

Abstract
Transporters of the SLC34 family (NaPi-IIa,b,c) catalyze uptake of inorganic phosphate (Pi) in renal and intestinal epithelia. The transport cycle requires three Na+ ions and one divalent Pi to bind before a conformational change enables translocation, intracellular release of the substrates, and reorientation of the empty carrier. The electrogenic interaction of the first Na+ ion with NaPi-IIa/b at a postulated Na1 site is accompanied by charge displacement, and Na1 occupancy subsequently facilitates binding of a second Na+ ion at Na2. The voltage dependence of cotransport and presteady-state charge displacements (in the absence of a complete transport cycle) are directly related to the molecular architecture of the Na1 site. The fact that Li+ ions substitute for Na+ at Na1, but not at the other sites (Na2 and Na3), provides an additional tool for investigating Na1 site-specific events. We recently proposed a three-dimensional model of human SLC34a1 (NaPi-IIa) including the binding sites Na2, Na3, and Pi based on the crystal structure of the dicarboxylate transporter VcINDY. Here, we propose nine residues in transmembrane helices (TM2, TM3, and TM5) that potentially contribute to Na1. To verify their roles experimentally, we made single alanine substitutions in the human NaPi-IIa isoform and investigated the kinetic properties of the mutants by voltage clamp and 32P uptake. Substitutions at five positions in TM2 and one in TM5 resulted in relatively small changes in the substrate apparent affinities, yet at several of these positions, we observed significant hyperpolarizing shifts in the voltage dependence. Importantly, the ability of Li+ ions to substitute for Na+ ions was increased compared with the wild-type. Based on these findings, we adjusted the regions containing Na1 and Na3, resulting in a refined NaPi-IIa model in which five positions (T200, Q206, D209, N227, and S447) contribute directly to cation coordination at Na1.
Introduction
Inorganic phosphate (Pi) levels in humans are tightly regulated to enable growth and metabolism and to prevent pathologies related to vascular calcification. Na+-coupled Pi transporters of the SLC34 solute carrier family (NaPi-II) are responsible for maintaining whole-body Pi homeostasis and mediate epithelial transport of Pi in intestine and kidney (1–3). Three isoforms catalyze the uphill transport of divalent phosphate (HPO42−) in the presence of a downhill Na+ electrochemical gradient. Electrogenic isoforms (NaPi-IIa and NaPi-IIb) are characterized by a transport stoichiometry (Na+:HPO42−) of 3:1 and show voltage-dependent cotransport rates, with one net positive charge translocated per cycle. In contrast, the stoichiometry of the electroneutral isoform (NaPi-IIc) is 2:1, its transport kinetics show no voltage dependence, and no net charge is translocated. This functional difference is postulated to arise because NaPi-IIc does not release the first-bound Na+ ion to the intracellular medium (4).
The interaction of one Na+ ion with the empty carrier is thought to be the critical step in initiating the transport cycle for both electrogenic and electroneutral isoforms (4,5). This leads to the cooperative binding of a second Na+ ion, followed by HPO42− and a third Na+ ion before translocation occurs (Fig. 1). For the electrogenic NaPi-IIa/b transporters, the voltage dependence of the overall transport kinetics can be satisfactorily explained by assuming that only the empty carrier reorientation (partial reaction 0↔1; Fig. 1), and the cation-carrier interactions between the first Na+ ion and the Na1 site are voltage dependent (partial reactions 1↔2, 0↔6; Fig. 1). By implication, these partial reactions must therefore involve charge displacements that are sensitive to the transmembrane (TM) electric field. Indeed, when rapid changes in membrane potential are applied to a membrane containing a large population of NaPi-IIa/b, charge movements manifesting as so-called presteady-state relaxations are readily detected. In the absence of external cations, these presteady-state relaxations, which are analogous to the gating charges observed in ion channel electrophysiology, arise from charges intrinsic to the protein, and additional charge is detected when cations are present (5).
Figure 1.
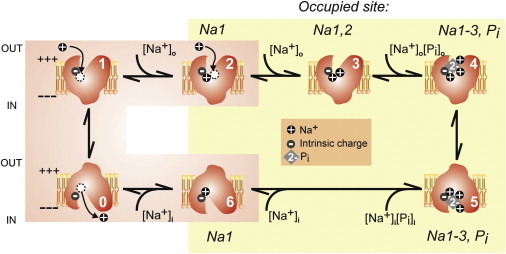
Kinetic scheme for the electrogenic NaPi-IIa/b isoforms. Schematics represent the conformational states identified by previous functional analyses to show the occupancy of the proposed substrate-binding sites. The physiologically relevant cotransport cycle (clockwise, starting from state 0) involves a voltage-dependent reorientation of the unloaded carrier (state 0) to state 1 (assuming a hyperpolarized membrane potential), and movement of a single Na+ ion to the proposed Na1 site (state 2), followed by the cooperative interaction of a second Na+ ion before divalent Pi and a final Na+ ion, resulting in occupancy of the Na2-Pi-Na3 sites (state 4). A major translocation event then occurs (transition 4–5) and substrates can be released to the cytosol (transition 5–6). The last Na+ unbinding event (transition 6–0) effectively frees the empty carrier intrinsic charge to allow reorientation of the empty carrier to state 1 under the influence of a hyperpolarizing TM potential, in readiness for the next cycle. For the electroneutral NaPi-IIc, the empty carrier reorientation is voltage independent and no detectable charge displacement is associated with any of the partial reactions.
For the electroneutral NaPi-IIc transporter, the order of substrate interactions remains the same, with two Na+ ions proposed to interact before Pi. However, it is thought that the first Na+ ion is not translocated and thus does not contribute to the transport stoichiometry. Moreover, unlike NaPi-IIa/b, the binding of the first Na+ ion to NaPi-IIc is not electrogenic, meaning that it does not lead to detectable presteady-state relaxations, yet a cooperative interaction of two Na+ ions still occurs (4). However, if a charged aspartic acid residue that is found in all electrogenic isoforms (D224 in hNaPi-IIa) is replaced by a glycine at the equivalent site in the electroneutral NaPi-IIc, the Pi cotransport becomes electroneutral (6). The converse substitution in NaPi-IIc restores electrogenic behavior, including presteady-state relaxations (7). This behavior implicates D224 in conferring voltage dependence to the transport cycle and potentially coordinating a Na+ ion at Na1. Moreover, kinetic studies of the electroneutral construct with restored electrogenicity (AAD-IIc) (8) established that whereas cation interactions originating from the external medium remained electroneutral, electrogenicity appeared to arise from the empty carrier and from cytosolic Na+ ion interactions (partial reactions 6↔0 and 0↔1; Fig. 1). Thus, D224 appears to be intimately associated with occupancy of Na1 from either side of the membrane.
Taking into account the highly conserved inverted repeat symmetry of all NaPi-II isoforms, we recently identified similar repeats in the x-ray crystallographic structure of a Na+-dicarboxylate transporter, VcINDY (9), which allowed us to generate a homology model of the human NaPi-IIa protein based on VcINDY in an outward-facing conformation (10). Only one Na+ ion site was identified in the 3.2 Å VcINDY crystal structure, and a second ion position was proposed based on pseudo-symmetry considerations (9), although, like NaPi-IIa/b, transport mediated by VcINDY is electrogenic, and the Na+/citrate stoichiometry is most likely 3:1 (11). Our structural model predicts sites for Pi and two Na+ ions similar to those in VcINDY, and when these are occupied, the model therefore corresponds to state 4 in the NaPi-IIa transport cycle without considering Na1 occupancy (Fig. 1). Importantly, mutations at the proposed Na2, Na3, and Pi sites abolished cotransport function, and the observation of Na+-dependent, presteady-state relaxations suggested that the hypothesized Na1 interaction still occurred (10).
Here, we investigate the Na1-binding site, whose position in the published structural model was not previously considered (10). We focus on eight residues close to D224, which, as mentioned above, has been identified as a critical determinant of electrogenicity and transport stoichiometry (6–8,12). We noted that in the model, D224 is located at the cytoplasmic end of TM3, i.e., deeper in the protein than the previously identified Na2-Pi-Na3 substrate site. This deep position would be consistent with a site that could allow cation interactions also from the cytosol and would be similar to the position of the Na1 site in the Na+-dependent aspartate transporter GltPh (13). Although it is not conclusively involved, this region appeared to be a reasonable starting point for identifying Na1-binding-site residues. Site-directed mutagenesis and functional analysis, including pre-steady state kinetics and Li+ substitution experiments, confirmed the functional relevance of four of the eight proposed amino acids in contributing to the Na1 site in NaPi-II transporters. Refinements to the model based on these data indicate how they may be arranged to form the site.
Materials and Methods
Molecular biology
Mutations causing single amino acid changes were introduced into human NaPi-IIa cDNA cloned into an oocyte expression vector using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies). Downscaled reactions were run according to the supplier’s instructions and primers were purchased from IDT Biotech (Integrated DNA Technologies). All mutations were confirmed by Sanger sequencing (Genevision, Newcastle, UK). The plasmids were linearized using XbaI, and RNA was transcribed using T3 RNA polymerase and the Message Machine kit (Ambion/ Life Technologies). The RNA was assessed by agarose gel electrophoresis and quantified by UV spectroscopy.
Oocytes
Oocytes were harvested from Xenopus laevis frogs (Xenopus Express France, Vernassal, France) following standard procedures according to Swiss Cantonal and Federal legislation. Defolliculated cells were incubated in modified Barth’s solution containing (in mM) 88 NaCl, 1 KCl, 0.41 CaCl2, 0.82 MgSO4, 2.5 NaHCO3, 2 Ca(NO3)2, and 7.5 HEPES, adjusted to pH 7.5 with TRIS, and supplemented with antibiotics (doxycycline and gentamicin, 5 mg/l). Oocytes were injected with 50 nl of RNA (0.2 μg/μl) and kept in modified Barth’s solution at 18°C for 3–5 days before analysis.
Solutions
The superfusing solutions contained (in mM) 100Na (100 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4 adjusted with TRIS) or 100Ch (the same as for the 100 Na solution but with equimolar replacement of NaCl with choline Cl). For intermediate Na+ concentrations, 100Na and 100Ch were appropriately mixed to maintain the same osmolarity. For the lithium-containing solutions (100Li and 50Na50Li), NaCl was replaced equimolarly with LiCl to give the required concentration. Inorganic phosphate (Pi) was added to the superfusate from 1 M K2HPO4 and KH2PO4 stocks that were mixed to give pH 7.4, and further diluted in H2O to give the required test concentration. All standard reagents were obtained from either Sigma-Aldrich or Fluka (Buchs, Switzerland).
Functional assays
Radioisotope uptake assays were performed according to standard procedures as described elsewhere (14) using 100Na solution and 1 mM cold Pi to which 32Pi (specific activity 10 mCi/mmol Pi) was added. Standard two-electrode voltage-clamp (TEVC) hardware was used for electrophysiology (Turbo TEC-10CX; NPI, Tamm, Germany) and controlled using pClamp 10 software (Molecular Devices, Sunnyvale, CA). Clampfit (Molecular Devices, Sunnyvale, CA) and Prism version 3.0, 5.0 (Graphpad Software, San Diego, CA) were used for postacquisition analysis. The protocol for determining the steady-state Pi activation used voltage steps from the standard holding potential (Vh = −60 mV) to voltages in the range of −160 mV to +40 mV. Presteady-state current relaxations were typically quantified for voltages in the range of −180 mV ≤ V ≤ 80 mV with four signal averages. Current relaxations were typically fitted with a multiple exponential function. The fastest component represented the linear capacitive charging of the oocyte membrane (typically 0.35−0.6 ms, depending on the oocyte). The slower component (typically >3 ms) was extrapolated to the peak of the capacitive transient, at which time we assumed the membrane was >90% charged, and numerically integrated to obtain the charge moved (Q) for a step from the holding potential to the test potential. The charge-voltage (Q-V) data were fitted with a Boltzmann function of the form given by Eq. 1:
| (1) |
where V0.5 is the voltage at which the charge is distributed equally between two hypothetical states, z is the apparent valency of an equivalent charge that moves through the whole of the membrane field, Qmax is the total mobile charge available, Qhyp is the charge of the hyperpolarizing limit and is a function of Vh, and e, k, and T have their usual meanings.
Western blotting and immunocytochemistry
Oocytes were fixed in 3% paraformaldehyde and sections were blocked in 1% BSA/PBS for 15 min at room temperature. NaPi-IIa antibody raised against a COOH-terminal peptide was diluted 1/400 in 0.02% Na-azide/PBS and slides were incubated overnight at 4°C. Sections were washed twice with hypertonic PBS (PBS with additional 18 g NaCl/L) and once in PBS for 5 min. Sections were incubated with secondary donkey anti-rabbit Alexa-488 antibody (Invitrogen) for 1 h, washed with PBS, and mounted with Glycergel (DAKO). Fluorescence was detected using a fluorescence microscope (Leica CTR600).
Bioinformatics
Sequences of NaPi-II transporters were identified from the NCBI nr20 database (dated February 22, 2013) using HHblits (15). Sequence conservation among the top 50 nonredundant sequences was analyzed using the Weblogo v3 webserver (16).
Structural modeling
In our previous work, we built a homology model of human NaPi-IIa (10) using a structure of VcINDY (9) (PDB ID: 4F35) as a template. In that work, we identified VcINDY as a possible template, albeit with a low confidence, using the HHpred server. We obtained further evidence of their fold similarity by comparing the individual repeats of VcINDY and NaPi-IIa using HHalign and AlignMe (either in PST or PS mode), indicating similarities with the region of the binding site of VcINDY, which was aligned to the conserved QSSS motif of NaPi-IIa (10). We obtained the same result when we aligned the family-averaged hydropathy profiles of the individual repeats using AlignMe (10). Here, the published homology model of human NaPi-IIa was refined in two stages, involving first TM2b and then TM5-6. In each stage, the alignment between NaPi-II and VcINDY was adjusted in regions with low scores according to ProQM analysis (see below) and/or to be consistent with the experimental evidence. In addition, helical constraints were introduced according to secondary structure predictions (10) for residues 202–205 (TM2b), 217–223 (TM3), 440–444, 448–452 (TM5a), 455–456, 461–462 (TM5b), and 493–505 (TM6).
The human NaPi-IIa model containing residues 97–249 and 346–505 (excluding the long extracellular loop, TM7, TM8, and both cytoplasmic terminal domains) was built using Modeller v9.12 (17), alignments were manipulated using Jalview (18), and models were assessed using Procheck (19) as a measure of the chemical fidelity of bonds and angles. ProQM (20) was used to quantify the agreement of the structural model with sequence conservation, secondary structure predictions, and TM predictions for individual residues, the entire model, and the template.
First, to position the side chains of residues Q206, D209, R210, and N227 in or close to the Na1-binding site according to the experimental data, we progressively adjusted the alignment of residues 202–214 in TM2b relative to the VcINDY template by one position at a time. We monitored agreement with the experiments by using the distance between the Cα atom of N227 and the Cα atom of Q206, D209, or R210, as well as the angle between the Cα atom of N227 and the Cα−Cδ bond of Q206, the Cα−Cγ bond of D209, or the Cα−Cζ bond of R210. A shift of five residues relative to the initial model resulted in the optimal arrangement of these residues to form the putative Na1 site (Table S1 in the Supporting Material).
Second, we adjusted the alignment of TM5 and TM6 progressively until we obtained a configuration that optimally positioned S447 proximal to and oriented toward Q206, and at the same time positioned T451 close to S417 near the Na2-Pi-Na3 site (Table S2). We monitored this adjustment using two Cα−Cα distances (i.e., between S417 and T451, and between S447 and Q206) and two angles (i.e., between the Cα atom of S419 and the Cα−Cδ bond of T451, and between the Cα atom of S447 and the Cα−Cγ bond of Q206).
In the refined alignment, the percentage of identical residues between the template and model was ∼14% in repeat unit 1 (RU1) and ∼8% in repeat unit 2 (RU2). The expected accuracy of a homology model with this level of sequence identity to the template is 1.5–4 Å in the Cα positions, assuming the correct alignment (21). In a final modeling stage, three Na+ ions and a Pi substrate were included. Harmonic upper-bound distance restraints of 3.3 Å were imposed between the Na+ ion at Na1 and side-chain O atoms of residues T200, Q206, D209, N227, and S447; the Na+ ion at Na2 and side-chain O atoms of residues S164, T195, S196, N199, and substrate Pi; the Na+ ion at Na3 and side-chain O atoms of residues Q417, S418, S419, T451, and T454; and the O atoms of Pi and side-chain O atom of residue S164, or side-chain N atom of N199. We selected the model with the lowest Modeller probability distribution function (Molpdf) score out of a set of 2000 models, i.e., the model that was in best agreement with all of the input constraints. In this model, only 0.7% of the residues are in disallowed regions (I142 and L143), 1.8% are in generously allowed regions of the Ramachandran plot, and all of these residues are located in loop regions away from the binding sites and helix packing interfaces. The total ProQM score increased from 0.555 in the published model (10) to 0.572 after the refinements in TM2 and TM5-6. ProQM scores can range from 0 to 1, with 1 being the most membrane-protein like, although typical structures have ProQM scores of ∼0.7 and the truncated template structure has a ProQM score of 0.643. The refined model is provided in the Supporting Material as Model S1.
Results
A putative Na1-binding region close to D224 in TM3
The universe of possible binding sites for Na+ in NaPi-IIa is large because the core of the protein is littered with serine, threonine, asparagine, and glutamine residues that are good candidates for cation coordination (22). Therefore, to identify Na1, we narrowed down the options by focusing on the region near D224 (see above) (6–8,12). Additional support for the importance of this region comes from the reported role of a position one turn away on TM3, namely, N227, mutations of which led to impaired substrate affinities that could be attributable to altered Na1 interactions (6) (Fig. 2).
Figure 2.
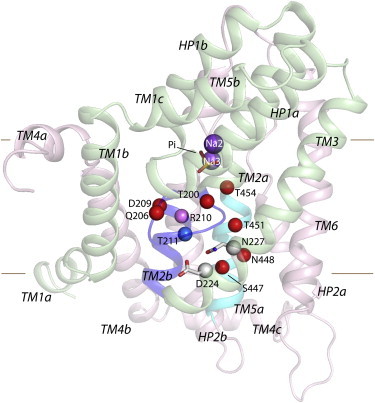
Candidate Na1-binding-site residues in a published structural model of human NaPi-IIa based on VcINDY. The model included two Na+ ions (purple spheres) and one Pi anion (yellow and red sticks), and was generated using the alignment described previously (10). The protein backbone is represented as cartoon helices with RU1 colored green and RU2 in pink, and is viewed from along the plane of the membrane from the face furthermost from the dimer interface in the template. TM2b and TM5a are colored blue and cyan, respectively. Candidate Na1-binding-site residues are represented as spheres at their Cα positions. Residues whose mutation had an effect on transport activity, apparent affinities for substrate or ions, or steady-state currents are colored red or pink. The residue whose mutation did not alter any of these parameters (T211) is colored blue. Residues D224 and N227 are colored gray, and their side chain and Cα atoms are represented as sticks and spheres, respectively.
The most reliable aspects of the published NaPi-IIa model are its overall fold, the positioning of the residues in TM3, and the regions around the Na2, Na3, and Pi sites, especially the helical hairpins (HP1 and HP2), all of which agree well with various experimental data (10). However, because of the low sequence identity between NaPi-IIa and its template, VcINDY (∼10%), other predicted regions that to date have not been validated in detail experimentally, such as the more peripheral helices, may be less reliable. Relatively minor adjustments to the underlying sequence alignment between NaPi-IIa and VcINDY could shift the orientation and vertical positioning of specific residues in the model while still maintaining the overall fold. Therefore, we broadened our search for putative Na1-site residues to any position along the cytoplasmic halves of the two helices that are adjacent to TM3 in the published model, i.e., TM2b and TM5a (10) (Fig. 2). This strategy identified nine hydroxylic, carboxylic, or amide-containing residues that could potentially contribute to Na1, namely, T200, Q206, D209, and T211 in TM2; N227 in TM3; and S447, N448, T451, and T454 in TM5 (Fig. 2). We then individually mutated these residues to alanine or to less severe substitutions (Q206N, D209E, and N227Q), to examine their impact on transport function (Table 1). Furthermore, we also included R210 from TM2 in our mutagenesis study because as a positively charged residue in a putative cation-binding region, it may play a critical functional role, as suggested by previous work (10).
Table 1.
Overview of human NaPi-IIa constructs
| Transmembrane Segment | Construct |
Expression |
Transport Function |
Kinetics |
|||
|---|---|---|---|---|---|---|---|
| WB |
IC |
32Pi |
TEVC |
Steady State |
Pre-Steady State |
||
| WT | + | + | + | 1.0 | + | + | |
| TM2 | T200A | + | + | + | 0.14 ± 0.02 | + | − |
| Q206A | ND | ND | ND | 0.35 ± 0.05 | + | + | |
| Q206N | ND | ND | ND | 0.79 ± 0.09 | + | + | |
| D209A | ND | ND | ND | 0.35 ± 0.05 | + | + | |
| D209E | ND | ND | ND | 1.11 ± 0.14 | + | + | |
| R210A | ND | ND | + | 0.37 ± 0.04 | + | + | |
| T211A | ND | ND | ND | 1.14 ± 0.01 | + | + | |
| TM3 | N227A | + | + | ND | ND | − | ND |
| N227Qa | + | ND | + | + | + | ND | |
| TM5 | S447A | + | + | + | 0.58 ± 0.13 | + | + |
| N448A | + | + | − | − | − | − | |
| T451A | + | + | − | − | − | + | |
| T454A | + | + | − | − | − | + | |
For TEVC transport function, values indicate mean ± SE fraction of Pi-induced current at −60 mV relative to WT for n >5 cells per construct. WB, western blot (total oocyte lysate); IC, immunocytochemistry; +, experimental verification; −, undetected/unresolved; ND, not done.
See Virkki et al. (6).
Functional characterization: steady-state response to Pi
We expressed the mutated transporters in Xenopus laevis oocytes and first screened them for electrogenic function using a TEVC. Seven constructs with mutations in TM2 and one in TM5 (Table 1) gave detectable responses to test applications of 1 mM Pi (in the presence of 100 mM Na+ (100Na solution)) in the form of an inwardly directed change in the holding current (typically with absolute Pi-induced currents in the range of −20 nA to −150 nA) when voltage clamped at −50 mV. The response to 1 mM Pi (at −60 mV) relative to the wild-type (WT) response varied among constructs (Table 1) and the largest deviation was documented for T200A, which displayed only 14% of the WT response. These changes in the magnitude of the response could result from reduced surface expression, altered kinetics, or both. In the case of T200A, the membrane staining was comparable to that observed for the other constructs, suggesting that membrane expression was not compromised (Fig. S1). Oocytes injected with the cRNA coding for N227A (TM3) developed leaky membranes 2–3 days after injection, which precluded a detailed functional analysis. Three mutants with substitutions in TM5 (N448A, T451A, and T454A) showed no Pi-induced inward currents and were further assayed for 32P uptake (to test for the possibility of electroneutral transport) and subjected to presteady-state assays (to detect charge displacements associated with the empty carrier and cation movements), western blotting, and immunocytochemistry (Table 1). For these constructs, immunocytochemistry confirmed that in each case the protein was expressed in the membrane (Fig. S1), although the uptake data for these mutants were indistinguishable from those obtained for noninjected control oocytes (Fig. S2).
We further characterized the functional mutants by measuring the Pi-dependent change in holding current (IPi) as a function of membrane potential under four different superfusing conditions (100Na + 1 mM Pi, 100Na + 0.1 mM Pi, 50Na + 1 mM Pi, and 50Na50Li + 1 mM Pi) and compared their behavior with that of the WT (Figs. 3 and 4). Mutations affecting the architecture of Na1 were expected to have significant consequences for the current-voltage (I-V) relation and/or the sensitivity to Li+, and the superfusion conditions were chosen to best highlight deviations in these properties from WT behavior. Data were normalized to the response to 1 mM Pi in 100Na at −100 mV for each construct to aid comparison. For the WT, IPi with 100Na + 0.1 mM Pi, 50 Na+1 mM Pi, and 50Na50Li + 1 mM Pi was ∼50% of the response in 100Na + 1 mM Pi at all membrane potentials in the test range (−160 mV to +40 mV; Fig. 3, upper left) and similar behavior was found for mutant T211A (Fig. 3, lower center), which suggested that this position is not a critical determinant of the kinetic profile.
Figure 3.
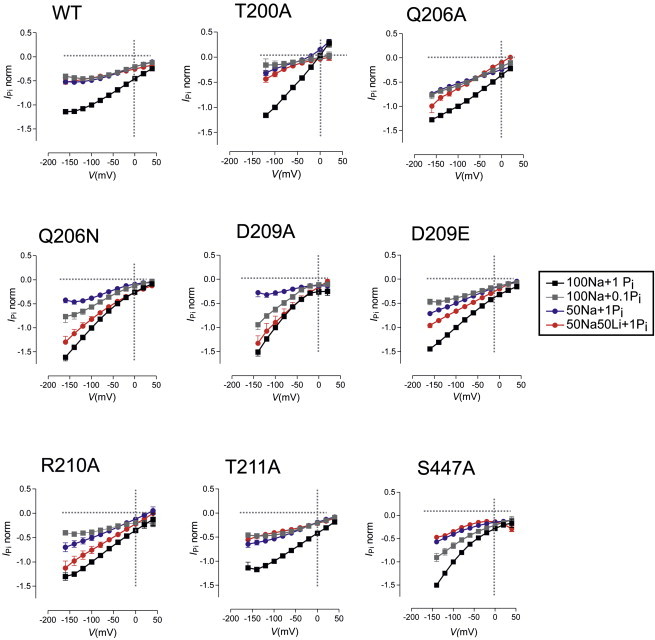
Current-voltage (I-V) data for the WT and functional mutants with substitutions at sites in TM2. Each panel shows the normalized Pi-dependent current (IPi) under four superfusion conditions: 100Na + 1 mM Pi (black squares); 100Na + 0.1 mM Pi (gray squares), 50Na + 1 mM Pi (blue circles), and 50Na50Li + 1 mM Pi (red circles). Data were pooled from more than three batches of oocytes and each point represents the mean ± SE of n > 10 oocytes. Data for each oocyte were normalized to the control response to 1 mM Pi in 100Na at −100 mV. Data points are joined for graphical clarity.
Figure 4.
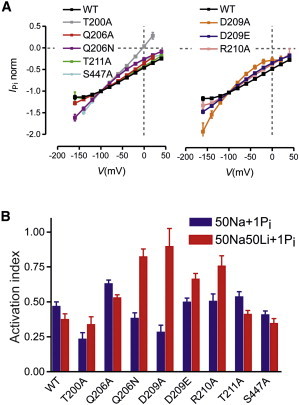
Steady-state properties of TM2 mutants. (A) Normalized I-V data for WT and mutants. Each data set is the IPi for 1 mM Pi in 100 Na normalized to the response at −100 mV. (Left) WT and mutants involving substitution of polar residues. (Right) WT and mutants involving substitution of acidic or basic residues. Data points have been joined for graphical clarity. (B) The effect of cation replacement at −100 mV is depicted as the ratio of IPi with 1 mM Pi superfusion of either 50Na + 1 mM Pi (blue bars) or 50Na50Li + 1 mM Pi to the control response in 100 Na+ 1 Pi (red bars). Each bar shows the mean ratio ± SE for n > 5 oocytes expressing the indicated construct.
In contrast, all other functional mutants showed deviations from the WT profile (i.e., altered voltage dependence and relative changes in IPi for the different superfusion conditions), and these deviations were unique for each mutation position. Moreover, the form of the changed I-V profiles depended on the type of substitution, as can be readily seen by comparing Q206A with Q206N, or D209A with D209E. Whereas the alanine substitution at Q206 resulted in a marginal change in the response to 100Na + 1 mM Pi, the more conservative Q→N substitution significantly altered the voltage dependence, which became more curvilinear, and concomitantly increased the response when Li+ ions were substituted for 50% of the external Na+. In contrast, the alanine substitution of D209 caused comparable changes to Q206N, whereas the conservative D→E substitution at 209 had a less dramatic effect on the electrogenic profile. Interestingly, like D209A and Q206N in TM2, the only mutant in TM5 (S447A) that showed cotransport (IPi; Fig. 3) and 32P uptake (Fig. S2) also showed a curvilinear relationship in response to 1 mM Pi. However, for the other superfusing conditions, especially 50Na50Li, its response pattern was dissimilar to that of D209A and Q206N.
These qualitative comparisons between the WT and mutant I-V profiles revealed that the mutagenesis had resulted in both common changes to their profiles (voltage dependence) and unique differences that we might expect given their unique structural disposition. In particular, they provided the first indication that the introduced changes at 206, 209, and 447 had possibly affected Na1. To analyze these electrogenic profiles in more detail and gain insight into their role in the Na1 interactions, we next focused on specific features of voltage dependence, apparent substrate affinities, and cation substitution by extracting relevant data from the I-V profiles.
Steady-state voltage dependence
Superposition of the I-V data, normalized to −100 mV in response to 100Na + 1 mM Pi (i.e., close to saturation for both substrates for the WT (23)), revealed that substitutions in TM2 (206, 209, and 210) and TM5 (447) caused variable hyperpolarizing shifts relative to the WT data (Fig. 4 A). These data are depicted separately for mutants in which either a polar residue (left panel) or charged residue (right panel) was replaced, and the WT data are shown in each case for comparison. The normalized I-V data for T211A were similar to those for the WT, indicating that this site was not critical for defining the steady-state voltage dependence, as already noted. Mutant T200A showed a significant outward component of current at depolarizing potentials, whereas the I-V relations for the other constructs showed only inward currents over the range of potentials tested. Replacement of external Cl− by MES reduced this outward component, which suggested that it was in part due to an inward Cl− flux in T200A similar to that reported previously, which may arise from a response of endogenous channels to Pi (24). The hyperpolarizing shifts were most pronounced for D209A and weaker for Q206N, D209E, R210A, and S447A. Given that the voltage dependence of NaPi-IIa arises from the partial reactions associated with the empty carrier and the first Na+ ion interaction, these data suggested that the mutagenesis at 206, 209, 210, and 447 had altered the kinetics of one or more partial reactions that link states 6, 0, 1, 2, and 3 (Fig. 1). However, from these data alone we were unable to discriminate between changes to the empty carrier kinetics and/or Na+ interactions.
Apparent affinities for Pi and Na+ at −100 mV
We applied a two-point screening assay to determine whether a mutation had significantly altered the apparent affinity for Pi (K0.5Pi), by defining a Pi activation index (see Supporting Results, Fig. S3, and Table S3) as the ratio of IPi induced by 0.1 mM Pi to that induced by 1 mM Pi, always in the presence of 100 mM Na (100Na solution; Fig. S3 C) (25). For the WT voltage clamped at −100 mV, this ratio lies typically in the range of 0.4–0.5, consistent with the reported K0.5Pi of ∼0.1 mM (6,23). An increase or decrease in this ratio compared with the WT value would indicate that in the presence of 100 mM Na+, K0.5Pi had decreased or increased, respectively. Similarly, we defined a Na+ activation index as the ratio of the response to 1 mM Pi in the presence of 50 mM Na (50Na solution) to that in the presence of 100 mM Na+ (see Supporting Results). For the WT at −100 mV, this ratio was found to be ∼0.5, consistent with the reported K0.5Na of ∼50 mM (6,23). This ratio also reflects how the maximum transport rate depends on external [Na+] under saturating [Pi].
For mutants Q206A, Q206N, D209A, and S447A, the Pi activation indices were larger than the WT value (Fig. S3 C). According to our two-point screening assay, the altered activation indices would correspond to an ∼25% decrease in the apparent K0.5Pi, and therefore indicated that the mutagenesis had resulted in a marginal effect on the apparent Pi affinities. In contrast, T200A showed a significantly reduced Pi-activation index, which suggested that its apparent K0.5Pi had increased by ∼8-fold. For the Na+-activation assay, T200A also showed a significantly reduced activation index, whereas for D209E, R210A, and S447A, the indices were closer to the WT value (23,26).
We concluded that, with the exception of T200A, only marginal changes in apparent substrate affinities resulted from the mutagenesis at 447 and at the other sites in TM2.
Cation selectivity
In the final steady-state assays, we examined the response of the mutants to 1 mM Pi when 50% of the external Na+ was replaced with Li+ (50Na50Li solution). Li+ is proposed to substitute for the first Na+ ion, but not for Na+ bound to Na2 and Na3 (14). Thus, mutations that affect Na1 might be expected to show altered electrogenic behavior in the presence of Li+. For the WT, we observed a small decrease in the relative activity when we compared the Pi responses in 50Na and 50Na50Li solutions, and the response to 1 mM Pi was ∼50% of that observed in 100Na (Fig. 4 B), consistent with previous findings for WT human NaPi-IIa (I.C.F. and C. Ghezzi, unpublished data). In contrast, the ratio of the response to 1 mM Pi in 50Na50Li relative to 100Na revealed that the mutagenesis at positions 206, 209, and 210 resulted in a significantly increased electrogenic response compared with the same ratio measured for 50Na (Q206N, D209A, and R201A; Fig. 4 B). The ratio reflects the ability of Li+ to substitute for Na+ at a given membrane potential (−100 mV); thus, if Li+ substituted fully for Na+, the ratio would be unity. Given that Li+ is hypothesized to interact with NaPi-IIa at the Na1 site, our data directly implicate positions 206 and 209 in coordinating a cation at Na1. Since R210 is positively charged, it is extremely unlikely to coordinate the Na+ ion directly. Nevertheless, these data suggest that position 210 is close enough that the R→A mutation affects either the electrostatics or sterics of Na1. It should be noted, however, that when Li+ fully replaced Na+ as the cosubstrate (100Li solution), no significant Pi-induced change in the holding current was observed for the mutants, which confirmed that Na+ was still required to complete the transport cycle (data not shown), as previously reported for the WT (27).
Functional characterization: presteady-state responses
Presteady-state analysis allowed us to quantify the charge displacements associated with conformational state changes between states 6 and 2 (Fig. 1) in the absence of external Pi, i.e., transitions between states associated with the empty carrier and cation interactions preceding Pi binding, as well as transitions following its cytosolic release (transition 6↔0). We resolved presteady-state relaxation currents induced by voltage steps for six mutants with substitutions in TM2 and three mutants with substitutions in TM5 (Table 1). The activity of T200A was too small for reliable presteady-state analysis, and for N448A we were unable to resolve relaxations that could be separated from the capacitive charging transient, even though the protein was detected at the membrane (Table 1; Fig. S1). Integration of the relaxation currents after baseline correction and exclusion of the capacitive component yielded presteady-state Q-V distributions that we fit with a single Boltzmann function (Eq. 1, Materials and Methods) to obtain an estimate of the maximum moveable charge (Qmax), the mid-point voltage (V0.5), and the apparent valence (z). Both Qmax and V0.5 were used as indicators of possible changes to the occupancies of the states relevant to cation interactions.
Representative presteady-state relaxation currents evoked by voltage steps in the range of −180 mV to +80 mV from the −60 mV holding potential (Vh) are depicted in Fig. 5 A for the WT and for two mutants that showed very different phenotypes (D209E and T454A). Quantification of the relaxation currents is shown as Q-V relationships for each construct for superfusion in the absence of Na+ ions (0Na) and with 100 mM Na+ (100Na). No relaxations were detected in the typical noninjected (NI) control oocyte for either of the superfusates (shown here for 100Na; Fig. 5 A). For the WT NaPi-IIa, presteady-state relaxation currents were detected under both superfusing conditions, as previously reported (23), and with 0Na the charge movements were approximately symmetrically distributed for equal positive and negative steps around Vh. With 100 mM Na+ present, the distribution became more asymmetrical with respect to the baseline record at Vh, such that more charge movement was evoked with depolarizing steps, reflecting the release of Na+ back into the external medium.
Figure 5.
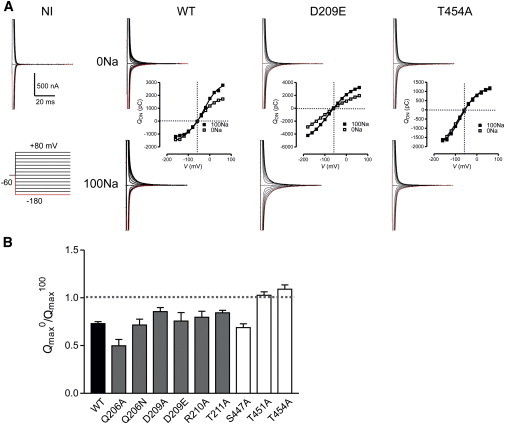
Presteady-state relaxations. (A) Representative examples of presteady-state relaxations evoked by voltage steps in the range of −180 mV to +80 mV from a −60 mV holding potential for a typical noninjected (NI) oocyte, the WT, and mutants D209E and T454A. Upper data traces correspond to superfusion in 0 Na solution, and lower traces correspond to superfusion in 100Na solution. For each construct, Q-V data are shown for the two superfusates. Gray dashed lines help to visualize the charge distribution about the holding potential. Continuous lines are fits to a single Boltzmann function (Eq. 1). The parameters for the fits for the individual cells shown are summarized in Table S4. (B) Ratio of the estimated total charge predicted from the Boltzmann equation fit to data obtained by superfusion with 0Na to that obtained by superfusion with 100Na (Qmax0/Qmax100) for each construct. Data are shown as mean ± SE for n > 5 cells. Black, WT; gray, mutants in TM2; white, mutants in TM5.
The behavior was very different for mutant D209E, which showed a more asymmetrically distributed charge movement in 0Na, with more charge evoked by hyperpolarizing steps from Vh. Moreover, like the WT, in the presence of 100Na, additional charge movement was evoked, although in contrast to the WT, the charge increase for D209E was detected for both hyper- and hypopolarizing steps relative to Vh. Mutant T454A also showed a significant asymmetry of charge movement with respect to Vh; however, there was little difference in the magnitude of the charge evoked, whether or not Na+ was present, apart from a small depolarizing shift in the Q-V in 100Na (see below).
To quantify these findings further, we first considered the effect of the mutation on the Boltzmann fit parameters Qmax and V0.5 when changing from 0Na to 100Na solutions. For the WT, superfusion with 100Na increases Qmax by ∼30% (23) (Fig. 5 A), and we quantified this as the ratio of Qmax in 0Na to Qmax in 100Na (Qmax0/Qmax100) (Fig. 5 B). This ratio was used as an indication of whether the mutagenesis had altered the electrogenic properties of the apparent Na+ ion interaction with the TM electric field before Pi binding. Such alterations may reflect changes in the ability of Na+ to bind in its site or changes in the ability of the protein to respond to Na+ binding. Like the WT, all mutants with substitutions in TM2, as well as S447A in TM5, showed an increase in total charge movement when the superfusate was 100Na, although the relative increase varied according to the site and substituted residue type. In contrast, T451A and T454A in TM5 showed little difference in Qmax whether external Na+ ions were present or not, and therefore exhibited distinct behavior from the other mutants (see Fig. 5 A).
Significant shifts in the V0.5 were observed in the pooled data for the mutants relative to the WT, and these shifts also depended on the superfusion conditions (Fig. 6 A). As expected from the steady-state analysis, T211A behaved similarly to the WT. Four of the mutants with substitutions in TM2 (Q206A, Q206N, D209A, and D209E) and one mutant in TM5 (S447A) that showed obvious hyperpolarizing shifts in their steady-state I-V relations (Fig. 4 A) also displayed significant hyperpolarizing shifts in V0.5 when superfused with 100Na. In the absence of external Na+ (0Na), we also found significant deviations of V0.5 from the WT value (Q206N, D209E, R210A, and S447A), which indicated that the mutagenesis had altered the distribution of occupancies among states 0, 1, and 6 (Fig. 1). This was particularly obvious for mutants R210A and S447A, for which we determined V0.5 values approaching −100 mV for each construct (c.f. WT: −65 ± 7 mV). As noted above, we also observed a small Na+-dependent depolarizing shift in V0.5 for the two TM5 mutants (T451A and T454A) when Na+ ions were present, even though Qmax0/Qmax100 was close to unity (Fig. 5, A and B). Their behavior suggested that Na+ ions still interact with the protein, but only weakly with the TM electric field (see Discussion).
Figure 6.
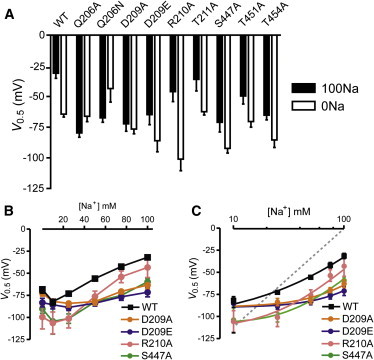
Quantification of midpoint voltage (V0.5) for WT and mutants. (A) Comparison of V0.5 for superfusion in 100Na (solid bars) and 0Na (open bars) for each construct. Each bar represents the mean ± SE for n ≥ 4 cells from at least two donor animals. (B) Dependence of V0.5 on external [Na+] for mutants of two neighboring charged sites (D209 and R210) in TM2. Data points are joined by lines. (C) The same data as in (B) plotted on a semilog scale. The continuous lines are best fits to the data points predicted by a four-state model that accounts for the empty carrier and sequential Na+ binding partial reactions (see Discussion). The dashed line represents the theoretical limiting slope of 116 mV/decade expected at high [Na+].
Based on the altered steady-state voltage dependence of IPi (Fig. 3), we hypothesized that the three mutants in TM2 (D209A, D209E, and R210A) involving removal or modification of the existing charged residues, and S447A in TM5, which showed a similar curvilinear IPi-V relationship, should also show correspondingly altered Na+ interactions in a presteady-state analysis. Therefore, we investigated the dependence of V0.5 on external [Na+] in detail for these mutants (Fig. 6 B). Previous studies have shown that with high external [Na+], there is a near linear relationship between V0.5 and log10[Na+], the slope of which reflects the cooperativity of interaction between Na+ ions and the empty carrier (5,14,23). This slope approaches ∼116 mV/decade for two Na+ ions interacting with the empty carrier (5,28). When the V0.5 versus [Na+] data were replotted on a semi-log scale (Fig. 6 C), it was obvious that the limiting slope behavior was severely compromised for D209A/E and S447A. In contrast, although R210A had a strongly hyperpolarized V0.5 in 0Na, its V0.5 approached that of the WT as [Na+] increased. This suggested that a direct involvement in Na1 coordination is less likely for this position.
In summary, the presteady-state assays corroborate the steady-state findings and provide further support for the association of sites Q206, D209, and S447 with cation interactions at Na1. The removal of charge at position 210 affected the empty carrier equilibrium; however, at a sufficiently high [Na+] the WT state distribution was almost reached, suggesting that R210 is not directly associated with Na1 in NaPi-IIa.
Refinement of the structural model of human NaPi-IIa
Unlike Q206, D209, and S447, which are strongly implicated in contributing to the Na1 site, T211 can be excluded from Na1 (Figs. 3 and 4), consistent with the weak sequence conservation of threonine at this position (Fig. S4). The T200A mutation, on the other hand, caused significant changes in the apparent affinity for both Na+ and Pi (Figs. 4 B and S3 C), consistent with the strong sequence conservation of this position (Fig. S4). Moreover, although the signal was too small to allow presteady-state currents to be analyzed, the voltage dependence of phosphate-induced currents was also altered by T200A, especially in the form of outward currents at depolarizing potentials (Fig. 4 A), reminiscent of previous observations for N227Q (23). The origin of these outward currents is not clear. An inward chloride leak has been ruled out in the case of N227Q (23), whereas in chloride-free solution, the phosphate-induced outward currents were suppressed for T200A. In any case, the similarity of the phenotypes of N227Q and T200A suggests that these two residues play a similar role in the voltage-dependent steps of transport, and therefore may be proximal in the protein structure.
Given the high confidence in the positioning of TM3 in the structural model of NaPi-IIa based on the agreement with accessibility measurements, it is reasonable to assume that D224 and N227 are indeed >10 Å from the Na2-Pi-Na3-binding regions, i.e., the tips of HP1 and HP2 (Fig. 2). Taking this assumption and all of the above data into account, we refined the structural model of human NaPi-IIa so that Q206 and D209 are on the same face of TM2b and point toward TM3, while T211 is farther away (see Materials and Methods; Fig. 7 B; Table S1). In this model, T200 remains midway across the membrane near the cytoplasmic end of TM2a, just before L2ab, with its backbone carbonyl pointing into the Na1 site, and its side chain can potentially orient either downward to Na1 or upward toward the Na2-Pi-Na3 cluster.
Figure 7.
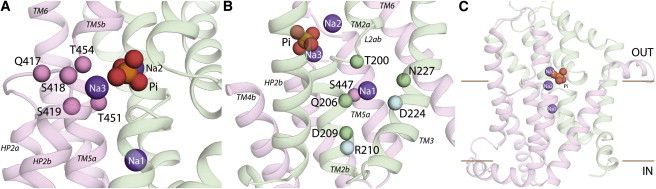
Refined structural model of human NaPi-IIa bound to three Na+ ions and one Pi anion. The Na+ ions are shown as purple spheres and the Pi is shown as orange and red spheres. Residues comprising RU1 and RU2 are colored in green and pink, respectively. (A) Close-up of the Na3-binding site, viewed from the extracellular side of the membrane into the binding site. The Cα atoms of several residues predicted to form the Na3 site or to coordinate Pi are highlighted (spheres). (B) Predicted Na1-binding site. The Cα atoms of the residues whose side chain and/or backbone atoms are predicted to form the Na1 site are shown as green spheres. The Cα atoms belonging to residues R210 and D224 are highlighted as light blue spheres. (C) Overview of the human NaPi-IIa model represented as cartoon helices, viewed from along the plane of the membrane.
The refinement of TM2b involved adjusting the sequence alignment of NaPi-IIa with VcINDY by five positions (Table S1; Fig. S5), which shortened the previously rather long loop L2ab between TM2a and TM2b (cf. Figs. 2 and 7). These adjustments resulted in other improvements. For example, several evolutionarily variable, ionizable residues, including D212, R214, and R215 (Fig. S4), were repositioned into the cytoplasmic loop (cf. Fig. S6, A and C), while R210 was moved away from the Pi-binding site (Fig. 7), consistent with the lack of effect of an R210A mutation on the Pi apparent affinity (10). The model quality score (ProQM) in the region of TM2b decreased somewhat after the refinement (Fig. S7), indicating a slightly worse model. This may be because conserved residues in the cytoplasmic end of TM3 now point away from the core of the protein (Fig. S6), which is atypical. Nevertheless, a comparison with VcINDY suggests that in the complete protein these residues will pack against the peripheral helices TM7 and TM8, which are not modeled due to the absence of a reliable template (Fig. S8). To test the effect of removing peripheral helices on the ProQM score of the remaining core helices, we compared the ProQM scores of the full-length structure of VcINDY with the scores obtained for the segments used as a template, i.e., after removing TM1-3, TM7-8, and the other protomer (Fig. S9). Significant reductions in the ProQM scores at TM5, TM9, and HP2b in the truncated structure of VcINDY (Fig. S9 A) reflect the removal of packing interactions with TM7, the other protomer, and TM3, respectively (Fig. S9, B and C). Therefore, a similar effect may explain the reduction in the ProQM profiles for TM2b/TM3 of the new NaPi-IIa model, in which those residues are more exposed (Fig. S8). Importantly, in the refined model, the residues from TM2 and TM3, which were implicated experimentally in Na1 binding, are clustered together. Moreover, without imposing specific constraints on their orientation, the side chain of R210 can readily contact D224 (Fig. 7 B), consistent with their similar phenotypes in altering the voltage dependence of the empty carrier, as well as their likely involvement in either the return step (Fig. 1, 0→1) of the empty transporter or release of the Na1 into the cytoplasm.
Informed by the functional and biochemical characterization of mutants S447, N448, T451, and T454, we also modified the alignment in the region of TM5 and TM6 (Fig. S5), placing T451 and T454 farther to the extracellular side and closer to the predicted Na3 site, and placing S447 closer to the other putative Na1-binding residues (Fig. 7 A; Table S2). The resultant structural model has a dramatically improved ProQM quality score in this region (Fig. S7 B), in part due to the repositioning of three arginines into the cytosol instead of facing the hydrophobic core of the membrane in TM6 (cf. Fig. S6, B and D). In accordance with PSIPRED secondary structure predictions (Fig. S5) and the strong helical conservation pattern (Fig. S6 D), this arginine-rich segment was modeled as a helix. Note that in the model, this helix extends into the cytoplasm because there are no additional constraints to fix its relative orientation (Fig. S6 D), although its strong amphipathic character (Fig. S5) suggests that in reality this segment will form a helix that lies parallel to the membrane plane along the lipid headgroups.
In summary, the experimentally guided refinements led to a structurally reasonable model of the core of human NaPi-IIa in which the overall fold is maintained (Supporting Model S1). Compared with the earlier model, TM2 and TM5-6 change by >5 Å, and the loops L2ab and L5ab are significantly shorter (Fig. S10). This new model illustrates how T451 and T454 from TM5 may contribute to the predicted Na3- or Pi-binding sites (Fig. 7 A) together with Q417, S418, and S419 from the HP2ab loop, whereas the binding site for Na1 would be formed by T200, Q206, D209, N227, and S447 (Fig. 7 B). Moreover, the model predicts that R210 and D224 point toward each other and are located on the cytoplasmic side of the Na1 site (Fig. 7, B and C).
Discussion
In this work, we improved upon our previous homology model of the human isoform of NaPi-IIa (SLC34A1) (10) to incorporate the first Na+ ion-binding site (Na1). The binding of Na+ at Na1 is the initiator of all subsequent steps in the transport cycle, and the kinetics of binding are a critical determinant of the overall transport rate and the electrogenic properties. Our strategy for defining the coordinating residues for Na1 involved bioinformatic prediction of candidate positions based on the existing structural model, functional investigation of the effects of single point substitutions at the predicted positions, and further model refinement in the light of the functional data. In addition to using conventional criteria for selecting potential Na+ coordinating residues (serine, threonine, asparagine, glutamine, aspartate, and glutamate), as seen in available Na+-coupled transporter structures (29–31), we also took into account two salient functional properties of NaPi-II proteins.
First, previous functional studies on both WT and mutated constructs have revealed the importance of the Na1-cation interaction in determining the electrogenic properties of the NaPi-II transport cycle. The aspartic acid at 224, which is conserved in all electrogenic isoforms, is a key requirement for the 3:1 stoichiometry and transport electrogenicity, and, by implication, the translocation of the first Na+ ion through the protein (4,6–8). Therefore, D224 became the focal point for identifying other side chains in close proximity. Second, previous kinetic studies have highlighted the involvement of the Na1 interaction with an uncoupled leak, and proposed that in the absence of substrate (Pi), Na+ ions participate in a uniport mode that involves cycling between states 0 and 6 (Fig. 1) (23,24,26,32,33). This phenomenon is only observed for the electrogenic isoforms. Although they were not functionally explored in this study because of their small magnitude, the existence of uncoupled leak currents also implies that the Na1 site must allow accessibility from either side of the membrane with minimal rearrangements, and we also used this feature as a criterion for selecting the initial binding region.
Once we had proposed the positions, the goals of the functional assays were twofold. First, we aimed to distinguish between mutations at positions that were more likely to be involved with Na1 and those that contributed to the Na2-Pi-Na3-binding region, as well as those with little functional significance, by comparison with the WT. Second, for the Na1-related mutations, we aimed to further distinguish between effects on the empty carrier kinetics and those on the Na+ interactions, and thereby gain more insight into the structure-function basis for electrogenicity, by using presteady-state assays. We then used the interpretation of the functional data for the final model refinement.
Discrimination between Na1 and Na2-Pi-Na3 sites using steady-state assays
In our previous study (10), Ala or Cys substitutions at three positions in TM2 and HP1 (S164, T195, and S196) and two positions in HP2 (S418 and S419), which we associate with Na2-Pi-Na3 coordination, caused a total loss of cotransport function, yet evidence for cation interactions at Na1 was still detected in the form of presteady-state relaxations in the presence of 100 mM Na+. In contrast, the mutagenesis in the study presented here was well tolerated by the transporter, as evidenced by the complete cotransport cycle remaining intact in most cases (Table 1). Notable exceptions were three positions in TM5 (N448, T451, and T454), for which we detected no transport activity from the respective mutants. In the case of T451 and T454, presteady-state relaxations were also resolved (see below), suggesting that those side chains were more likely to be associated with the Na2-Pi-Na3-binding region. The basis for the lack of any functional activity for N448A, which was expressed in the oocyte membrane, is unclear. In all, only one mutant showed WT-like behavior (T211A), and this was taken into account in subsequent model refinement (see below).
In the steady-state cotransport mode, the kinetics of the transport cycle for WT NaPi-IIa (Fig. 1) have been extensively described in terms of macroscopic phenomenological parameters, such as apparent substrate affinities derived from standard TEVC assays (23,26). These parameters are a function of the rate constants associated with all of the hypothesized partial reactions in the transport cycle (Fig. 1), and therefore deviations from the WT macroscopic kinetic profile resulting from mutagenesis might be caused by changes in the rates associated with more than one partial reaction, leading to ambiguous interpretations. Thus, with three partial reactions involving external Na+ interactions for the cotransport cycle, any one or all of them might be involved in determining K0.5Na and K0.5Pi.
With the exception of T200A, the two-point assay for substrate activation (Fig. S3) indicated that the mutagenesis at all considered positions did not cause large changes in the apparent substrate affinities (Fig. S3 C). Nevertheless, small but significant deviations were observed that would require a more detailed analysis, beyond the scope of this study, to identify the partial reactions affected. For example, in the Pi-activation assay (Fig. S3 C), Q206A/N, D209A, and S447A all showed increased activation indices, consistent with an increased K0.5Pi. This increase could arise indirectly from changes to the Na+ interactions preceding Pi binding, the Pi binding step, or a combination thereof. Indeed, for Q206A the Na+ activation index was also increased, consistent with an altered Na+ interaction involving partial reactions (1↔2 and 2↔3; Fig. 1), whereas for Q206N, D209A, and S447A, the Na+ activation index was not markedly altered compared with the WT. Because deviations from WT behavior for the activation indices might be attributable to changes in more than one partial reaction in the overall cotransport cycle, we were careful to avoid overinterpreting these data. This was particularly true for the Na+-inactivation data, which reflect the combined effect of all five cation- and substrate-dependent partial reactions (Fig. 1), including cation and substrate release to the cytosol. Of all the mutants investigated, T200A showed the largest deviation from WT behavior. Its very low functional activity allowed us to perform only basic steady-state assays, from which we concluded that K0.5Na and K0.5Pi were significantly reduced based on the smaller activation indices (Fig. S3). Similar effects were also reported for the N→C substitution at 199 (32) and the N→Q substitution at 227 located on TM3 (6). Given the limited kinetic analysis, we are unable assign this phenotype to either a direct effect on Na1 interactions or more global effects involving the Na2-Pi-Na3 site. As positions 199 and 200 are located between the proposed Na1- and Na2-Pi-Na3-binding regions, with N227 close by (Fig. 7, A and B), we speculate that these positions may play a critical role in the coupling between the two regions, specifically in the observed apparent cooperativity of Na2 occupancy. Furthermore, the fact that the N→A substitution at position 227 was not tolerated by the oocytes suggests a critical role for the side chain at 227 and corroborates the previous finding that only a N→Q substitution yielded functional activity (6).
Despite the potential limitations of steady-state assays for resolving effects unique to a specific partial reaction, the I-V data (Fig. 3) revealed two significant deviations from WT behavior that implicated the involvement of positions 206, 209, and 210 from TM2, and 447 from TM5 in coordinating a cation at Na1.
First, alanine substitution at 209, 210, and 447 resulted in the same qualitative hyperpolarizing shift in the voltage dependence of IPi as observed for the more conservative Q→N substitution at 206. This result is consistent with the first Na+ interaction being a strong determinant of voltage dependence, and also underscores how elements of both structural repeats (RU1 and RU2) are involved in cation coordination. Similar shifts in the steady-state voltage dependence were reported in two structure-function studies on the rat NaPi-IIa (34) and flounder NaPi-IIb (12). In the former study, Cys was substituted at two conserved sites (corresponding to I449 and A461 in TM5 of hNaPi-IIa) (34). In the latter study, an A→C substitution was also made at site 175 (corresponding to 203 in TM2 of hNaPi-IIa) (12), and simulations of the A175C kinetics could readily account for the observed shift in voltage dependence by effectively increasing the dissociation constant for the first Na+ binding partial reaction (1↔2; Fig. 1) (12). In our structural model, these conserved positions are involved in the packing of helices TM2 and TM5 with their neighbors, suggesting unspecified indirect effects of these mutations and supporting the notion that the molecular determinants of the steady-state voltage dependence are shared between functionally important elements of the symmetry-equivalent repeat units.
Second, Li+ is proposed to bind only at Na1, where it competes with Na+ ions for occupancy when both are present in the external medium (14). This is a useful property because it allowed us to discriminate between Na1 interactions and those involving Na1 and Na2 when only Na+ was present. Simulations based on an extensive kinetic analysis of the flounder NaPi-IIb isoform predict that the dissociation constant associated with partial reaction 2↔3 (Fig. 1)—leading to occupancy of Na2 by a Na+ ion—is significantly reduced when Na1 is occupied by Li+. Moreover, Li+ occupancy of Na1 was predicted to reduce the inward translocation rate of the fully loaded carrier to account for the smaller IPimax of the WT at hyperpolarizing potentials (see Fig. 3 C) (14). In this study, superfusion with the 50Na50Li solution increased the response to 1 mM Pi for Q206N, D209A/E, R210A, and S447A, exceeding the response with 50Na. In contrast, for the WT there was only a marginal change in IPi over a wide voltage range when superfusing with either solution (Fig. 3). This behavior established that Li+ ions interact with the mutated constructs and are more readily tolerated as a driving cation compared with the WT. Although our data were consistent with the notion that mutagenesis directly impacts only the Na1 site, and 100% replacement of Na+ with Li+ resulted in no electrogenic activity or 32P uptake (data not shown) (14), we cannot exclude the possibility that for the mutants, both Na1 and Na2 sites might bind Li+ and result in an overall cotransport stoichiometry of 2 Li+:1 HPO42−:1 Na+.
Presteady-state assays focus on Na1 and the empty carrier
The presteady-state assays performed in the absence of external Pi allowed us to restrict the number of occupied conformational states and obtain additional insights into the functional consequences of the mutagenesis. Voltage steps applied to the membrane move the transporters between state 6 (extreme depolarizing potentials) and state 1 (0Na) or 3 (100Na) (extreme hyperpolarizing potentials), with a distribution between the possible states at intermediate potentials that depends on the kinetics of the partial reactions. The Q-V data were fit with a single Boltzmann function, which is strictly valid only for a two-state system (35). Here, data resolution limitations and fit uncertainties precluded fitting with a more complex function; however, despite this limitation, two parameters from the single Boltzmann fit analysis (Qmax and V0.5; Eq. 1) provided useful indicators of the effects of mutagenesis.
For all mutants assayed, except for T451A and T454A, there was an increase in estimated total displaceable charge (Qmax) when external Na+ was present, with the Qmax0/Qmax100 ratio being comparable to the WT estimate (Fig. 5 B). Therefore, we can conclude that the mutagenesis did not alter the amount of charge displacement attributable to Na+ ions under the influence of the TM electric field. In this context, it should be noted that Qmax100 describes both the movement of the empty carrier and the additional charge due to Na+ ion displacement. Constructs T451A and T454A, for which we detected no cotransport activity, also showed no significant dependence of Qmax on Na+. This result indicated that Na+ movements to the Na1 and Na2 sites were severely compromised, consistent with the lack of transport activity, although we cannot exclude the possibility that the charge movement attributable to Na+ ions remained undetected due to the resolution limitations of the system. We note that the molecular rearrangements that accompany the Na+ interaction with Na1 remain unresolved and will require knowledge about the structure of NaPi-IIa (or homologs thereof) in states other than the equivalent of state 4 (Fig. 1).
The hyperpolarizing shift in the steady-state voltage dependence of IPi was reflected in corresponding changes in the presteady-state charge distributions of the mutants (i.e., a hyperpolarizing shift of the midpoint voltage, V0.5; Fig. 6 A) when 100 mM Na+ was present externally (Q206A/N, D209A/E, and S447A). This was in accord with the notion that the kinetics of transitions between states associated with the empty carrier (states 0 and 1) and when Na1 (states 2 and 6) or Na2 (state 3) is occupied (Fig. 1) are the main determinants of the voltage dependence of the overall cotransport cycle. In the absence of external Na+, there was more variation in the empty carrier equilibrium potential (V0.5) among the constructs, which suggested that the mutagenesis had also affected the distribution between states 6, 0, and 1, to an extent that depended on the site mutated. Mutants D209E and R210A (TM2), and S447A (TM5) showed significant hyperpolarizing shifts in V0.5 when no external Na+ was present. This indicated that at the −60 mV holding potential, at which the WT would be equally likely to be in state 0 or 1, the inward orientation of the empty carrier (state 0) was more favored for these mutants (Fig. 1). It should be noted that since we had no direct access to the oocyte cytosol to manipulate the intracellular [Na+], we cannot fully exclude the possible occupancy of Na+ bound from the inside (state 6), although indirect evidence from kinetic simulations and the low internal [Na+] for typical oocytes (<10 mM (36)) would be consistent with a low occupancy of state 6 for the WT.
For the two nontransporting mutants in TM5 (T451A and T454A), charge movement was detected in the absence of external Na+. This indicated that the conformational change accompanying the empty carrier reorientation (0↔1; Fig. 1) in response to changes in the TM field still occurred, although for T454A, V0.5 was further shifted in the hyperpolarizing direction relative to the WT, consistent with an inward facing orientation of the empty carrier being favored, as also found for D209E, R210A, and S447A. When Na+ was present in the external medium, the ∼+20 mV depolarizing shift of V0.5 seen for both T451A and T454A could be explained by an electroneutral, or weakly electrogenic, interaction of Na+ with Na1. Given the profound effect on function that mutagenesis had at these sites, we propose that the side chains at 451 and 454 are critical for coupling the Na1 occupancy to the subsequent binding at Na2 rather than directly participating in cation coordination at Na1. Indeed, their general phenotype was consistent with that of mutations at other sites in TM5 that we propose to be nearer the Na2-Pi-Na3-binding region (10).
Two of the positions explored in this study were charged (D209 and R210), and the finding that electrogenic activity remained after charge removal indicates that the charge per se at these positions was not critical for overall function and might argue against their direct involvement at Na1. This contrasts with the removal of negative charge at 224, which results in electroneutral cotransport (6,7). Nevertheless, since the alanine substitution at each position, or merely changing the side-chain length (D209E), significantly altered the electrogenic properties of the mutants, we explored their presteady-state kinetics in more detail by determining the dependence of V0.5 on external [Na+]. This dependence has been used to investigate the apparent affinities of the Na1- and Na2-binding sites in the Na+-dependent sugar transporter SGLT1 (28). Similar to what was found for SGLT1, analysis of presteady-state data from WT NaPi-IIa and NaPi-IIb yields a relationship between V0.5 and log10[Na+] that is consistent with a sequential binding model for Na+. At high [Na+], this relationship is linear and approaches a maximum slope of ∼116 mV/decade (5,14,23,28). Over the range of external [Na+] used in our assays, the data for D209A and D209E showed no evidence of limiting slope behavior, which suggested that the Na+ interactions had been compromised by mutagenesis at position 209. For R210A, the empty carrier V0.5 was also shifted in the hyperpolarizing direction relative to the WT; however, its V0.5 value tended to approach that of the WT at high [Na+]. Based on this behavior, we concluded that R210 was not critical for Na1 coordination, but that the charge at this site was important for defining the equilibrium between the empty carrier states. Interestingly, the polar-to-nonpolar substitution at 447 also resulted in a comparable hyperpolarizing shift to that of R210A for the 0Na superfusion condition. However, this mutant showed a weaker depolarizing shift in V0.5 when Na+ was present that lay between the WT and D209A/E values, suggesting an intermediate effect on Na1 interactions.
We attempted to gain further insight into the significance of these data by fitting them with the implicit function obtained by modeling a four-state sequential binding scheme describing the dependence of V0.5 on [Na+] (corresponding to transitions between states 0, 1, 2, and 3; Fig. 1). The variables in this model comprise the ratio of forward and backward zero voltage rates for each partial reaction and associated apparent valences (14,28) (Supporting Results). Although our ability to discriminate between effects only at Na1 or only at Na2 was limited by having only five nonzero [Na+] data points and a maximum [Na+] = 100 mM, the fit parameters were nevertheless consistent with significantly increased dissociation constants for the first Na+ binding partial reaction compared with the WT for D209A/E and S447A, whereas for R210A the change was less dramatic. These findings further confirmed the likely participation of D209 and S447 in cation coordination at Na1. Changes were also observed for the dissociation constant associated with the Na2 occupancy partial reaction that would suggest that the mutagenesis had also impacted the cooperativity of Na+ interaction.
Taken together, the steady-state and presteady-state functional data strongly implicated Q206, D209, D224, and S447 as likely candidates for coordinating Na+ at Na1. The positive charge at 210 appeared to be a critical determinant of the kinetics of the 0↔1 transition after cytosolic release of Na+ and coupling to the Na2 site.
A structural model of human NaPi-IIa including all four binding sites
The low sequence identity between human NaPi-IIa and its only available template structure, VcINDY, means that although the overall fold is likely to be correct, key regions should be optimized with the guidance of specific experimental data. Preliminary data implicated D224 from TM3 in the role of Na1, and, helpfully, the vertical positioning of the residues in this TM helix was one of the most reliable aspects in the published model (10) because of the clear agreement with the TM prediction from OCTOPUS (residues 224–244) and experimental accessibility data (6). That Na1 is likely to be fairly close to the cytoplasmic surface can be inferred from the leak mode observed in the absence of substrate for the electrogenic forms of SLC34. Therefore, we focused our efforts on residues close to D224 and on the cytoplasmic halves of TM2 and TM5, i.e., the only two helices contacting TM3 in this fold. This strategy allowed us to identify nine candidate positions for the Na1 site, only three to five of which are likely to be directly involved. Remarkably, the experimental data described above delineates the roles of most of the nine tested residues.
For the available template, only one alignment of TM2b between NaPi-IIa and VcINDY was compatible with a Na1 site in which D206 and N209 coordinate the ion, but T211 does not. This adjustment had the added advantage that a conspicuously long loop in the membrane (L2ab), which previously could not be unambiguously modeled, was significantly shortened, and several charged residues could be repositioned into a more suitable aqueous environment (Fig. S6). The distinct behavior of mutations at T451 and T454 was reminiscent of previous results for residues in the Na2-Pi-Na3-binding region of NaPi-IIa. Although the published data do not delineate which residues contribute to the Na2, Na3, or Pi sites in NaPi-IIa, they nevertheless provide strong support for the involvement of several residues in HP1 (S164, T195, and S196), HP2 (Q417, S418, and S419), and TM2 (N199) in contributing to this binding region (10). The similar data for T451A and T454A suggest that these two residues in TM5 also contribute to this binding region, so we positioned T451 and T454 close to the tip of HP2 by adjusting the alignment of TM5, shortening the L5ab loop, and improving the model of TM6.
We note that S462, which we previously concluded might be part of the Na2-Pi-Na3-binding region, and might coordinate Na3, is now moved farther to the periplasmic end of TM5b. Our earlier proposal was based on the observation that Pi transport by an S462C mutant of rat NaPi-IIa was inhibited by an MTS reagent (34). However, it should be noted that similar observations have been made for positions remote from binding sites (e.g., the helical hairpin HP2 in glutamate transporters) if the repacking of that segment is required for transport (37). Therefore, we propose that an alternative role should be assigned to S462, and that T451 and T454 may be positioned close to the Na2-Pi-Na3 cluster.
As described above, the type of side chain at 224 plays a critical role in determining charge translocation and electrogenicity by NaPi-II family members (6–8,12). After refinement, the NaP-IIa model predicts a possible salt-bridge between R210 (TM2) and D224 (TM3), with both residues close to the Na1 site, but on the cytoplasmic side thereof. Although a clear structural interpretation of the electrophysiological data is difficult, we speculate that when D224 is a Gly, as in NaPi-IIc, R210 does not interact with TM3, reducing the repulsion of R210 to the Na1 ion and allowing the ion to remain bound during the 0↔1 transition (Fig. 1). In addition, in NaPi-IIa/b, formation of the salt-bridge by D224 may be voltage dependent; thus, breaking of this bridge could provide a pathway for the ion to leak in the absence of Pi (24). Whatever the mechanism, it is clear that these two residues are involved in distinguishing between the electrogenic and electroneutral isoforms. Therefore, it will be essential to examine in further detail how those positions and their valency relate to the voltage dependence of transport.
In conclusion, the fact that residues from three distinct TM segments (TM2, TM3, and TM5) come together to form the Na1 site, as predicted by our models, provides strong support for the predicted fold of NaPi-II transporters and the choice of VcINDY as a template. Moreover, the refinements of TM2, TM5, and TM6 in this structural model and the identification of the Na1 site residues provide a firmer foundation for the future design of experiments to define functionally critical aspects of transport by the SLC34 family of proteins, as well as of VcINDY homologs from the SLC13 family such as NaDC1 and NaDC3 (Fig. S11).
Author Contributions
C.F.-F.: bioinformatics and modeling. L.R.F.: analysis of computational data. A.W.: molecular biology. M.P.: molecular biology, electrophysiology, and data analysis. I.C.F.: electrophysiology and data analysis. T.K.: tracer assays and immunohistochemistry. L.R.F., A.W., and I.C.F.: experimental design and writing of the manuscript.
Acknowledgments
We thank Eva Hänsenberger for oocyte preparation, Mehdi Taslimifar (Interface Group, Institute of Physiology, University of Zürich) for valuable assistance in MATLAB modeling, and members of the Theoretical Molecular Biophysics Unit of the National Heart, Lung and Blood Institute for useful discussions.
This work was supported by The Dunhill Medical Trust (AW SA10/0210), the Swiss National Science Foundation (I.C.F.), the Hartmann Müller Foundation (I.C.F.), and the Intramural Research Program of the NIH, National Institute of Neurological Disorders and Stroke (L.R.F.).
Footnotes
Cristina Fenollar-Ferrer and Ian C. Forster contributed equally to this work.
Andreas Werner and Lucy R. Forrest contributed equally to this work.
Contributor Information
Ian C. Forster, Email: iforster@access.uzh.ch.
Andreas Werner, Email: andreas.werner@newcastle.ac.uk.
Lucy R. Forrest, Email: lucy.forrest@nih.gov.
Supporting Material
References
- 1.Biber J., Hernando N., Forster I. Phosphate transporters and their function. Annu. Rev. Physiol. 2013;75:535–550. doi: 10.1146/annurev-physiol-030212-183748. [DOI] [PubMed] [Google Scholar]
- 2.Forster I.C., Hernando N., Murer H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Aspects Med. 2013;34:386–395. doi: 10.1016/j.mam.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Lederer E. Renal phosphate transporters. Curr. Opin. Nephrol. Hypertens. 2014;23:502–506. doi: 10.1097/MNH.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghezzi C., Murer H., Forster I.C. Substrate interactions of the electroneutral Na+-coupled inorganic phosphate cotransporter (NaPi-IIc) J. Physiol. 2009;587:4293–4307. doi: 10.1113/jphysiol.2009.175596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forster I.C., Hernando N., Murer H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr. Top. Membr. 2012;70:313–356. doi: 10.1016/B978-0-12-394316-3.00010-7. [DOI] [PubMed] [Google Scholar]
- 6.Virkki L.V., Forster I.C., Murer H. Functionally important residues in the predicted 3(rd) transmembrane domain of the type IIa sodium-phosphate co-transporter (NaPi-IIa) J. Membr. Biol. 2005;206:227–238. doi: 10.1007/s00232-005-0796-x. [DOI] [PubMed] [Google Scholar]
- 7.Bacconi A., Virkki L.V., Forster I.C. Renouncing electroneutrality is not free of charge: switching on electrogenicity in a Na+-coupled phosphate cotransporter. Proc. Natl. Acad. Sci. USA. 2005;102:12606–12611. doi: 10.1073/pnas.0505882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patti M., Ghezzi C., Forster I.C. Conferring electrogenicity to the electroneutral phosphate cotransporter NaPi-IIc (SLC34A3) reveals an internal cation release step. Pflugers Arch. 2013;465:1261–1279. doi: 10.1007/s00424-013-1261-9. [DOI] [PubMed] [Google Scholar]
- 9.Mancusso R., Gregorio G.G., Wang D.N. Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter. Nature. 2012;491:622–626. doi: 10.1038/nature11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fenollar-Ferrer C., Patti M., Forrest L.R. Structural fold and binding sites of the human Na+-phosphate cotransporter NaPi-II. Biophys. J. 2014;106:1268–1279. doi: 10.1016/j.bpj.2014.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulligan C., Fitzgerald G.A., Mindell J.A. Functional characterization of a Na+-dependent dicarboxylate transporter from Vibrio cholerae. J. Gen. Physiol. 2014;143:745–759. doi: 10.1085/jgp.201311141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghezzi C., Meinild A.K., Forster I.C. Voltage- and substrate-dependent interactions between sites in putative re-entrant domains of a Na(+)-coupled phosphate cotransporter. Pflugers Arch. 2011;461:645–663. doi: 10.1007/s00424-011-0948-z. [DOI] [PubMed] [Google Scholar]
- 13.Yernool D., Boudker O., Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 14.Andrini O., Meinild A.K., Forster I.C. Lithium interactions with Na+-coupled inorganic phosphate cotransporters: insights into the mechanism of sequential cation binding. Am. J. Physiol. Cell Physiol. 2012;302:C539–C554. doi: 10.1152/ajpcell.00364.2011. [DOI] [PubMed] [Google Scholar]
- 15.Remmert M., Biegert A., Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 16.Crooks G.E., Hon G., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Šali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 18.Waterhouse A.M., Procter J.B., Barton G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laskowski R.A., Macarthur M.W., Thornton J.M. PROCHECK—a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 20.Ray A., Lindahl E., Wallner B. Model quality assessment for membrane proteins. Bioinformatics. 2010;26:3067–3074. doi: 10.1093/bioinformatics/btq581. [DOI] [PubMed] [Google Scholar]
- 21.Forrest L.R., Tang C.L., Honig B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys. J. 2006;91:508–517. doi: 10.1529/biophysj.106.082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harding M.M. The architecture of metal coordination groups in proteins. Acta Crystallogr. D Biol. Crystallogr. 2004;60:849–859. doi: 10.1107/S0907444904004081. [DOI] [PubMed] [Google Scholar]
- 23.Virkki L.V., Forster I.C., Murer H. Substrate interactions in the human type IIa sodium-phosphate cotransporter (NaPi-IIa) Am. J. Physiol. Renal Physiol. 2005;288:F969–F981. doi: 10.1152/ajprenal.00293.2004. [DOI] [PubMed] [Google Scholar]
- 24.Andrini O., Ghezzi C., Forster I.C. The leak mode of type II Na(+)-P(i) cotransporters. Channels (Austin) 2008;2:346–357. doi: 10.4161/chan.2.5.6900. [DOI] [PubMed] [Google Scholar]
- 25.Ehnes C., Forster I.C., Murer H. Structure-function relations of the first and fourth extracellular linkers of the type IIa Na+/Pi cotransporter: II. Substrate interaction and voltage dependency of two functionally important sites. J. Gen. Physiol. 2004;124:489–503. doi: 10.1085/jgp.200409061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forster I., Hernando N., Murer H. The voltage dependence of a cloned mammalian renal type II Na+/Pi cotransporter (NaPi-2) J. Gen. Physiol. 1998;112:1–18. doi: 10.1085/jgp.112.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Virkki L.V., Biber J., Forster I.C. Phosphate transporters: a tale of two solute carrier families. Am. J. Physiol. Renal Physiol. 2007;293:F643–F654. doi: 10.1152/ajprenal.00228.2007. [DOI] [PubMed] [Google Scholar]
- 28.Loo D.D., Jiang X., Wright E.M. Functional identification and characterization of sodium binding sites in Na symporters. Proc. Natl. Acad. Sci. USA. 2013;110:E4557–E4566. doi: 10.1073/pnas.1319218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamashita A., Singh S.K., Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl—dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 30.Verdon G., Oh S., Boudker O. Coupled ion binding and structural transitions along the transport cycle of glutamate transporters. eLife. 2014;3:e02283. doi: 10.7554/eLife.02283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zdravkovic I., Zhao C., Noskov S.Y. Atomistic models of ion and solute transport by the sodium-dependent secondary active transporters. Biochim. Biophys. Acta. 2012;1818:337–347. doi: 10.1016/j.bbamem.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 32.Köhler K., Forster I.C., Murer H. Identification of functionally important sites in the first intracellular loop of the NaPi-IIa cotransporter. Am. J. Physiol. Renal Physiol. 2002;282:F687–F696. doi: 10.1152/ajprenal.00282.2001. [DOI] [PubMed] [Google Scholar]
- 33.Köhler K., Forster I.C., Murer H. Transport function of the renal type IIa Na+/P(i) cotransporter is codetermined by residues in two opposing linker regions. J. Gen. Physiol. 2002;120:693–705. doi: 10.1085/jgp.20028645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambert G., Forster I.C., Murer H. Cysteine mutagenesis reveals novel structure-function features within the predicted third extracellular loop of the type IIa Na(+)/P(i) cotransporter. J. Gen. Physiol. 2001;117:533–546. doi: 10.1085/jgp.117.6.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bezanilla F., Villalba-Galea C.A. The gating charge should not be estimated by fitting a two-state model to a Q-V curve. J. Gen. Physiol. 2013;142:575–578. doi: 10.1085/jgp.201311056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sciortino C.M., Romero M.F. Cation and voltage dependence of rat kidney electrogenic Na(+)-HCO(−)(3) cotransporter, rkNBC, expressed in oocytes. Am. J. Physiol. 1999;277:F611–F623. doi: 10.1152/ajprenal.1999.277.4.F611. [DOI] [PubMed] [Google Scholar]
- 37.Qu S., Kanner B.I. Substrates and non-transportable analogues induce structural rearrangements at the extracellular entrance of the glial glutamate transporter GLT-1/EAAT2. J. Biol. Chem. 2008;283:26391–26400. doi: 10.1074/jbc.M802401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.