

ABSTRACT
Nemaline myopathy (NM) is one of the most common forms of congenital myopathy, and affects either fast myofibers, slow myofibers, or both. However, an animal model for congenital myopathy with fast-myofiber-specific atrophy is not available. Furthermore, mutations in the leiomodin-3 (LMOD3) gene have recently been identified in a group of individuals with NM. However, it is not clear how loss of LMOD3 leads to NM. Here, we report a mouse mutant in which the piggyBac (PB) transposon is inserted into the Lmod3 gene and disrupts its expression. Lmod3PB/PB mice show severe muscle weakness and postnatal growth retardation. Electron microscopy and immunofluorescence studies of the mutant skeletal muscles revealed the presence of nemaline bodies, a hallmark of NM, and disorganized sarcomeric structures. Interestingly, Lmod3 deficiency caused muscle atrophy specific to the fast fibers. Together, our results show that Lmod3 is required in the fast fibers for sarcomere integrity, and this study offers the first NM mouse model with muscle atrophy that is specific to fast fibers. This model could be a valuable resource for interrogating myopathy pathogenesis and developing therapeutics for NM as well as other pathophysiological conditions with preferential atrophy of fast fibers, including cancer cachexia and sarcopenia.
KEY WORDS: Leiomodin-3, Nemaline myopathy, Mouse, Fast-myofiber atrophy
Highlighted Article: A leiomodin-3 mouse mutant generated by insertion of the piggyBac transposon exhibits nemaline myopathy with fast-myofiber-specific atrophy.
INTRODUCTION
Nemaline myopathy (NM) accounts for 17% of cases of congenital myopathy, with an estimated incidence of 1 in 50,000 individuals (Maggi et al., 2013; Wallgren-Pettersson, 1990). The characteristic features of NM include muscle weakness and the presence of rod-like structures (nemaline bodies) in skeletal muscle fibers (North et al., 1997). Based on age of onset and severity of symptoms, NM is clinically defined into six forms: severe congenital, Amish, intermediate congenital, typical congenital, childhood-onset and late-onset (Wallgren-Pettersson and Laing, 2000), although there is considerable overlap among the different forms. Accordingly, the clinical presentation of NM varies from lethality in early childhood to slow progression of milder defects in adults (Ryan et al., 2001). The type of myofiber that shows atrophy in individuals with NM also varies, from fast or slow fibers alone to both. Interestingly, preferential atrophy of fast fibers is also a common feature of other myopathy conditions, including cancer cachexia (Mendell and Engel, 1971) and sarcopenia (Lexell, 1995). However, an animal model for congenital myopathy with fast-myofiber-specific atrophy is not available.
Mutations in ten genes have been identified in individuals with NM (Agrawal et al., 2007; Donner et al., 2002; Gupta et al., 2013; Johnston et al., 2000; Laing et al., 1995; Nowak et al., 1999; Pelin et al., 1999; Ravenscroft et al., 2013; Sambuughin et al., 2010; Yuen et al., 2014). NM is inherited in an autosomal dominant or autosomal recessive manner depending on the mutation. The overall penetrance of NM is unknown, although cases of potential incomplete penetrance and sexual dimorphism have been reported (Agrawal et al., 2004; Nowak et al., 1999). Seven of the NM causative genes encode sarcomere thin filament proteins or regulators of their assembly, which suggests that disorganization of the thin filament could cause NM. Most recently, mutations in the gene encoding leiomodin-3 (LMOD3) on human chromosome 3, a newly identified protein localized to sarcomere thin filaments, have been detected in 21 NM patients from 14 families (Yuen et al., 2014). The mutant LMOD3 alleles are inherited in an autosomal recessive manner. Consistently, knockdown of Lmod3 in zebrafish and Xenopus resulted in disorganization of thin filaments and muscle weakness (Nworu et al., 2015; Yuen et al., 2014). However, a mammalian Lmod3 mutant model will be a valuable resource for interrogating the underlying pathogenesis of NM and for the development of therapeutics.
Here, we describe a mouse mutant in which the homologous Lmod3 gene on mouse chromosome 6 is disrupted by a piggyBac (PB) transposon insertion. The Lmod3PB/PB mutant animals display severe muscle weakness in addition to growth retardation. Furthermore, Lmod3PB/PB muscle fibers display disorganization of sarcomere and the presence of NM bodies. Finally, Lmod3 deficiency causes atrophy specifically in fast myofibers. Together, our study shows that Lmod3-deficient mice display NM and offers the first mouse model for congenital myopathy with fast-myofiber atrophy.
RESULTS
Generation of Lmod3-deficient mice
Using PB-mediated germline mutagenesis, we generated mouse mutants, each of which carries a single PB transposon insertion (Ding et al., 2005) (S.D., X.W. and T.X., unpublished data). In one mutant, PB is inserted into the second intron of the Lmod3 gene (Lmod3PB) (Fig. 1A). Quantitative RT-PCR revealed that Lmod3 mRNA expression is reduced to less than 1% of the wild-type control in homozygous mutants (Fig. 1B). Consistent with this, western blotting showed that Lmod3 protein is undetectable in Lmod3PB/PB mice (Fig. 1C). These results indicate that Lmod3 expression is dramatically downregulated in Lmod3PB/PB mice. However, the expression of Lmod2, the other muscle-specifically expressed member of the Leiomodin gene family, was comparable between Lmod3PB/PB mice and wild-type controls (supplementary material Fig. S1).
RESOURCE IMPACT.
Background
Nemaline myopathy (NM) is one of the most common forms of congenital myopathy, a group of muscle disorders present at birth. The characteristic features of NM include muscle weakness and the presence of rod-like structures (nemaline bodies) in skeletal muscle fibers. The type of myofiber showing atrophy in individuals with NM varies from fast or slow fibers alone to both. However, animal models of congenital myopathy with fast-myofiber-specific atrophy are not available. Recently, mutations in the gene encoding leiomodin-3 (LMOD3) have been detected in individuals with NM. However, the molecular mechanism via which loss of LMOD3 leads to NM is still unclear. Furthermore, preferential atrophy of fast fibers is also a common feature of other myopathy conditions, including cancer cachexia and other aging- or drug-induced myopathies. Currently no therapy is available to treat NM or other forms of fast-fiber atrophy.
Results
In the present study, the authors describe a mouse mutant in which the piggyBac (PB) transposon is inserted into the Lmod3 gene to disrupt its expression. Mutant Lmod3PB/PB mice show severe muscle weakness and postnatal growth retardation. Moreover, the authors discovered disorganized sarcomeric structures and nemaline bodies in the skeletal muscles of the mutant mice. Interestingly, mutant animals exhibit atrophy specifically in fast myofibers, a unique clinical feature shown by only a subgroup of individuals with NM as well as other myopathy-affected individuals.
Implications and future directions
This study shows the first mouse mutant of NM that exhibits fast-myofiber-specific atrophy. The Lmod3PB/PB mouse is thus a unique mammalian model to study disease mechanisms and to dissect how LMOD3 mutations can lead to NM. In addition, this model could prove helpful to develop therapeutics for both congenital and acquired myopathies that are specifically associated with fast-myofiber atrophy.
Fig. 1.

Disruption of Lmod3 expression causes growth retardation in Lmod3PB/PB mice. (A) Schematic representation of the genomic region of the Lmod3 gene and the position of PB insertion. Black box: exon. White box: untranslated region (UTR). Arrows: primers for quantitative RT-PCR. (B) Quantitative RT-PCR analysis of Lmod3 mRNA from TA muscles of 5-week-old mice with indicated genotypes. n=5∼7. (C) Western blotting of 5-week-old mouse TA muscles with antibodies against Lmod3 and β-tubulin (as loading control). (D) Growth curves of male Lmod3+/+ mice (n=5), Lmod3PB/+ mice (n=11) and Lmod3PB/PB mice (n=6). (E) A picture of three 1-week-old male littermates with genotype labeled. ^, # or * indicates P<0.05; ^^, ## or ** indicates P<0.01; ### or *** indicates P<0.001.
Lmod3PB/PB mice show growth retardation and muscle weakness
Lmod3PB/PB mice were born at expected frequency with normal weight (Fig. 1D and supplementary material Fig. S2; data not shown). However, both sexes of Lmod3PB/PB homozygous, but not heterozygous, mice showed growth retardation as early as 1 week of age (Fig. 1D,E and supplementary material Fig. S2). By week 4, Lmod3PB/PB mice weighed 50% less than wild type (Fig. 1D and supplementary material Fig. S2), although all of them survived into adulthood.
Autopsy revealed that Lmod3PB/PB mice are much leaner than wild type (Fig. 2A). Quantitative EchoMRI analysis confirmed that lean mass, but not fat mass, were dramatically decreased in Lmod3PB/PB mice (Fig. 2B). The grip-strength assay showed that forelimb grip strength of Lmod3PB/PB mice was less than 50% of that of Lmod3+/+ controls (Fig. 2C). These results indicate that Lmod3 deficiency results in a skeletal muscle defect.
Fig. 2.
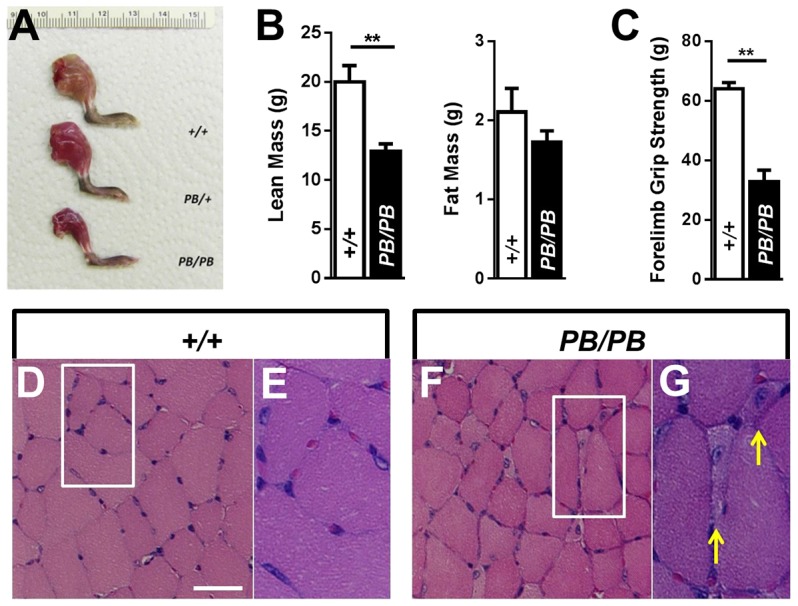
Lmod3PB/PB mice show muscle weakness due to atrophy of myofibers. (A) A picture of unskinned hindlimbs of three 4-week-old male littermates with genotype labeled. (B) Lean mass and fat mass of 5-week-old Lmod3PB/PB males (black bar, n=10) and Lmod3+/+ males (white bar, n=5) measured by EchoMRI. ** indicates P<0.01. (C) Forelimb grip strength of 4-week-old Lmod3PB/PB males (black bar, n=4) and Lmod3+/+ males (white bar, n=5). (D-G) H&E staining of TA muscles from a 5-week-old Lmod3PB/PB male (F,G) and a wild-type male littermate control (D,E). Scale bar: 50 µm. Yellow arrows: atrophic muscle fibers with internalized nuclei.
We further examined the Lmod3PB/PB muscle defect histologically in cross-sections of multiple muscles stained with hematoxylin and eosin (H&E), and observed atrophic myofibers in tibialis anterior (TA), gastrocnemius and quadriceps muscles of Lmod3PB/PB mice, but not controls (Fig. 2D-G and supplementary material Fig. S3A-F). In addition, internalized nuclei were detected in these small myofibers (Fig. 2G and supplementary material Fig. S3B,D,F). However, soleus muscles in Lmod3PB/PB mice appeared normal (supplementary material Fig. S3G,H).
Internalized myonuclei is one of the characteristics of regenerating fibers. However, the atrophic fibers with internal nuclei in the Lmod3PB/PB TA muscles were negative when stained with an embryonic myosin heavy chain antibody, a marker for active regenerating fibers (supplementary material Fig. S4A-E). Furthermore, typical histopathological features of degenerative muscles, including fiber necrosis and cellular infiltration, were not observed in the H&E staining of the Lmod3PB/PB muscles (Fig. 2F and supplementary material Fig. S3). These results suggest that the fibers with internal nuclei in the Lmod3PB/PB muscles are not undergoing degeneration or regeneration.
Disorganized sarcomeric structure and nemaline bodies are present in Lmod3PB/PB muscles
To evaluate the sarcomeric structure in Lmod3PB/PB muscles, we examined the sarcomeres in the longitudinal sections of the TA muscles by simultaneous phalloidin staining for the F-actin and α-actinin antibody staining to label the Z-line. In comparison to the highly regular pattern in the Lmod3PB/+ control (Fig. 3A), the phalloidin labeling in many of Lmod3PB/PB myofibers was narrower and highly disorganized (Fig. 3E). Consistent with the phalloidin staining result, the Z-lines were widened and disorganized in many of Lmod3PB/PB myofibers (Fig. 3F). In total, 62.6±2.2% of the Lmod3PB/PB TA myofibers displayed a disorganized sarcomere structure. Although, in many of the mutant myofibers, the sarcomere structure is too disorganized to measure the length of the thin filaments, we noted that the less disorganized ones are shorter than the control.
Fig. 3.
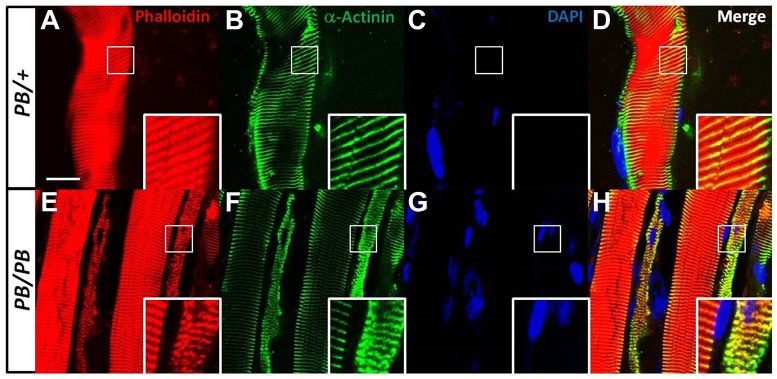
Disorganized sarcomere structure in Lmod3PB/PB muscles. Longitudinal sections of 4-week-old TA muscles from Lmod3PB/PB or Lmod3PB/+ mice stained with phalloidin (A,E), α-actinin (B,F), DAPI (C,G) or merged (D,H). Scale bar: 20 µm. Inlets show magnification of marked regions.
To visualize the nemaline bodies at the light microscopic level, we performed modified Gomori trichrome staining on multiple muscles. Nemaline bodies were present predominantly in the atrophic fibers as small dark dots in TA, quadriceps and gastrocnemius muscle in Lmod3PB/PB mice, but not controls (Fig. 4A,B and supplementary material Fig. S5A-C). Such signals were not found in soleus, a type-I-predominant muscle (supplementary material Fig. S5D).
Fig. 4.
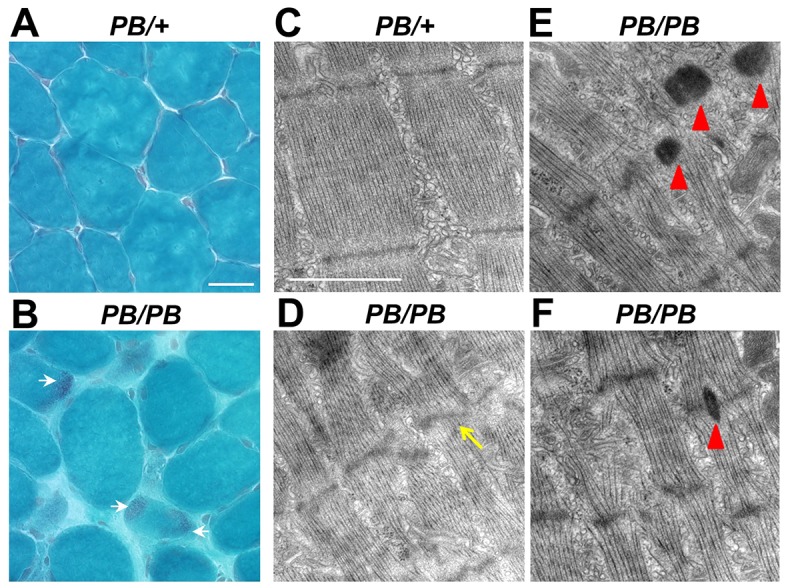
Disorganized Z-line and nemaline bodies in Lmod3PB/PB muscles. (A,B) Modified Gomori trichrome staining on 4-week-old TA muscles from Lmod3PB/+ and Lmod3PB/PB mice. Scale bar: 50 µm. White arrows: nemaline bodies. (C-F) Electron microscopy images of EDL muscles from 6-week-old Lmod3PB/+ (C) and Lmod3PB/PB (D-F) mice. Scale bar: 1 µm. Yellow arrow: Z-line streaming. Red arrowheads: nemaline bodies.
To investigate the ultrastructure of the sarcomere, transmission electron microscopy was performed on extensor digitorum Longus (EDL) and TA muscles (Fig. 4C-F and supplementary material Fig. S6). The Z-lines in Lmod3PB/+ sarcomeres were thin and well-organized (Fig. 4C and supplementary material Fig. S6A), whereas, in Lmod3PB/PB myofibrils, we found many widened Z-lines with local Z-line streaming (Fig. 4D and supplementary material Fig. S6B). More importantly, electron-dense nemaline bodies were present at the location of the Z-line in Lmod3PB/PB myofibrils (Fig. 4E-F and supplementary material Fig. S6C,D).
Disruption of Lmod3 causes atrophy that is specific to fast myofibers
Interestingly, in Lmod3PB/PB mice, only some of the myofibers were significantly smaller in size compared with controls (Fig. 2F,G and supplementary material Fig. S3). To examine whether the group of small myofibers in Lmod3PB/PB muscles belonged to a specific fiber type, the sections were stained with antibodies against different isoforms of myosin heavy chain. The sizes of type-IIb-positive (fast) fibers in Lmod3PB/PB muscles (TA, soleus and quadriceps) were much smaller than the Lmod3PB/+ controls (Fig. 5A-F,S, supplementary material Fig. S7A,B,G and Fig. S8A,B,G). The sizes of type-I-positive (slow) and type-IIa-positive (intermediate) fibers in the mutant type-IIb-predominant muscles (TA and quadriceps) were larger than those in the controls (Fig. 5G-R,T-U and supplementary material Fig. S8H,I). However, in soleus, a type-I-predominant muscle, the sizes of type I fibers in the mutants were similar to those in the controls (supplementary material Fig. S7H) and type IIa fibers were slightly smaller (supplementary material Fig. S7I). Furthermore, there were more type I fibers in type-IIb-predominant muscles in Lmod3PB/PB mice than in the Lmod3PB/+ controls (Fig. 5V and supplementary material Fig. S8J). Together, these data revealed that disruption of Lmod3 results in atrophy specifically in fast myofibers.
Fig. 5.

Disruption of Lmod3 causes atrophy specifically to fast myofibers. (A-R) Cross-sections of 4-week-old TA muscles from Lmod3PB/+ and Lmod3PB/PB mice stained with the antibodies indicated. Scale bar: 200 µm. (S-U) Size distribution of cross-sectional area (CSA) of type-IIb (S), type-I (T) and type-IIa (U) myofibers in TA muscles from 4-week-old Lmod3PB/+ and Lmod3PB/PB mice (n=4). Panels S-U show P<0.001 in Kolmogorov–Smirnov test. (V) Relative ratio between the number of myofibers of a specific fiber type to the number of total myofibers in TA muscles from Lmod3PB/+ and Lmod3PB/PB mice (n=4). *P<0.05.
In summary, Lmod3PB/PB mice recapitulate the muscular phenotypes shown in individuals with NM, including severe muscle weakness and the presence of nemaline bodies. Importantly, the Lmod3-deficient mice exhibit atrophy that is specific to fast myofibers, which makes it a unique animal model for NM and muscle atrophy.
DISCUSSION
Recently, mutations in the LMOD3 gene have been detected in a group of NM patients (Yuen et al., 2014). Subsequently, the expression of the Lmod3 homologous gene was knocked-down in zebrafish and Xenopus by antisense morpholino (MO) (Nworu et al., 2015; Yuen et al., 2014). Both models display sarcomere disorganization and muscle dysfunction, which confirmed that Lmod3 plays an evolutionarily conserved role in muscle biology. In addition, the visibility of embryogenesis makes them useful models for studying Lmod3 function during embryonic myofibrillogenesis. However, neither of the models definitively shows the appearance of nemaline bodies, the hallmark of the NM disease. Furthermore, the relative short lifetime of MO RNA limits their potential for exploring Lmod3 function in adult muscles and in different fiber types. Therefore, a mammalian Lmod3 mutant model will be a valuable resource for interrogating the underlying pathogenesis of NM and developing therapeutics.
Previously, we and others reported that the piggyBac (PB) transposon is able to transpose efficiently in the mouse germline and could be an efficient mutagen for insertional mutagenesis (Ding et al., 2005; Landrette et al., 2011; Ni et al., 2013; Rad et al., 2010). Here, we report a PB is inserted into the second intron of the Lmod3 gene and almost abrogates Lmod3 expression in homozygous mice. Lmod3PB/PB mice exhibit a series of NM-like phenotypes, including severe muscle weakness, muscle fiber atrophy, disorganization of the sarcomere structure and the presence of nemaline bodies, recapitulating the clinical presentations recently reported from the NM patients carrying mutations in the LMOD3 gene (Yuen et al., 2014). Our results also indicate that PB-mediated germline mutagenesis in mice is a powerful genetic approach to discover human disease genes and generate new disease mouse models. During the revision of our manuscript, another Lmod3 mutant mouse model with NM was generated by TALEN mutagenesis (Cenik et al., 2015). The phenotypes reported by Olsen and colleagues are similar to ours except that they also observed atrophy in soleus muscle. One possible explanation is a difference in genetic backgrounds, because the TALEN-induced mutant is in a C57BL/6×C3H mixed background, whereas ours is in a C57BL/6 background.
Several mouse models of NM have been developed either by knocking out (KO) an endogenous gene (e.g. Nebulin-KO, Klhl40-KO, Cfl2-KO) or overexpressing a mutant protein via transgenesis (Tg) or knock-in (KI) [e.g. Tg(ACTA1D286G), KI(ACTA1H40Y), Tg(TPM3M9R)] (Agrawal et al., 2012; Bang et al., 2006; Corbett et al., 2001; Garg et al., 2014; Ravenscroft et al., 2011a,b; Witt et al., 2006). Generally these mice display defects in sarcomere structure, muscle weakness and nemaline bodies, although with various age of onsets and severities. However, characterization of Lmod3PB/PB mice revealed specific atrophy of fast fibers. The phenotype has not been previously reported in other mouse models for NM or other congenital myopathy. In human skeletal muscles, LMOD3 is localized near the pointed end of the thin filaments (Yuen et al., 2014). However, it was observed that its location from the Z-disk is differentially far away in fast myofibers in comparison to slow myofibers, raising the possibility that LMOD3 might play different roles in the two fiber types (Yuen et al., 2014). Alternatively, differential expression of functionally redundant molecules in the fast and slow fibers could be the reason for the Lmod3PB/PB phenotype, because Tmod4 has been shown to be able to replace Lmod3 function in Xenopus (Nworu et al., 2015). Notably, atrophy of fast fibers and hypertrophy of slow fibers are also found in individuals with NM type 6 (NM6) (Olivé et al., 2010). The KBTBD13 gene is mutated in individuals with NM6 and encodes a muscle-specific substrate adaptor of a ubiquitin ligase (Sambuughin et al., 2012). It is not clear whether sarcomere malfunction contributes to NM6 pathogenesis. Given the similar fiber-type-restricted phenotype, it is possible that sarcomere defects resulting from dysregulation of Lmod3 could be involved in the pathogenesis of NM6. Interestingly, preferential atrophy of fast fibers is a common feature of pathophysiological conditions, including cancer cachexia and sarcopenia, as well as glucocorticoid-induce myopathy (Lexell, 1995; Mendell and Engel, 1971; Schakman et al., 2013). Thus, Lmod3PB/PB mice offer a unique mammalian model for studying myopathy mechanisms and for developing therapeutics.
MATERIALS AND METHODS
Animals
The founder Lmod3PB/+ mouse was generated by random germline transposition of PB[Act-RFP], a PB transposon, on the C57BL/6J background (Jackson Laboratories, USA) (Ding et al., 2005). All animal experimental protocols used in this study were reviewed and approved by Yale Institutional Animal Care and Use Committee. The data presented are from male mice unless mentioned otherwise.
Quantitative RT-PCR
Total RNA was isolated from TA muscles from 5-week-old mice by using TRIzol reagent (Invitrogen, USA) and used in iScript reaction (Bio-Rad, USA) to synthesize the cDNA. Quantitative RT-PCR with iTaq Universal SYBR Green Supermix (Bio-Rad, USA) was performed on StepOne system (Applied Biosystems, USA) to determine gene expression using the relative standard-curve method. β-actin was used as an internal control. Primers targeting Lmod3 were: 5′-CAATGTCGCCTACCTTAAACCT-3′ and 5′-TGCTGTTCTAGGTGACTCTGCT-3′. Primers targeting β-actin were: 5′-GGCTGTATTCCCCTCCATCG-3′ and 5′-CCAGTTGGTAACAATG-CCATGT-3′.
Western blotting
TA muscles were dissected and lysed in RIPA buffer with Complete Mini protease inhibitors (Roche, USA) at 4°C. Total protein was quantified by the BCA Assay (Pierce, USA). Protein samples were separated by SDS/PAGE according to standard western blotting procedures and transferred to nitrocellulose membranes (Bio-Rad, USA), followed by blocking with 5% skim milk for 1 h. Blots were incubated with primary antibodies to Lmod3 (1:1000; HPA036034; Sigma, USA), Lmod2 (1:400; AP10364a; Abgent, USA) or β-tubulin (1:4000; T5168; Sigma, USA). Anti-rabbit and anti-mouse IgG antibodies conjugated to horseradish peroxidase were used (1:5000; Jackson ImmunoResearch Laboratories, USA) for protein detection, and signal was visualized using enhanced chemiluminescence (Perkin-Elmer, USA).
Body composition
Lean mass and fat mass were determined by EchoMRI-100 analyzer according to the manufacturer's instruction (EchoMRI LLC, USA).
Grip strength
Forelimb grip strength was measured with a grip-strength meter according to the manufacturer's instruction (Columbus Instruments, USA).
Histochemistry
Cross-sections (10 µm) of isopentane-frozen muscles were stained with H&E or modified Gomori trichrome with standard histochemical techniques (Sheehan and Hrapchak, 1987). Light microscopic images were captured using a Zeiss AxioPhot microscope with an AxioCam 105 color camera.
Electron microscopy
The mice were cardiacally perfused with Karnovsky fixative solution. TA and EDL muscles were fixed in 3% glutaraldehyde in 0.1 M cacodylate buffer pH 7.4 for 60 min, washed 3×10 min in 0.1 M cacodylate buffer, post-fixed with 1% potassium ferrocyanide reduced OsO4 for 3 h on ice, dehydrated through graded methanol, and embedded in EMbed 812 resin. Ultrathin sections (60 nm) were contrasted with uranyl acetate and lead citrate, and viewed in a Hitachi H600 TEM.
Immunofluorescence
Longitudinal or cross-sections (10 µm) of isopentane-frozen muscles were subjected to immunofluorescence assay according to standard methods. Primary antibodies were to: α-actinin (A7811, Sigma-Aldrich, USA), laminin (L9393, Sigma-Aldrich, USA), myosin heavy chain IIb (BF-F3, Developmental Studies Hybridoma Bank, USA), myosin heavy chain I (BA-D5, Developmental Studies Hybridoma Bank, USA), myosin heavy chain IIa (SC-71, Developmental Studies Hybridoma Bank, USA), embryonic myosin heavy chain (F1.652, Developmental Studies Hybridoma Bank, USA). Rhodamine phalloidin (R415, Invitrogen, USA) and Alexa-Fluor-405, -488 or -633 secondary antibodies (Invitrogen, USA) were used for immunofluorescence detection. Images were captured using a Leica TSC SP8 confocal laser scanning microscope with Leica Application Suite Advanced Fluorescence 4.0. Number or size of the muscle myofibers were quantified using the ImageJ program (National Institutes of Health, USA).
Statistics
Data were presented as mean±s.e.m. For statistical analysis, two-tailed unpaired Student's t-tests were used unless otherwise stated. P<0.05 was considered significant. Prism 6 (GraphPad Software, USA) was used for plotting. Error bars represent s.e.m.
Supplementary Material
Acknowledgements
We thank Yanfeng Tan, Yueying Chen and He Tan for technical support, and 973 and 863 working group and Xu lab members for discussion.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
L.T., S.D., L.S. and T.X. designed the project; L.T., S.D., Y.Y., T.-r.L. and Y.L. did the experiments and collected data; L.T., S.D., X.W., L.S. and T.X. analyzed the data; L.T., S.D. and T.X. prepared the manuscript; T.X. and L.S. supervised all aspects of the work.
Funding
This work was supported in part by Chinese Key Projects for Basic Research (973) [2013CB945301, 2013CB945304], Chinese Hi-tech Research and Development Project of China (863) [2012AA022401], and National Institutes of Health [U01HG004073]. T.X. is a Howard Hughes Medical Institute Investigator.
Supplementary material
Supplementary material available online at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.019430/-/DC1
References
- Agrawal P. B., Strickland C. D., Midgett C., Morales A., Newburger D. E., Poulos M. A., Tomczak K. K., Ryan M. M., Iannaccone S. T., Crawford T. O. et al. (2004). Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations. Ann. Neurol. 56, 86-96. 10.1002/ana.20157 [DOI] [PubMed] [Google Scholar]
- Agrawal P. B., Greenleaf R. S., Tomczak K. K., Lehtokari V.-L., Wallgren-Pettersson C., Wallefeld W., Laing N. G., Darras B. T., Maciver S. K., Dormitzer P. R. et al. (2007). Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am. J. Hum. Genet. 80, 162-167. 10.1086/510402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal P. B., Joshi M., Savic T., Chen Z. and Beggs A. H. (2012). Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 21, 2341-2356. 10.1093/hmg/dds053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang M.-L., Li X., Littlefield R., Bremner S., Thor A., Knowlton K. U., Lieber R. L. and Chen J. (2006). Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell Biol. 173, 905-916. 10.1083/jcb.200603119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B. K., Garg A., McAnally J. R., Shelton J. M., Richardson J. A., Bassel-Duby R., Olson E. N. and Liu N. (2015). Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J. Clin. Invest. 125, 1569-1578. 10.1172/JCI80115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett M. A., Robinson C. S., Dunglison G. F., Yang N., Joya J. E., Stewart A. W., Schnell C., Gunning P. W., North K. N. and Hardeman E. C. (2001). A mutation in alpha-tropomyosin(slow) affects muscle strength, maturation and hypertrophy in a mouse model for nemaline myopathy. Hum. Mol. Genet. 10, 317-328. 10.1093/hmg/10.4.317 [DOI] [PubMed] [Google Scholar]
- Ding S., Wu X., Li G., Han M., Zhuang Y. and Xu T. (2005). Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122, 473-483. 10.1016/j.cell.2005.07.013 [DOI] [PubMed] [Google Scholar]
- Donner K., Ollikainen M., Ridanpää M., Christen H.-J., Goebel H. H., de Visser M., Pelin K. and Wallgren-Pettersson C. (2002). Mutations in the beta-tropomyosin (TPM2) gene--a rare cause of nemaline myopathy. Neuromuscul. Disord. 12, 151-158. 10.1016/S0960-8966(01)00252-8 [DOI] [PubMed] [Google Scholar]
- Garg A., O'Rourke J., Long C., Doering J., Ravenscroft G., Bezprozvannaya S., Nelson B. R., Beetz N., Li L., Chen S. et al. (2014). KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Invest. 124, 3529-3539. 10.1172/JCI74994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V. A., Ravenscroft G., Shaheen R., Todd E. J., Swanson L. C., Shiina M., Ogata K., Hsu C., Clarke N. F., Darras B. T. et al. (2013). Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am. J. Hum. Genet. 93, 1108-1117. 10.1016/j.ajhg.2013.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. J., Kelley R. I., Crawford T. O., Morton D. H., Agarwala R., Koch T., Schäffer A. A., Francomano C. A. and Biesecker L. G. (2000). A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am. J. Hum. Genet. 67, 814-821. 10.1086/303089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing N. G., Wilton S. D., Akkari P. A., Dorosz S., Boundy K., Kneebone C., Blumbergs P., White S., Watkins H. and Love D. R. (1995). A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy NEM1. Nat. Genet. 10, 249 10.1038/ng0695-249a [DOI] [PubMed] [Google Scholar]
- Landrette S. F., Cornett J. C., Ni T. K., Bosenberg M. W. and Xu T. (2011). piggyBac transposon somatic mutagenesis with an activated reporter and tracker (PB-SMART) for genetic screens in mice. PLoS ONE 6, e26650 10.1371/journal.pone.0026650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexell J. (1995). Human aging, muscle mass, and fiber type composition. J. Gerontol. A. Biol. Sci. Med. Sci. 50, 11-16. [DOI] [PubMed] [Google Scholar]
- Maggi L., Scoto M., Cirak S., Robb S. A., Klein A., Lillis S., Cullup T., Feng L., Manzur A. Y., Sewry C. A. et al. (2013). Congenital myopathies--clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul. Disord. 23, 195-205. 10.1016/j.nmd.2013.01.004 [DOI] [PubMed] [Google Scholar]
- Mendell J. R. and Engel W. K. (1971). The fine structure of type II muscle fiber atrophy. Neurology 21, 358-365. 10.1212/WNL.21.4.358 [DOI] [PubMed] [Google Scholar]
- Ni T. K., Landrette S. F., Bjornson R. D., Bosenberg M. W. and Xu T. (2013). Low-copy piggyBac transposon mutagenesis in mice identifies genes driving melanoma. Proc. Natl. Acad. Sci. USA 110, E3640-E3649. 10.1073/pnas.1314435110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North K. N., Laing N. G. and Wallgren-Pettersson C. (1997). Nemaline myopathy: current concepts. The ENMC International Consortium and Nemaline Myopathy. J. Med. Genet. 34, 705-713. 10.1136/jmg.34.9.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak K. J., Wattanasirichaigoon D., Goebel H. H., Wilce M., Pelin K., Donner K., Jacob R. L., Hübner C., Oexle K., Anderson J. R. et al. (1999). Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat. Genet. 23, 208-212. 10.1038/13837 [DOI] [PubMed] [Google Scholar]
- Nworu C. U., Kraft R., Schnurr D. C., Gregorio C. C. and Krieg P. A. (2015). Leiomodin 3 and Tropomodulin 4 have overlapping functions during skeletal myofibrillogenesis. J. Cell Sci. 128, 239-250. 10.1242/jcs.152702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivé M., Goldfarb L. G., Lee H.-S., Odgerel Z., Blokhin A., Gonzalez-Mera L., Moreno D., Laing N. G. and Sambuughin N. (2010). Nemaline myopathy type 6: clinical and myopathological features. Muscle Nerve 42, 901-907. 10.1002/mus.21788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelin K., Hilpelä P., Donner K., Sewry C., Akkari P. A., Wilton S. D., Wattanasirichaigoon D., Bang M.-L., Centner T., Hanefeld F. et al. (1999). Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc. Natl. Acad. Sci. USA 96, 2305-2310. 10.1073/pnas.96.5.2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rad R., Rad L., Wang W., Cadinanos J., Vassiliou G., Rice S., Campos L. S., Yusa K., Banerjee R., Li M. A. et al. (2010). PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science 330, 1104-1107. 10.1126/science.1193004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft G., Jackaman C., Sewry C. A., McNamara E., Squire S. E., Potter A. C., Papadimitriou J., Griffiths L. M., Bakker A. J., Davies K. E. et al. (2011a). Actin nemaline myopathy mouse reproduces disease, suggests other actin disease phenotypes and provides cautionary note on muscle transgene expression. PLoS ONE 6, e28699 10.1371/journal.pone.0028699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft G., Jackaman C., Bringans S., Papadimitriou J. M., Griffiths L. M., McNamara E., Bakker A. J., Davies K. E., Laing N. G. and Nowak K. J. (2011b). Mouse models of dominant ACTA1 disease recapitulate human disease and provide insight into therapies. Brain 134, 1101-1115. 10.1093/brain/awr004 [DOI] [PubMed] [Google Scholar]
- Ravenscroft G., Miyatake S., Lehtokari V.-L., Todd E. J., Vornanen P., Yau K. S., Hayashi Y. K., Miyake N., Tsurusaki Y., Doi H. et al. (2013). Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am. J. Hum. Genet. 93, 6-18. 10.1016/j.ajhg.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M. M., Schnell C., Strickland C. D., Shield L. K., Morgan G., Iannaccone S. T., Laing N. G., Beggs A. H. and North K. N. (2001). Nemaline myopathy: a clinical study of 143 cases. Ann. Neurol. 50, 312-320. 10.1002/ana.1080 [DOI] [PubMed] [Google Scholar]
- Sambuughin N., Yau K. S., Olivé M., Duff R. M., Bayarsaikhan M., Lu S., Gonzalez-Mera L., Sivadorai P., Nowak K. J., Ravenscroft G. et al. (2010). Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am. J. Hum. Genet. 87, 842-847. 10.1016/j.ajhg.2010.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambuughin N., Swietnicki W., Techtmann S., Matrosova V., Wallace T., Goldfarb L. and Maynard E. (2012). KBTBD13 interacts with Cullin 3 to form a functional ubiquitin ligase. Biochem. Biophys. Res. Commun. 421, 743-749. 10.1016/j.bbrc.2012.04.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schakman O., Kalista S., Barbé C., Loumaye A. and Thissen J. P. (2013). Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 45, 2163-2172. 10.1016/j.biocel.2013.05.036 [DOI] [PubMed] [Google Scholar]
- Sheehan D. C. and Hrapchak B. B. (1987). Theory and Practice of Histotechnology. Columbus, OH: Battelle Memorial Institute. [Google Scholar]
- Wallgren-Pettersson C. (1990). Congenital Nemaline Myopathy: A Longitudinal Study. Helsinki: University of Helsinki. [Google Scholar]
- Wallgren-Pettersson C. and Laing N. G. (2000). Report of the 70th ENMC International Workshop: nemaline myopathy, 11-13 June 1999, Naarden, The Netherlands. Neuromuscul. Disord. 10, 299-306. 10.1016/S0960-8966(99)00129-7 [DOI] [PubMed] [Google Scholar]
- Witt C. C., Burkart C., Labeit D., McNabb M., Wu Y., Granzier H. and Labeit S. (2006). Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J. 25, 3843-3855. 10.1038/sj.emboj.7601242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen M., Sandaradura S. A., Dowling J. J., Kostyukova A. S., Moroz N., Quinlan K. G., Lehtokari V.-L., Ravenscroft G., Todd E. J., Ceyhan-Birsoy O. et al. (2014). Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Invest. 124, 4693-4708. 10.1172/JCI75199 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.