

Abstract
The regulation of T-cell-mediated immune responses depends on the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) on T-cell receptors. Although many details of the signaling cascades are well understood, the initial mechanism and regulation of ITAM phosphorylation remains unknown. We used molecular dynamics simulations to study the influence of different compositions of lipid bilayers on the membrane association of the CD3ϵ cytoplasmic tails of the T-cell receptors. Our results show that binding of CD3ϵ to membranes is modulated by both the presence of negatively charged lipids and the lipid order of the membrane. Free-energy calculations reveal that the protein-membrane interaction is favored by the presence of nearby basic residues and the ITAM tyrosines. Phosphorylation minimizes membrane association, rendering the ITAM motif more accessible to binding partners. In systems mimicking biological membranes, the CD3ϵ chain localization is modulated by different facilitator lipids (e.g., gangliosides or phosphoinositols), revealing a plausible regulatory effect on activation through the regulation of lipid composition in cell membranes.
Introduction
The T-cell receptor (TCR) recognizes both self and foreign peptides embedded in the peptide-binding groove of major histocompatibility complex (MHC) molecules (1). TCR recognition of the peptide-MHC complex is an essential step in initiating the host adaptive immune response. Although the α and β subunits of the TCR are essential for peptide/MHC recognition, intracellular signaling events are mediated by paired invariant subunits (CD3 ζζ, CD3 γϵ, and CD3 δϵ) (2,3). Each of these chains contains one (CD3 γ, δ, and ϵ) or three copies (CD3 ζ) of a well-conserved signaling domain, the immunoreceptor tyrosine-based activation motif (ITAM) (4,5). Upon TCR engagement with peptide/MHC, the two canonical tyrosines located within the ITAMs are phosphorylated by Src-family protein tyrosine kinases (PTKs), specifically Lck and Fyn. In the biphosphorylated state, the ITAM recruits the tandem SH2 domains of ZAP-70 tyrosine kinase (6).
Structurally, ITAM-containing chains belong to a family of proteins called intrinsically disordered proteins (IDPs) (7,8), which have polypeptide stretches that lack a well-defined three-dimensional conformation under physiological conditions. Several studies have shown that the cytoplasmic domains of at least some ITAM chains are not suspended in the cytosol, but rather are bound to the inner leaflet of the plasma membrane (3–5,9). Using fluorescence spectroscopy, Xu et al. (10) showed that membrane binding of the murine CD3ϵ cytoplasmic domain is mediated primarily by clusters of basic residues and is dramatically affected by the nature of the surrounding lipids. In general, membrane binding of ITAMs is much more favorable to anionic lipids, including phosphatidylserine (PS), phosphatidilglycerol (PG), and phosphatidylinositol (PI) species, such as phosphatidylinositol 4,5-bisphosphate (PIP2) (2). NMR data (3,10) also indicate that this binding mode is dynamic, with a clear shift during TCR triggering in the equilibrium from the lipid-bound to the lipid-unbound state, rendering the ITAM tyrosines accessible.
Here, we investigated the molecular mechanism responsible for the association of the disordered ITAM-bearing cytoplasmic tail of human CD3ϵ to lipid bilayers of differing compositions. We apply molecular dynamics simulations at two levels of resolution: all-atom (AA) simulations allow us to obtain a detailed description of the binding process, and a coarse-grained (CG) model provides insight into the thermodynamics at longer timescales. Through these combined approaches, we show that interactions of the CD3ϵ peptide with membranes are markedly influenced by charge and by lipid packing state. To our knowledge, this work provides new insight into the potential influence of ordered lipid cell membrane domains (e.g., rafts) on cell signaling.
Materials and Methods
System setup
Membrane simulations of CD3ϵ interactions were conducted at both AA and CG levels of resolution. Three independent membranes were built using negatively charged palmitoyloleoyl PG (POPG), unsaturated palmitoyloleoyl PC (POPC), and saturated dipalmitoyl PC (DPPC) lipids (Fig. S1 in the Supporting Material). Each membrane consisted of 150 lipids per leaflet and was solvated with 20,000 water molecules. POPG charges were neutralized by an equal amount of sodium ions. One single CD3ϵ chain was added in close contact to the surface of the membranes and equilibrated before production time. The initial coordinates of the CD3ϵ were taken from the NMR structure of the mouse CD3ϵ cytoplasmic tail (10). Nonconserved residues were manually mutated to resemble the human T-cell surface glycoprotein CD3ϵ chain precursor (NCBI reference sequence NP-000724.1). The resulting sequence of the cytoplasmic tail peptide considered in this study is WSKNRKAKAKPVTRGAGAGGRQRGQNKERPPPVPNPDYEPIRKGQRDLYSGLNQRRI, with or without phosphorylation of the two tyrosines.
The coarse-grained systems were similarly built consisting of the same amount of lipids, water molecules, and CD3ϵ chain. For the potential of mean force (PMF) calculation, however, the equilibrated configuration of the AA POPG simulation was transformed into CG beads using a center-of-mass approach (11). Posteriorly, a water slab was added in the z direction to allow further elongation of the peptide during the free-energy calculation. POPG lipids were exchanged by either POPC or DPPC for the different calculations. The transmembrane region of the CD3ϵ was simulated with the builder tool of the chimera molecular viewer (12) using as a template the sequence of human CD3ϵ. The helix was posteriorly inserted and equilibrated in a POPG membrane. The equilibrated configuration was transformed into CG beads and attached to the cytoplasmic tail. In a similar way, the extracellular domain (PDB 1SY6) was attached to the transmembrane region and equilibrated. Finally, we preequilibrated bilayers composed of different lipid species to study the preferential solvation and colocalization of the CD3ϵ chain. Lipids were randomly inserted in the bilayer and fully solvated with CG water beads. Details of the bilayer building and the system setup can be found in the Supporting Materials and Methods.
Computational details
Simulations were performed using the GROMACS 4.5 molecular dynamics package (13). The lipids and proteins were modeled based on the parameters set for aliphatic chains, which are part of the GROMOS 54a7 force field (14). The SPC water model was used to model the solvent (15). Temperature was maintained at 300 K by weak coupling of the solvent and solute separately to a Berendsen heat bath (16), with a relaxation time of 1.0 ps. The pressure of the systems also was controlled by weak coupling with a relaxation time of 1.0 ps. Parameters for the CG lipids, glycolipids, and CD3ϵ chain were taken from the MARTINI model (17,18). The polarizable water model (19) was used in combination with the rest of the parameters. More details of the computation can be found in the Supporting Materials and Methods, including a complete description of the PMF approach.
In the initial set of simulations, we directly probed the interaction of a single human cytoplasmic CD3ϵ epsilon (PubMed access NP_000724.1) chain with three different lipid bilayers, using a united-atom force field (GROMOS 54A7) (14). Here, we considered only a single lipid species (POPG, POPC, or DPPC) and did not mix different lipids. In this case, bilayers were easily equilibrated during the timescale of our simulations. As an example, Fig. S2 shows the area per lipid dependency during 500-ns simulations. In the second part of our study, we used CG simulations to address the membrane that mimics a biological membrane by considering mixtures of different lipids. Lipid mixtures of similar composition have previously been considered using the MARTINI CG force field, and these mixtures have shown convergence (20). Regardless, we demonstrate this convergence using CG simulations, (Figs. S3 and S4). AA and CG simulations of pure lipid bilayers of POPG, DPPC, and POPC and CG simulations of disordered domains were performed at 300 K. The lipid mixtures containing both ordered and disordered domains were simulated at 298 K. The MARTINI CG force field (18) employed in our simulations has been largely applied to the study of lipid phases and lipid transition temperatures (21–23). With this force field, the liquid ordered-liquid disordered (Lo-Ld) transition temperature for the DPPC/cholesterol mixture was determined to be around 310 K. Therefore, a good choice to fully represent the main physical properties of the Lo and Ld domains is 298 K, as has been reported elsewhere (20).
Results
Binding of CD3ϵ cytoplasmic tail to a model membrane
First, we sought a detailed mechanistic description of the binding of the human CD3ϵ cytoplasmic tail (lacking both extracellular and transmembrane regions) to membranes uniformly composed of POPG. At the initiation of the MD simulation, the cytoplasmic tail of the CD3ϵ chain was placed close to the water-membrane interface. The equilibrated configuration of the CD3ϵ chain upon binding to a POPG bilayer is depicted in Fig. 1 B. During 500 ns of simulation, the CD3ϵ chain was observed to spontaneously interact deeply with the POPG bilayer. The peptide rapidly adopted a buried configuration, where all the basic residues (arginines and lysines) were found to form hydrogen bonds with the negatively charged regions of the membrane, especially the phosphate groups. Notably, the two tyrosine residues present in the ITAM motif (Fig. 1 A, green) were found in close interaction with the glycerol moiety and the aliphatic chains of POPG. The interaction, however, did not affect the morphology of the POPG membrane; the area per lipid and the membrane thickness were comparable to those in a control simulation without the CD3ϵ chain (data not shown). These findings suggest that the area between the lipid headgroups allowed the basic arginines and the hydrophobic tyrosines to deeply interact with the phosphate groups and the aliphatic chains, respectively. The featured bound configuration is also depicted in the calculated electron-density profile from the trajectory (Fig. 1 B). Clearly, the polypeptide is colocalized close to the phosphate groups (orange line) and the glycerol moieties (red line) of the membrane. The tyrosines of the two ITAM regions, however, are found close to the region of the aliphatic tails (green line). We set up two independent simulations of 500 ns each under the same conditions, obtaining similar results for both cases. Despite our limited timescale, our results are consistent with experimental findings (3,10), showing not only the specific interactions but also the configuration of the CD3ϵ upon binding (Fig. S5).
Figure 1.
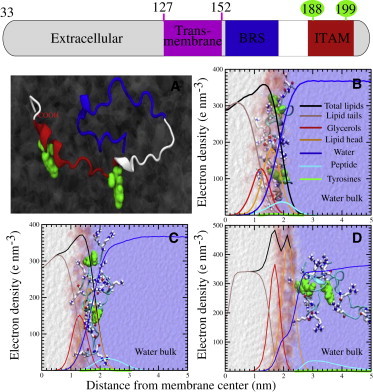
Mode of binding of the cytoplasmic tail of CD3ϵ to different lipid model membranes at AA resolution. The extracellular, transmembrane, and cytoplasmic regions of CD3ϵ as found in the UniProtKB database are depicted at the top of the figure. (A) Representation of the secondary structure of the cytoplasmic domain of CD3ϵ, showing the two ITAM tyrosines (green spheres). The ITAM and BRS regions are shown in red and blue, respectively. (B–D) The protein can adopt different configurations upon binding. The most stable conformer is largely favored by charged interactions between the basic residues (stick representation) of the protein and the negative headgroups of the POPG lipid (B). Extra stabilization is provided by the internalization of ITAM tyrosines into the hydrophobic core of the membrane. Replacing POPG by a zwitterionic lipid (POPC) renders the protein in a more solvent-exposed configuration (C). However, the tight packing of more ordered lipids (DPPC) dramatically affects the binding process (D). The latter is illustrated by the localization of the protein in the bulk water at the end of the simulation. Background electron-density profiles provide a clear structural description of the protein (cyan) and its interaction with the membrane (lipid headgroups (orange), the glycerol moiety (red), and the hydrophobic aliphatic tails (brown)). Solvent accessibility and localization of the ITAM residues are represented by the blue and green lines, respectively. Total density of the bilayers is represented by solid black lines. To see this figure in color, go online.
Binding of CD3ϵ cytoplasmic tail is affected by the nature of the lipids
We also studied the differential binding of the cytoplasmic tail of the CD3ϵ chain after modifying the nature of the lipids present in the membrane. After substituting POPG with a bilayer of zwitterionic, unsaturated POPC, the CD3ϵ chain was also observed to interact with lipids. However, now the key tyrosines were unable to deeply penetrate the bilayer. Furthermore, most of the basic arginines were found to interact with the surrounding water molecules. Although not fully buried, the peptide remained along the interface, and some interactions with phosphate groups of POPC were observed during the simulation (Fig. 1 C). Notice here that the electron-density profile suggests that POPG and POPC share morphological similarities. However, the localization of the peptide suggests that the positive amine groups present in the choline head of POPC pose a high penalty for interaction with the peptide.
The POPG and POPC bilayers represented Ld membranes, based on their unsaturated fatty acid moieties. In the next set of simulations, the CD3ϵ cytoplasmic tail was placed next to a more ordered lipid, fully saturated DPPC. The CD3ϵ chain was rapidly expelled from the water-lipid interface. In contrast to the bound conformation observed in both POPG and POPC bilayers, in DPPC, the peptide was preferentially located in the water bulk subphase in most simulations (Fig. 1 D). Note that the temperature of our simulations (300 K) was below the melting temperature of the DPPC lipids in these simulations, which results in a gel-like conformation. Although it may represent a stringent condition, it is apparent that the tight packing of ordered lipids does not allow the phosphate groups to be fully exposed to the surface and renders the lipid interactions with the arginines of the CD3ϵ chain highly unfavorable. Also, the same preferences in membrane association were observed with unbiased CG simulations of the three independent systems (POPG, POPC, and DPPC). The cytoplasmic tail was placed at 10 nm from the normal to a fully solvated lipid bilayer and 10 independent simulations of 10 μs were carried out with each of the bilayers. In all 10 simulations, the CD3ϵ cytoplasmic tail associates with POPG. In contrast, the association with POPC membrane was seen only 30% of the time, and no association with DPPC membrane was observed.
Thermodynamics of the CD3ϵ cytoplasmic tail binding to model membranes
To better understand the molecular driving forces during the binding process, we quantified the energetics of the interaction between the CD3ϵ tail and the different lipid components. Although our AA simulations clearly indicated a difference in the CD3ϵ binding mode for different lipids, use of this approach to follow the entire binding-unbinding process would be computationally costly, requiring much longer times and involving large free-energy barriers. Instead, we considered a CG model based on the last version of the polarizable MARTINI force field (18). Our initial CG setup is based on the last configuration of the AA CD3ϵ chain interacting with the POPG bilayer.
Coordinates were transformed into pseudo-CG positions, and the system was reequilibrated (see Materials and Methods). Fig. 2 summarizes the results of a PMF calculation (see Supporting Material), that directly quantifies the preferential binding of the CD3ϵ chain between POPG, POPC, and DPPC. We slowly dragged the cytoplasmic tail from the C-terminus toward the water bulk subphase to avoid the generation of large forces during the pulling and to account for the contribution of every single residue to the detachment (see Supporting Material). As clearly depicted, the presence of POPG lipids renders an energy barrier ΔGdett = ∼125 kJ mol−1 during the initial steps of the CD3ϵ pulling. Interestingly, the maximum is found when the C-terminus of the peptide protrudes ∼2 nm from the membrane center. A close-up of the process (Fig. 2 B) shows that detachment of the residues located within the ITAM (Fig. 1 A) is responsible for the initial energy barrier (Fig. 2 B). This configuration is favored by the second tyrosine and the basic-rich stretch. However, stretching the peptide even more (∼8 nm from the center of the bilayer), renders the pulling process favorable. This step matches the detachment of the second tyrosine and the basic residues located in frame (Fig. 3 C). Overall, detachment of the ITAM region of the peptide (ΔGdett) is unfavorable and requires 125 kJ mol−1 for 9 nm stretching. A different energy landscape is observed for the interaction with either POPC or DPPC bilayers. Pulling the peptide out of the membrane is largely favorable along the chosen reaction coordinate.
Figure 2.
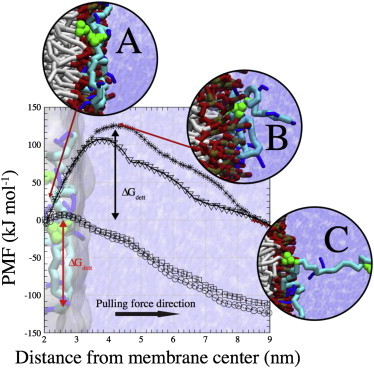
PMF for detachment of the cytoplasmic tail of CD3ϵ from POPG (crosses), POPC (squares), and DPPC (circles) model membranes. Using an umbrella sampling method, the C-terminus of CD3ϵ was slowly pulled out of the different membranes. The PMF for detachment of the activated CD3ϵ chain (phosphorylated ITAM tyrosines) was also probed (triangles). The order parameter represents the distance between the centers of mass of the bilayers and the C-terminus of the protein. Initially placed in close contact to the bilayer (A), the protein was steadily pulled from the bilayer. A high activation energy (ΔGdett = ∼125 kJ mol−1) is noticeable for detachment from the negatively charged POPG bilayer, structurally corresponding to exposure of several basic residues and one tyrosine of the ITAM region (B). As the order parameter increases, the free energy is lowered by detachment of the second ITAM tyrosine and consecutive basic residues close to the BRS (C). The peak in the energy (B) drops by ∼20 kJ mol−1 when the ITAM tyrosines are phosphorylated, presumably by their repulsion with the negatively charged lipids of a pure POPG bilayer. Detachment from either a POPC or DPPC bilayer does not pose any noticeable energetic barrier. To see this figure in color, go online.
Figure 3.
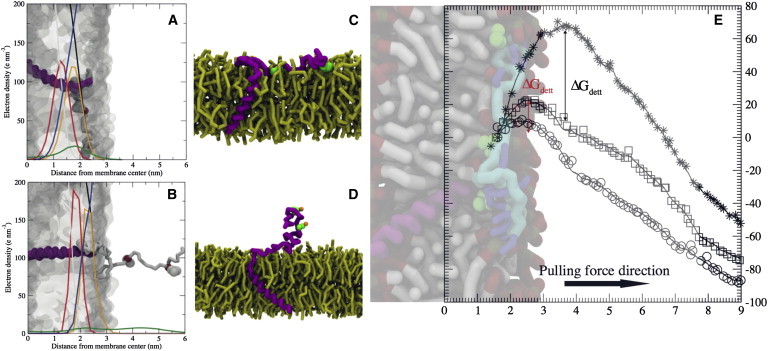
Influence of the transmembrane region of CD3ϵ on binding to model membranes. The helical transmembrane region (purple) was attached to the cytoplasmic CD3ϵ tail (light gray) and posteriorly inserted into a bilayer lipid (transparent surface). (A) Representation of the uncleaved CD3ϵ after 40 μs effective CG simulation time interacting with a POPG bilayer. The cytoplasmic region resembles the typical configuration obtained at the AA level. Notice the structural localization of the ITAM tyrosines (van der Waals spheres representation) and their interaction with the membrane aliphatic tails. (B) Configuration of the uncleaved CD3 chain inserted into a DPPC bilayer, clearly showing the spontaneous detachment of the cytoplasmic tail. (C and D) Snapshots of an equilibrated structure of the CD3ϵ chain (transmembrane and cytoplasmic regions) in contact with a pure POPG bilayer (yellow). (C) The nonphosphorylated tyrosines (green spheres) located in the ITAM region were found to be embedded in the bilayer. (D) In contrast, the phosphorylated tyrosines were found to be exposed to bulk water (water spheres are omitted from the picture for better depiction). Phosphate groups are in orange. (E) PMFs for detachment of the CD3ϵ ITAM region from POPG (crosses), POPC (squares), and DPPC (circles) bilayers. Pulling reaction coordinates as in Fig. 2. A noticeable energy barrier is shown for detachment from either a POPG (ΔGdett= 70 kJ mol−1) or a POPC (ΔGdett = 25 kJ mol−1) bilayer. However the process is fully favorable for a DPPC membrane. To see this figure in color, go online.
In comparing results with three different lipid compositions, the different features found in the energy profiles for the detachment of the CD3ϵ cytoplasmic chain are notable. First, binding is favored by Coulombic interactions; they pose the initial barrier during the detachment of the CD3ϵ chain from the POPG bilayer but are not present in POPC membranes. As observed during the PMF calculations, the ITAM tyrosines of the CD3ϵ chain represent a very important anchoring point in the hydrophobic regions of the membrane. It is important to note that, as shown with the PMF in Fig. 2 (triangles), the anchoring effect of tyrosines is diminished when the canonical tyrosines in the ITAM motif are phosphorylated. In the case of phosphorylated tyrosines, the ITAM motif was found to be exposed to the solvent. Second, although the tyrosines were able to interact with the hydrophobic region of the POPC bilayer during the pulling process, they do not overcome the large repulsion of the choline headgroups during binding, as observed by the detachment from the POPC bilayer. Thus, during the binding process, the initial peptide-membrane recognition is favored by the interactions between the basic residues and the negatively charged groups of the lipids. Once the peptide is translocated to the interface, the hydrophobic unphosphorylated tyrosines in the ITAM motif become responsible for the anchoring and overall stability.
We further quantified the binding affinity of the CD3ϵ cytoplasmic tail for the POPG membrane using the CG force field. In these PMF calculations, the entire CD3ϵ cytoplasmic tail was pulled from its center of mass from its membrane-associated position (see Supporting Material). The affinity constant (standardized at 1 M) to a pure POPG bilayer was then computed as (24):
where NA is Avogadro’s number and ΔG is the calculated membrane-protein PMF as a function of distance r between the center of mass of the protein and the POPG lipid membrane. rave is the average total membrane area. These calculations provided a KA of 5 × 106 M−1, confirming that the binding is clearly enhanced by the presence of negatively charged lipids.
Influence of the transmembrane region of CD3ϵ on binding of its tail to model membranes
Next, we studied the effect of adding the transmembrane helical region to the cytoplasmic CD3ϵ tail that we have characterized so far. As shown in Fig. 3, the final configurations of the CD3ϵ chain attached to the transmembrane region did not differ from the untethered cytoplasmic chain. After simulations on an effective timescale of 40 μs, the structures of the cytoplasmic regions preserved the basic features observed in our previous simulations: deep attachment of the CD3ϵ cytoplasmic region to the POPG bilayers (Fig. 3 A), with the key tyrosines deeply embedded in the bilayer. The electron-density profile depicted in Fig. 3 B shows that the cytoplasmic regions remain in the bulk water in the case of DPPC bilayers. The two tyrosines are out of the membrane interface and highly solvated. We carried out additional simulations with phosphorylated ITAM tyrosines to ensure that the observed burial effects are only applicable to the nonactivated ITAM motifs. These simulations show that phosphorylation prevents the ITAM tyrosines from being fully embedded in a pure POPG bilayer. We show the differences between the equilibrated structures for both the nonphosphorylated and the phosphorylated chains in Fig. 3, C and D. As can be observed, the basic residue stretch (BRS) region extensively interacts with the bilayer in both cases. However, the nonphosphorylated tyrosines are able to deeply interact with the hydrophobic tails of the bilayer, whereas the phosphorylated tyrosines avoid this interaction and are exposed to the solvent.
Also, we calculated the energy profile for detachment from the three different lipid membranes, as done for the untethered CD3ϵ cytoplasmic tail. Although in general the energies show the same trend for the tethered cytoplasmic tail, the magnitudes clearly differ from those for the untethered CD3ϵ chain, especially in the case of detachment from the POPG bilayer. The tethered CD3ϵ chain renders an energy barrier of ∼70 kJ mol−1 (Fig. 3 E). Note that this represents a drop of ∼65 kJ mol−1 when compared to the untethered CD3ϵ chain (Fig. 2). Furthermore, and contrary to the results obtained for the cleaved peptide, detachment of the CD3ϵ chain is unfavorable in the POPC bilayer by ∼20 kJ mol−1.
A more detailed inspection of the configurations from the simulations revealed that the membrane thickness was affected upon insertion of the transmembrane region (Fig. S6). As depicted by the electron-density profile, the membrane thickness was decreased by nearly 0.5 nm. Altogether, the peaks representing the positions of the headgroups, glycerol, aliphatic tails, and protein are shifted, and clearly indicate a more disordered state. Apparently, as revealed by the PMFs, a decrease in the thickness of bilayers can antagonize the binding process. On the one hand, the thickness in POPG increases the accessibility to the water molecules and sodium ions, which drastically affects the solvation shell around the phosphate groups and the basic residues of the peptide (electron density provided in Fig. S6). As a result, the interactions between the lipids and the peptide are diminished. On the other hand, the increase in area per lipid expansion in POPC not only increases the solvation accessibility but also allows the peptide to bypass the screening of the amine groups in the headgroup. Furthermore, the presence of closer water molecules provides a shield around the positive regions of the lipids.
Localization of the entire CD3ϵ chain in the Ld domains of biological membranes
To this point, our simulations were restricted to a lipid model membrane composed of a single lipid species and partial constructs of CD3ϵ. To extend our investigation to a more biologically relevant system, we studied the preferential partitioning of the whole CD3ϵ protein (composed of extracellular, transmembrane, and cytoplasmic domains) in bilayers composed of mixed lipids. First, we looked at the preferential interactions in a polydispersed bilayer composed of POPC, POPG, palmitoyloleoyl phosphatidylethanolamine (POPE), palmitoyloleoyl phosphatidylserine, and PI. The mixture should resemble the properties of an Ld state, and the lipids present have been previously reported to interact with the CD3ϵ chain (4,25). Starting from a random configuration, the protein is rapidly solvated by the different lipid species. Furthermore the ITAM located in the cytoplasmic tail is locally embedded and associated with the lipids of the bilayer. As the simulation proceeds, PI lipids cluster closely near the BRS of the cytoplasmic tail (shown in Fig. S7 for a simulation of 5 μs). We further quantified the degree of interaction by calculating the protein preferential partitioning (Table S1) and lipid radial distribution function. The calculations revealed that interactions with POPC and POPE are least favored, whereas PI represents ∼50% of the total solvation, even above the negatively charged POPG, which accounted for ∼30% of the solvation. This is consistent with experimental data for the critical roles of the phosphoinositides in cell signaling (26) and suggests that favored receptor-lipid associations are likely not restricted to simple mutual charge interaction. It follows that decoration of the inositol headgroup with phosphates at the 3, 4, and 5 positions may alter BRS-lipid binding.
Localization of the entire CD3ϵ chain in the mixed ordered and disordered domains of biological membranes
Finally, we examined the potential for preferential partitioning of the CD3ϵ chain when incorporated into membranes composed of a mix of Lo-Ld membrane domains (Fig. 4). We found that even when simulations were initialized with well-mixed DPPC, cholesterol (Chol), diundecanoyl PC (DUPC), and POPG (4:3:2:1 molar ratio), Lo and Ld domains can spontaneously form (Fig. S8) and can coexist at a longer timescale. Characterization of these domains revealed that the Lo membrane domain is mainly composed of the fully saturated DPPC lipid and Chol, whereas the Ld domain is enriched in the doubly saturated DUPC lipid and the negatively charged POPG lipid. Furthermore, this domain has reduced Chol content. Fig. 4 A illustrates the case where the protein was initially placed in the Lo domain (green). Over a duration of 5–10 μs, the protein localizes in the Lo-Ld boundary region. A close-up of the last frame reveals that the transmembrane helix is enriched in POPG lipids (orange), closely interacting with the BRS stretch. We observed a similar behavior after starting the simulation with the protein colocalized in the Ld domain (data not shown). These results suggest a clear preferential association within the boundary of the two domains regardless of the initial condition. In addition, we considered an entire CD3ϵ chain containing phosphorylated ITAM tyrosines in a membrane mimic composed of a mix of Lo and Ld domains (Fig. S9). We do not observe any difference in the localization of the cytoplasmic chains, specifically the localization of the phosphorylated tyrosines.
Figure 4.
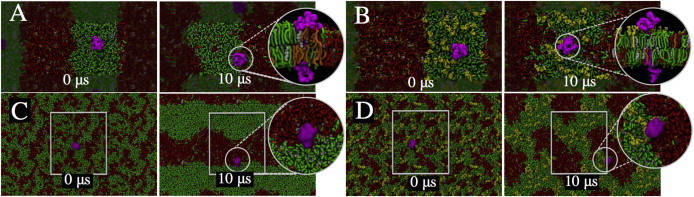
Preferential sorting of the entire CD3ϵ chain in mixed ordered and disordered domains of biological membranes. The whole CD3ϵ chain (magenta), consisting of extracellular, transmembrane, and cytoplasmic domains, was inserted into a preequilibrated Lo-Ld membrane patch composed of DPPC (green), cholesterol (Chol) (white), DUPC (red), and POPG (orange) in a 4:3:2:1 molar ratio. (A) Although CD3ϵ is initially placed in the Lo domain, it localizes in the Lo-Ld boundary domain of the membrane patch at the end of the 10 μs CG simulation (40 μs effective time). (Inset) The protein is highly solvated in the negatively charged POPG lipid (orange). (B) However, interaction with the ganglioside GM1 (light green) prevents diffusion of the protein toward the Ld domain. (C and D) The CD3ϵ protein was also inserted into a randomly distributed lipid membrane patch and simulated for 10 μs. At the end of the simulation, and similar to previous simulations, the protein is seen to reside in the Lo-Ld region (C) or in close contact with a GM1 nanocluster (inset) (D). To see this figure in color, go online.
We next probed the chemical equilibrium in the bilayer of the protein by introducing additional complexity. The fully saturated ganglioside GM1 was added to the external leaflet of the membrane at 1% of the total membrane composition. After placing the protein in the Lo domain (Fig. 4 B), contrary to the previous results, the protein remained mainly associated with Lo lipids (DPPC and GM1). Interestingly, the extracellular domain of the chain makes very specific interactions with the gangliosides that are observed on the timescale of our simulations. A full description of the residue interactions with GM1 is provided in Fig. S10. Although it is unclear whether these associations would be favored when CD3ϵ is complexed with other chains of the TCR complex, it is of interest that the result is the opposite of what was found before adding GM1 to the membrane. It suggests that the extracellular domain association of membrane proteins can improve association within the Lo domain. To avoid any bias resulting from the initial system conditions and the limited domain size, we also carried out different sets of simulations where the protein was inserted in a larger and nonequilibrated membrane with a random lipid mix. As seen in Fig. 4 C, after 10 μs of equilibration the lipids were clearly segregated into independent Lo and Ld domains. Furthermore, and in line with our previous setup, the protein was found within the boundary of the two domains (enclosed in circle). Also similar to previous simulations, the protein was closely associated with a GM1 nanocluster (Fig. 4 D). It is very likely that the timescales of simulations considered in this study are not adequate to observe the CD3ϵ fully embedded in the Lo domain. Taken together, our results show that CD3ϵ, when incorporated as a monomer in a lipid bilayer, preferentially localizes within the boundary between the Lo and Ld domains. Addition of the ganglioside GM1 shifts the protein’s preferential equilibrium toward the Lo domain. The latter, however, may require longer timescales and larger energetic barriers to overcome.
Discussion
A key molecular process in adaptive immunity is the recognition of the MHC-peptide complex by the TCR. T-cells survey the surface of antigen-presenting cells, such as dendritic cells, for engagement with the peptide-bound MHC complex (pMHC). When the receptor on the surface of the T-cell interacts with pMHC, it triggers intracellular signaling that leads to specific effector functions such as cytokine production and T-cell proliferation. A central question about T-cell triggering has been how the TCR-pMHC engagement gets transmitted to the intracellular signaling (ITAM) motifs in the CD3 subunits. Models related to receptor clustering and protein conformational changes have been put forth (27–32). However, the affinity between TCR and pMHC is weak, and the engagement lacks significant conformational changes. Therefore, T-cell triggering may not be a simple process of protein-protein interaction, and it is likely that membrane-mediated mechanisms are involved. Even though numerous models of T-cell triggering have been proposed, only a few of these models have suggested a role for membrane in T-cell activation. Unfortunately, little is known about the initial intracellular activation of TCR-pMHC at the molecular level, and this lack of information has limited the efforts to deduce the mechanistic details.
In this study, we have probed the molecular aspects of the interaction between the membrane and a signaling subunit of the TCR with the intention of shedding some light on the potential role of the membrane in the early events that follow the engagement of TCR-pMHC. Signaling is initiated by phosphorylation of the canonical tyrosines in the ITAM motif in the cytoplasmic tails of the CD3 and ζ subunits. Next, other relevant proteins bind to those phosphorylated sites and propagate signaling (Fig. 5 A). We considered one of those subunits containing the signaling ITAM motif, CD3ϵ. First, we elucidated the molecular aspects of association of the cytoplasmic tail of CD3ϵ with the membrane. Second, we identified specific interactions with membrane components that are responsible for the association. It is likely that the initial activation involves localization into specific domains in the membrane. Past studies showed that lipid-raft regions were likely at the interface between antigen-presenting cells and T-cells (33,34), and TCRs and Lck may also thus localize in this region. Then, the question is, what mechanism drives these molecules to localize in lipid-raft regions, and how does their presence influence intracellular signaling? With such questions in mind, we looked at the localization of CD3ϵ in a biological membrane mimic containing ordered and disordered domains.
Figure 5.
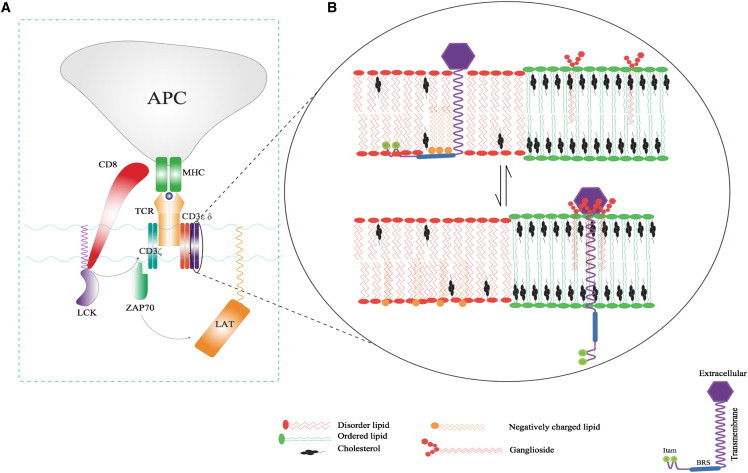
A schematic description of the proposed model for initial activation of the TCR. (A) During formation of the immunological synapsis, an MHC molecule reports an antigenic peptide to the TCR. Intracellular signaling is initiated by phosphorylation of the ITAM motifs by Lck. Subsequent downstream signaling involving LAT is initiated by Zap70, which binds to the CD3ζ chain after phosphorylation of the ITAM motifs. (B) A schematic diagram capturing the proposed mechanism in which the nature of lipid composition drives the initial activation. In the basal state (upper), CD3ϵ is localized in disordered regions (POPG, POPE, POPS, and PI). During this state, the ITAM motifs are buried in the membrane. The immunological synapsis associated with TCR-MHC complex formation causes rearrangement of membrane components, leading to changes in membrane state and formation of lipid-raft-like ordered domains. This change in lipid composition exposes the ITAM motifs for phosphorylation by kinases (lower). As shown in the scheme, we propose this mechanism based on the behavior of the CD3ϵ chain in different membrane states as characterized in this study. To see this figure in color, go online.
First, we elucidated the mechanism for the modes of binding to model membranes at atomistic resolution. Our unbiased and biased simulations suggest that the CD3ϵ tail preferentially binds to negatively charged PG-like lipids. In addition, binding is improved by the presence of the canonical ITAM tyrosines. These tyrosines are embedded in the bilayer upon binding and provide additional stabilization to the bound CD3ϵ tail. In contrast, zwitterionic lipids (e.g., PC lipids) provide a less favorable environment for binding as a result of two cooperative mechanisms: 1) the external shell of local positive charges (choline groups); and 2) the tight lipid packing in more ordered lipids such as DPPC. This tight packing shields the negative phosphate groups and reduces the available space between the headgroups. As a result, the basic residues are not able to reach the surface of the bilayer, and the hydrophobic residues do not interact deeply in the bilayer. However, the substantial adherence to negatively charged lipids helps to overcome the above two unfavorable aspects of binding. Thus, although our study was restricted to certain lipids, our results provide a generic mechanism that may be applicable to other ITAM-containing motifs proximal to cell membranes.
The differential association of CD3ϵ chain to membranes is supported by NMR and fluorescence resonance energy transfer measurements (3,6). Microdialysis-based fluorescence resonance energy transfer experiments performed by Xu et al. (10) showed an approximately eightfold greater binding preference for pure POPG over pure POPC lipid bilayers. Also, Shi et al. (35) demonstrated that when POPG was replaced by POPC lipids, the binding preference decreased by approximately ninefold. AA MD simulations show a similar preference in the electron-density profile in a qualitative manner. More quantitative agreement is seen with both biased and unbiased CG simulations. Thermodynamic calculations from this study suggest that upon binding, the partial detachment of the CD3ϵ chain from negatively charged lipid bilayers involves a high energetic penalty. Considering the different energy profiles for detachment, for the same pulling distance, the CD3ϵ chain exhibits an order-of-magnitude preference for detachment from pure POPC lipid bilayers over POPG bilayers, consistent with the above measurements. The differential affinity for pure lipid bilayers was further confirmed by unbiased CG simulations. We found that association was reduced by 30% for POPC compared to POPG, whereas association was not seen in the case of DPPC lipids. Mass-spectrometry-based measurements reported on the partitioning of the CD3ϵ chain using a molar partition coefficient, K, of 6 × 107 M−1, which is the proportionality factor between the mole fraction of protein bound to a phospholipid vesicle membrane and the molar concentration of protein free in the bulk aqueous phase (36). Thus, the computed constant of 5 × 106 M−1 for affinity to a pure POPG bilayer is in reasonable quantitative agreement with the above value.
Our simulations captured the mechanistic details responsible for the overall stability of the CD3ϵ cytoplasmic tail in PG-like lipids, including the key role played by the basic residues. Interestingly, hydrophobic residues (including the canonical tyrosines in the ITAM) are important to the attachment to the cell membrane. At the molecular level, simulations showed that the secondary structure of the cytoplasmic tail is modulated by the interaction with the lipid bilayer, especially in the case of disordered lipids. The CD3ϵ tail exhibits stretches of α-helices when interacting with negatively charged POPG (Fig. S5, upper) and zwitterioninc POPC (Fig. S5, middle). In contrast, the peptide is more disordered when interacting with the ordered DPPC lipid. Xu et al. (10) reported the nuclear Overhauser effect spectrum of the CD3ϵ chain in close interaction with a POPG/DHPC lipid mixture. Among all the features reported, the averaged structure exhibits a short stretch of α-helices. Our results are also supported by the findings of Duchart et al. (3) in the structure of the TCR ζ chain. In solution, the cytoplasmic tail was unstructured. However, upon binding to lipid micelles, the peptide exhibited changes in the configuration, as is evident by the change in the chemical shift of the peptide. Here, we also observed that the conserved ITAM tyrosines are buried and interacting with the hydrophobic region of a negatively charged membrane, in line with the nuclear Overhauser effect measurements (10).
We find that the entire CD3ϵ chain (containing extracellular, transmembrane, and cytoplasmic domains) discriminates between different lipids present in biological membranes and forms sub-nano-complexes. In our simulations, we observed that the CD3ϵ chain preferentially colocalizes with PI lipids in a biorelevant system resembling an Ld domain, as suggested by DeFord-Watts et al. (2). Through a phospholipid binding assay, it was shown that the BRS stretch was strongly attracted to single- and double-phosphorylated derivatives. In brief, enzyme-linked immunosorbent assay plates were coated with different acidic lipids, and peptides were then added. The specificity toward PI is seen even when it is present at a low molar ratio. In the simulations, the interaction region is spatially located within the BRS sequence and provides a mechanism for stabilization of the protein in Ld domains. Indeed, it explains why mutations or deletion of this region impairs TCR activation and leads to cell death (25). Thus, the CD3ϵ chain shows a degree of specificity for lipid composition, and that may be critical for modulating the activation of the ITAM region. Slight changes in the ratio of different lipid species present in the membrane may serve as a switch during the initial steps of TCR activation. Even though a few studies have shown that detachment of the cytoplasmic tail from the membrane can be triggered by Ca2+ (35), it is later in the sequence than the initiating ITAM phosphorylation (37). Rather, we suggest that effects induced by the subsequent rise in cytoplasmic Ca+2 amplify TCR signaling by maximally exposing the ITAM motif to binding partners.
The entire CD3ϵ chain preferentially localizes in the boundary region between the Lo and Ld domains when incorporated as a monomer. So far, no transmembrane peptides or proteins have been reported to spontaneously partition into the Lo domains in model membranes, irrespective of the amino acid sequence (38–42). CD3ϵ is not an exception. In general, the driving force is the enthalpic penalty associated with the presence of a cylindrical object (the transmembrane region) inside the ordered lipid phase (42). In the case of CD3ϵ, the cost could be even higher, since the negatively charged lipids are localized in the disordered region. Moreover, the presence of PI or any phosphorylated derivatives, which are typically localized in the Ld region, can intensify the preferential partitioning of CD3ϵ in the disordered domain. A key finding from this work is that the preferential localization of the CD3ϵ chain can be modulated by the ganglioside GM1, a marker for Lo regions of membrane. We show that the chemical potential of the CD3ϵ chain is affected by the addition of GM1, which can shuttle the CD3ϵ chain into the Lo domain by means of specific interactions with the extracellular domain. A similar observation has been described in detail for a different helical peptide (43). In our simulations, upon addition of GM1, the CD3ϵ chain was shuttled into close contact with the Lo-Ld boundary region.
Based on our findings, we propose a model for preferential colocalization and modulation of the CD3ϵ chain between the Lo and Ld phases that may contribute to the underlying mechanisms of activation of the ITAM-containing disordered chains in different immune receptors (e.g., T- and B-cell receptors). This model is schematically captured in Fig. 5 B. In the basal state, the tyrosines of CD3ϵ and other TCR ITAM sequences are sequestered from phosphorylation due to interaction with negatively charged lipids. Subsequently, the transduction of the immune signaling and the active synthesis and metabolism of lipids (e.g., PI (26)) might influence the tendency for CD3ϵ to collect at nanoscale Lo-Ld boundaries and alter the degree to which the ITAM stretch is deeply embedded and inaccessible to kinases for phosphorylation and docking. Our proposed mechanism relies on changes in the membrane state after formation of the TCR-MHC complex. Although simulations of the entire multimeric TCR complex are presently not feasible, it is intriguing to speculate that the activation of TCRs and other ITAM-bearing immunoreceptors may lead to reorganization and relocalization of the different lipid components and rearrangement of the membrane domains. It is quite possible that such a change in membrane state can be driven by an increase in the local high concentration of TCRs.
Our proposed mechanism for the initial activation of the TCR and other ITAM-bearing immunoreceptors not only summarizes our computational results but also is consistent with the involvement of Lo domains during immune signaling (33,44–46). We observe the shift in localization of the CD3 chain toward the Lo-Ld interface from the Ld domain within the timescales of simulations. To fully characterize the localization of the CD3 chain in Lo domains, one needs to consider factors such as protein concentration, overall behavior of the TCR oligomer, and spatial aspects of membrane domains, which can influence the chemical potential responsible for localization. In living cells, the differences between the Lo and Ld domains are less pronounced due to a more complex lipid composition and protein concentration (47) hampering the tight packing of the Lo domain. Our simulations show that if it were a monomer, CD3ϵ would interact preferentially with GM1. Since membrane domains are indeed dynamic entities, it is highly probable that localized enrichment of GM1 may lead to increased interaction of the CD3ϵ chain in the TCR with the ordered domain. In fact, a recent study shows that GM1 is upregulated upon TCR activation in vivo (48), which may strengthen the driving force for the relocalization of the immunoreceptor. If activation of the TCR induces association with the Lo domain, as others have proposed (44,45), this would result in exposure of the ITAM motif in the cytoplasmic tail and concomitant phosphorylation of tyrosines.
Conclusions
Here, we suggest a previously unknown molecular mechanism for T-cell activation based on the preferential binding and localization of the intrinsically disordered cytoplasmic signaling tail of CD3ϵ. The binding is easily modified by the presence of different lipid species and by the physical state of the membrane. Moreover, in a biological membrane with Lo and Ld domains, we show that the CD3ϵ tail has specific binding sites, which are more effectively associated with glycolipid derivatives, highlighting their importance in immunoreceptor signaling. CD3ϵ is dually attracted by ordered lipids (GM1) and lipids from the disordered region (POPG). It is possible that such an interaction fractures the line tension, and the membrane responds by changing the distribution between the two domains, as shown in the localization of Ras targeting motifs to domain boundaries (49). The remarkable influence of a single, low-density transmembrane protein on the bilayer suggests that proteins are unappreciated drivers of plasma-membrane functions, likely to influence both membrane dynamics and overall architecture (50). The importance of lipid-protein interactions in controlling signal transduction represents a new frontier. Multiscale computational approaches such as those shown here will be needed to fully understand the molecular mechanisms driving immunoreceptor signaling in the context of biological membranes.
Acknowledgments
We thank Jennifer Macke for editing this article and Jianhui Tian for helping us with membrane simulations during the exploratory stage of this project.
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers P50GM065794 (Spatiotemporal Modeling Center (STMC) at the University of New Mexico) and R37GM035556 (Department of Energy through contract DE-AC52-06NA25396). S.G. and C.A.L. were also supported by STMC subcontracts to the New Mexico Consortium and the Center for Nonlinear Studies at the Los Alamos National Labs, respectively. S.G. was partially supported by an STMC subcontract to the New Mexico Consortium. C.A.L. was supported by the Center for Nonlinear Studies at the Los Alamos National Labs. LANL institutional computing was used by C.A.L. for carrying out MD simulations.
Supporting Material
Supporting Citations
References (51–58) appear in the Supporting Material.
References
- 1.Call M.E., Pyrdol J., Wucherpfennig K.W. Stoichiometry of the T-cell receptor-CD3 complex and key intermediates assembled in the endoplasmic reticulum. EMBO J. 2004;23:2348–2357. doi: 10.1038/sj.emboj.7600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeFord-Watts L.M., Dougall D.S., van Oers N.S. The CD3 ζ subunit contains a phosphoinositide-binding motif that is required for the stable accumulation of TCR-CD3 complex at the immunological synapse. J. Immunol. 2011;186:6839–6847. doi: 10.4049/jimmunol.1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duchardt E., Sigalov A.B., Schwalbe H. Structure induction of the T-cell receptor ζ-chain upon lipid binding investigated by NMR spectroscopy. ChemBioChem. 2007;8:820–827. doi: 10.1002/cbic.200600413. [DOI] [PubMed] [Google Scholar]
- 4.Sigalov A.B., Hendricks G.M. Membrane binding mode of intrinsically disordered cytoplasmic domains of T cell receptor signaling subunits depends on lipid composition. Biochem. Biophys. Res. Commun. 2009;389:388–393. doi: 10.1016/j.bbrc.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reth M. Antigen receptor tail clue. Nature. 1989;338:383–384. doi: 10.1038/338383b0. [DOI] [PubMed] [Google Scholar]
- 6.Hatada M.H., Lu X., Theriault K. Molecular basis for interaction of the protein tyrosine kinase ZAP-70 with the T-cell receptor. Nature. 1995;377:32–38. doi: 10.1038/377032a0. [DOI] [PubMed] [Google Scholar]
- 7.Liu J., Faeder J.R., Camacho C.J. Toward a quantitative theory of intrinsically disordered proteins and their function. Proc. Natl. Acad. Sci. USA. 2009;106:19819–19823. doi: 10.1073/pnas.0907710106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 9.Sigalov A.B. Membrane binding of intrinsically disordered proteins: critical importance of an appropriate membrane model. Self Nonself. 2010;1:129–132. doi: 10.4161/self.1.2.11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu C., Gagnon E., Wucherpfennig K.W. Regulation of T cell receptor activation by dynamic membrane binding of the CD3ϵ cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rzepiela A.J., Schäfer L.V., Marrink S.J. Reconstruction of atomistic details from coarse-grained structures. J. Comput. Chem. 2010;31:1333–1343. doi: 10.1002/jcc.21415. [DOI] [PubMed] [Google Scholar]
- 12.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 13.Pronk S., Páll S., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmid N., Eichenberger A.P., van Gunsteren W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 15.Berendsen H.J.C., Postma J.P.M., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. Springer; New York: 1981. pp. 331–342. [Google Scholar]
- 16.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 17.López C.A., Sovova Z., Marrink S.J. Martini force field parameters for glycolipids. J. Chem. Theory Comput. 2013;9:1694–1708. doi: 10.1021/ct3009655. [DOI] [PubMed] [Google Scholar]
- 18.Marrink S.J., Risselada H.J., de Vries A.H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 19.Yesylevskyy S.O., Schäfer L.V., Marrink S.J. Polarizable water model for the coarse-grained MARTINI force field. PLOS Comput. Biol. 2010;6:e1000810. doi: 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Risselada H.J., Marrink S.J. The molecular face of lipid rafts in model membranes. Proc. Natl. Acad. Sci. USA. 2008;105:17367–17372. doi: 10.1073/pnas.0807527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hakobyan D., Heuer A. Phase separation in a lipid/cholesterol system: comparison of coarse-grained and united-atom simulations. J. Phys. Chem. B. 2013;117:3841–3851. doi: 10.1021/jp312245y. [DOI] [PubMed] [Google Scholar]
- 22.Waheed Q., Tjörnhammar R., Edholm O. Phase transitions in coarse-grained lipid bilayers containing cholesterol by molecular dynamics simulations. Biophys. J. 2012;103:2125–2133. doi: 10.1016/j.bpj.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marrink S.J., Risselada J., Mark A.E. Simulation of gel phase formation and melting in lipid bilayers using a coarse grained model. Chem. Phys. Lipids. 2005;135:223–244. doi: 10.1016/j.chemphyslip.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Shoup D., Szabo A. Role of diffusion in ligand binding to macromolecules and cell-bound receptors. Biophys. J. 1982;40:33–39. doi: 10.1016/S0006-3495(82)84455-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deford-Watts L.M., Tassin T.C., van Oers N.S. The cytoplasmic tail of the T cell receptor CD3ϵ subunit contains a phospholipid-binding motif that regulates T cell functions. J. Immunol. 2009;183:1055–1064. doi: 10.4049/jimmunol.0900404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Meer G., de Kroon A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011;124:5–8. doi: 10.1242/jcs.071233. [DOI] [PubMed] [Google Scholar]
- 27.Bachmann M.F., Ohashi P.S. The role of T-cell receptor dimerization in T-cell activation. Immunol. Today. 1999;20:568–576. doi: 10.1016/s0167-5699(99)01543-1. [DOI] [PubMed] [Google Scholar]
- 28.Germain R.N. T-cell signaling: the importance of receptor clustering. Curr. Bio. 1997;7:R640–R644. doi: 10.1016/s0960-9822(06)00323-x. [DOI] [PubMed] [Google Scholar]
- 29.Reich Z., Boniface J.J., Davis M.M. Ligand-specific oligomerization of T-cell receptor molecules. Nature. 1997;387:617–620. doi: 10.1038/42500. [DOI] [PubMed] [Google Scholar]
- 30.Krogsgaard M., Prado N., Davis M.M. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol. Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- 31.Levin S.E., Weiss A. Twisting tails exposed: the evidence for TCR conformational change. J. Exp. Med. 2005;201:489–492. doi: 10.1084/jem.20050179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gil D., Schrum A.G., Palmer E. T cell receptor engagement by peptide-MHC ligands induces a conformational change in the CD3 complex of thymocytes. J. Exp. Med. 2005;201:517–522. doi: 10.1084/jem.20042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janes P.W., Ley S.C., Magee A.I. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 1999;147:447–461. doi: 10.1083/jcb.147.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pizzo P., Giurisato E., Viola A. Lipid rafts and T cell receptor signaling: a critical re-evaluation. Eur. J. Immunol. 2002;32:3082–3091. doi: 10.1002/1521-4141(200211)32:11<3082::AID-IMMU3082>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 35.Shi X., Bi Y., Xu C. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature. 2013;493:111–115. doi: 10.1038/nature11699. [DOI] [PubMed] [Google Scholar]
- 36.Sigalov A.B., Aivazian D.A., Stern L.J. Lipid-binding activity of intrinsically unstructured cytoplasmic domains of multichain immune recognition receptor signaling subunits. Biochemistry. 2006;45:15731–15739. doi: 10.1021/bi061108f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cronin S.J., Penninger J.M. From T-cell activation signals to signaling control of anti-cancer immunity. Immunol. Rev. 2007;220:151–168. doi: 10.1111/j.1600-065X.2007.00570.x. [DOI] [PubMed] [Google Scholar]
- 38.Fastenberg M.E., Shogomori H., London E. Exclusion of a transmembrane-type peptide from ordered-lipid domains (rafts) detected by fluorescence quenching: extension of quenching analysis to account for the effects of domain size and domain boundaries. Biochemistry. 2003;42:12376–12390. doi: 10.1021/bi034718d. [DOI] [PubMed] [Google Scholar]
- 39.Bacia K., Schuette C.G., Schwille P. SNAREs prefer liquid-disordered over “raft” (liquid-ordered) domains when reconstituted into giant unilamellar vesicles. J. Biol. Chem. 2004;279:37951–37955. doi: 10.1074/jbc.M407020200. [DOI] [PubMed] [Google Scholar]
- 40.Vidal A., McIntosh T.J. Transbilayer peptide sorting between raft and nonraft bilayers: comparisons of detergent extraction and confocal microscopy. Biophys. J. 2005;89:1102–1108. doi: 10.1529/biophysj.105.062380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammond A.T., Heberle F.A., Feigenson G.W. Crosslinking a lipid raft component triggers liquid ordered-liquid disordered phase separation in model plasma membranes. Proc. Natl. Acad. Sci. USA. 2005;102:6320–6325. doi: 10.1073/pnas.0405654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schäfer L.V., de Jong D.H., Marrink S.J. Lipid packing drives the segregation of transmembrane helices into disordered lipid domains in model membranes. Proc. Natl. Acad. Sci. USA. 2011;108:1343–1348. doi: 10.1073/pnas.1009362108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Jong D.H., Lopez C.A., Marrink S.J. Molecular view on protein sorting into liquid-ordered membrane domains mediated by gangliosides and lipid anchors. Faraday Discuss. 2013;161:347–363. doi: 10.1039/c2fd20086d. discussion 419–359. [DOI] [PubMed] [Google Scholar]
- 44.He H.T., Lellouch A., Marguet D. Lipid rafts and the initiation of T cell receptor signaling. Semin. Immunol. 2005;17:23–33. doi: 10.1016/j.smim.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Kabouridis P.S. Lipid rafts in T cell receptor signalling. Mol. Membr. Biol. 2006;23:49–57. doi: 10.1080/09687860500453673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janes P.W., Ley S.C., Kabouridis P.S. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 2000;12:23–34. doi: 10.1006/smim.2000.0204. [DOI] [PubMed] [Google Scholar]
- 47.Hyman A.A., Simons K. Cell biology. Beyond oil and water—phase transitions in cells. Science. 2012;337:1047–1049. doi: 10.1126/science.1223728. [DOI] [PubMed] [Google Scholar]
- 48.Cho J.H., Kim H.O., Sprent J. T cell receptor-dependent regulation of lipid rafts controls naive CD8+ T cell homeostasis. Immunity. 2010;32:214–226. doi: 10.1016/j.immuni.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janosi L., Li Z., Gorfe A.A. Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc. Natl. Acad. Sci. USA. 2012;109:8097–8102. doi: 10.1073/pnas.1200773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lillemeier B.F., Pfeiffer J.R., Davis M.M. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. USA. 2006;103:18992–18997. doi: 10.1073/pnas.0609009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hess B., Henk B., Fraaije J. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 52.van Gunsteren W.F., Berendsen H.J.C. Computer simulation of molecular dynamics: methodology, applications, and perspectives in chemistry. Angew. Chem. Int. Ed. Engl. 1990;29:992–1023. [Google Scholar]
- 53.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 54.Torrie G., Valleau J. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- 55.Hub J.S., De Groot B.L., van der Spoel D. g_wham—a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 2010;6:3713–3720. [Google Scholar]
- 56.Sigalov A.B. The SCHOOL of nature: II. Protein order, disorder and oligomericity in transmembrane signaling. Self Nonself. 2010;1:89–102. doi: 10.4161/self.1.2.11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sigalov A.B. Evolution of immunity: no development without risk. Immunol. Res. 2012;52:176–181. doi: 10.1007/s12026-011-8256-4. [DOI] [PubMed] [Google Scholar]
- 58.Sigalov A.B. Protein intrinsic disorder and oligomericity in cell signaling. Mol. Biosyst. 2010;6:451–461. doi: 10.1039/b916030m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.