for highly specific genome editing. Nature biotechnology. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 97.Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, Amora R, Miller JC, Leung E, Meng X, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Meyer BJ. Targeted genome editing across species using ZFNs and TALENs. Science. 2011;333:307. doi: 10.1126/science.1207773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature biotechnology. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fineran PC, Charpentier E. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–209. doi: 10.1016/j.virol.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 100.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera MD, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nature biotechnology. 2013:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 103.Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- 104.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic acids research. 2010;38:W462–468. doi: 10.1093/nar/gkq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sander JD, Zaback P, Joung JK, Voytas DF, Dobbs D. Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic acids research. 2007;35:W599–605. doi: 10.1093/nar/gkm349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zheng Q, Cai X, Tan MH, Schaffert S, Arnold CP, Gong X, Chen CZ, Huang S. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. BioTechniques. 2014;57:115–124. doi: 10.2144/000114196. [DOI] [PubMed] [Google Scholar]
- 109.Blanchard AP, Hood L. Sequence to array: probing the genome’s secrets. Nature biotechnology. 1996;14:1649. doi: 10.1038/nbt1296-1649. [DOI] [PubMed] [Google Scholar]
- 110.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, Ebert BL. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nature biotechnology. 2014;32:941–946. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nature communications. 2014;5:3964. doi: 10.1038/ncomms4964. [DOI] [PubMed] [Google Scholar]
- 113.Gonzalez F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV, Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell stem cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E, Lowe SW, Ventura A. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harms DW, Quadros RM, Seruggia D, Ohtsuka M, Takahashi G, Montoliu L, Gurumurthy CB. Mouse Genome Editing Using the CRISPR/Cas System. Current Protocols in Human Genetics. 2014;83:15.17.11–15.17.27. doi: 10.1002/0471142905.hg1507s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ma Y, Zhang X, Shen B, Lu Y, Chen W, Ma J, Bai L, Huang X, Zhang L. Generating rats with conditional alleles using CRISPR/Cas9. Cell research. 2014;24:122–125. doi: 10.1038/cr.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nature protocols. 2013;8:2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.O’Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014;516:263–266. doi: 10.1038/nature13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Guimaraes CP, Carette JE, Varadarajan M, Antos J, Popp MW, Spooner E, Brummelkamp TR, Ploegh HL. Identification of host cell factors required for intoxication through use of modified cholera toxin. The Journal of cell biology. 2011;195:751–764. doi: 10.1083/jcb.201108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nature reviews Molecular cell biology. 2006;7:568–579. doi: 10.1038/nrm1985. [DOI] [PubMed] [Google Scholar]
- 126.Pertea M. The human transcriptome: an unfinished story. Genes. 2012;3:344–360. doi: 10.3390/genes3030344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, Zhu W, Wu W, Chen R, Zhao Y. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic acids research. 2014;42:D98–103. doi: 10.1093/nar/gkt1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marrone AK, Beland FA, Pogribny IP. Noncoding RNA response to xenobiotic exposure: an indicator of toxicity and carcinogenicity. Expert opinion on drug metabolism & toxicology. 2014;10:1409–1422. doi: 10.1517/17425255.2014.954312. [DOI] [PubMed] [Google Scholar]
- 129.Ho TT, Zhou N, Huang J, Koirala P, Xu M, Fung R, Wu F, Mo YY. Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic acids research. 2014 doi: 10.1093/nar/gku1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhao Y, Dai Z, Liang Y, Yin M, Ma K, He M, Ouyang H, Teng CB. Sequence-specific inhibition of microRNA via CRISPR/CRISPRi system. Scientific reports. 2014;4:3943. doi: 10.1038/srep03943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature biotechnology. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature biotechnology. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Molecular therapy Nucleic acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, Joung JK. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature biotechnology. 2014 doi: 10.1038/nbt.3117. doi: 10.1038/nbt.3117. [Epub ahead of print] [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.GeCKO. GeCKO Basics: Choice of Vector System. 2014 http://genome-engineering.org/gecko/?page_id=31.
- 139.Gaytan BD, Loguinov AV, Lantz SR, Lerot JM, Denslow ND, Vulpe CD. Functional profiling discovers the dieldrin organochlorinated pesticide affects leucine availability in yeast. Toxicological sciences : an official journal of the Society of Toxicology. 2013;132:347–358. doi: 10.1093/toxsci/kft018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gaytan BD, Loguinov AV, Penate X, Lerot JM, Chavez S, Denslow ND, Vulpe CD. A genome-wide screen identifies yeast genes required for tolerance to technical toxaphene, an organochlorinated pesticide mixture. PloS one. 2013;8:e81253. doi: 10.1371/journal.pone.0081253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eggert US, Kiger AA, Richter C, Perlman ZE, Perrimon N, Mitchison TJ, Field CM. Parallel chemical genetic and genome-wide RNAi screens identify cytokinesis inhibitors and targets. PLoS biology. 2004;2:e379. doi: 10.1371/journal.pbio.0020379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kim Y, Sun H. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging cell. 2007;6:489–503. doi: 10.1111/j.1474-9726.2007.00302.x. [DOI] [PubMed] [Google Scholar]