

Abstract
Background
Secondary, phased small interfering RNAs (phasiRNAs) derived from protein-coding or noncoding loci (PHAS) are emerging as a new type of regulators of gene expression in plants. However, the evolution and function of these novel siRNAs in plant species remain largely unexplored.
Results
We systematically analyzed PHAS loci in 23 plant species covering major phylogenetic groups spanning alga, moss, gymnosperm, basal angiosperm, monocot, and dicot. We identified over 3,300 PHAS loci, among which ~1,600 were protein-coding genes. Most of these PHAS loci were novel and clade- or species-specific and showed distinct expression patterns in association with particular development stages, viral infection, or abiotic stresses. Unexpectedly, numerous PHAS loci produced phasiRNAs from introns or exon–intron junction regions. Our comprehensive analysis suggests that phasiRNAs predominantly regulate protein-coding genes from which they are derived and genes from the same families of the phasiRNA-deriving genes, in contrast to the dominant trans-regulatory mode of miRNAs. The stochastic occurrence of many PHAS loci in the plant kingdom suggests their young evolutionary origins.
Conclusions
Our study discovered an unprecedented diversity of protein-coding genes that produce phasiRNAs in a wide variety of plants, and set a kingdom-wide foundation for investigating the novel roles of phasiRNAs in shaping phenotype diversities of plants.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-015-0142-4) contains supplementary material, which is available to authorized users.
Keywords: Evolution, Function, Plant phasiRNAs, Protein-coding genes
Background
The workings of combinatorial genetic regulatory networks control how an organism grows and develops as well as responds to biotic and abiotic stresses. Distinct regulatory networks emerged during evolution likely have contributed to the diversification of biological phenotypes. Investigations over the last decade from many different organisms have uncovered the novel regulatory roles of numerous small, noncoding RNAs. These include microRNAs (miRNAs) and short interfering RNAs (siRNAs) that exert epigenetic regulation of gene expression at the transcriptional, posttranscriptional, and translational levels [1–3]. These discoveries have led to RNA-based new paradigms of genetic regulatory networks that control various biological processes including the emergence of biological diversity [4–6].
In plants, miRNAs can trigger the generation of secondary, phased siRNAs (phasiRNAs) from protein-coding or intergenic loci (PHAS) [7]. Some phased siRNAs can trans-regulate the expression of target genes, and are called trans-acting siRNAs (tasiRNAs or TAS) [8]. Early studies with Arabidopsis thaliana provided genetic evidence supporting that some tasiRNAs regulate gene expression critical to plant development [9, 10], and it has been shown that a deeply conserved TAS3 pathway is pivotal in developmental processes from moss to higher plants [11–14]. More recent studies showed the generation of phasiRNAs from mRNAs that encode many nucleotide binding site (NBS) and leucine-rich repeat (LRR) disease resistance proteins in legumes [15, 16] and Solanaceae [17, 18]. Some disease resistance gene-derived phasiRNAs have been shown to down-regulate the expression of genes in the same family [19]. Besides NBS-LRR disease resistance proteins, many other types of protein-coding genes have been identified as PHAS loci in plants, such as MYB transcription factors [16, 20, 21], pentatricopeptide repeat (PPR) genes [16, 20, 22, 23], transporter inhibitor response (TIR)/auxin F-box genes (AFB) [15, 22, 24, 25], and calcium ATPase transporters [23, 26].
These findings suggest potentially broad roles of phasiRNAs in plant evolution and function, but many fundamental questions remain outstanding: (1) How widespread are PHAS loci in the plant kingdom? (2) How many types of protein-coding genes can function as PHAS loci in the plant kingdom? (3) How conserved or unique are these PHAS loci in different plants? (4) What categories of genes do phasiRNAs regulate? and (5) What is the broad spectrum of biological processes that may be associated with or regulated by phasiRNA biogenesis? To address these questions, we performed a systematic search of PHAS loci in 23 plant species covering major phylogenetic groups including alga, moss, gymnosperm, basal angiosperm, monocot, and dicot as shown in Fig. 1. Our study uncovered a wide range of novel PHAS loci from protein-coding genes. Intriguingly, many phasiRNAs appear to be derived from introns or intron-exon junctions. While some PHAS loci occur broadly in plants, many are found in a limited number of species. Production of phasiRNAs from protein-coding genes is associated with developmental processes, plant–virus interaction, and abiotic stresses, demonstrating the highly dynamic nature of phasiRNA biogenesis regulation. Our data provide the most comprehensive kingdom-wide landscape to date of phasiRNA distribution and potential regulatory function in plants, which will serve as an important foundation for developing mechanistic studies on the evolution, biogenesis, and function of these novel siRNAs underlying many aspects of plant biology.
Fig. 1.
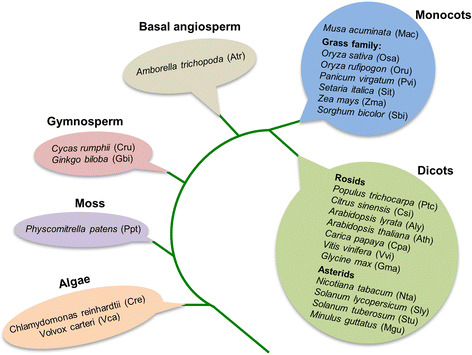
Plant species investigated in this study. These species belong to major groups with well-established phylogeny. Three-letter abbreviations of these species, shown in brackets, were used in the present study
Results and discussion
A broad search for PHAS loci in different plant species
We collected deep small RNA (sRNA) sequences of 23 plant species spanning from green algae to angiosperm groups from public resources (Fig. 1). These sRNAs were generated from organs or tissues at different developmental stages or under various growth conditions. The majority of the libraries contained 1–5 million high-quality cleaned sRNA sequences [27–33] (Additional file 1). To identify phasiRNA-generating loci, we aligned the sRNA sequences to the corresponding genome and cDNA reference sequences (Additional file 2). Based on the alignments, we identified PHAS loci from the 23 species using the method described in Xia et al. [23] (see Methods for details). The phasiRNA-generating loci mapped to the same protein-coding gene were further combined to represent a unique locus.
In total, we found 3,339 PHAS loci in all the 23 species, including 3,306 that generated 21-nucleotides (nt) and 33 that generated 24-nt phasiRNAs (Table 1). The 24-nt phasiRNA-generating loci were found exclusively in the grass family (Oryza rufipogon, Setaria italica, and Sorghum bicolor), and all of them were mapped to intergenic regions (Additional file 3). The number of 21-nt phasiRNA-generating loci in each plant group or species varied greatly (Additional file 4: Figure S1). Dicot species tended to have large numbers of PHAS loci, with many species having more than 100. In addition, approximately 75 % (1,122 out of 1,511 as listed in Table 1) of the PHAS loci mapped to annotated genic regions in all tested dicot species. The number of PHAS loci in monocot plants ranged from 13 to more than 900. In contrast to those in dicots, significantly fewer PHAS loci were mapped to genic regions in monocots. In a basal angiosperm species, Amborella trichopoda, there were 67 phasiRNA-generating loci, comparable to that in many dicot plants. In moss, Physcomitrella patens, there were four PHAS loci. In algae, Chlamydomonas reinhardtii had a comparable number of PHAS loci as in many higher plants whereas Volvox carteri generated phasiRNAs from only six loci. The complete list of 21-nt phasiRNA-generating loci is provided in Additional file 5. All the PHAS loci identified in the present study are stored in an interactive online database we developed [34]. The database provides a set of query interfaces and tools to analyze, visualize, and mine the phasiRNA data presented in this study.
Table 1.
Summary of PHAS loci identified from various plant species
| Species | Protein-coding gene | Intergenic | Total | |||
|---|---|---|---|---|---|---|
| Exon | Intron | Exon-intron | Subtotal | |||
| 21-nt phasiRNA generating loci | ||||||
| Populus trichocarpa (poplar) | 125 | 3 | 9 | 137 | 12 | 149 |
| Citrus sinensis (sweet orange) | 233 | 1 | 25 | 259 | 97 | 356 |
| Arabidopsis lyrata | 27 | 1 | 4 | 32 | 19 | 51 |
| Arabidopsis thaliana | 33 | 0 | 0 | 33 | 21 | 54 |
| Carica papaya (papaya) | 104 | 3 | 15 | 122 | 33 | 155 |
| Vitis vinifera (grape) | 132 | 24 | 77 | 233 | 123 | 356 |
| Nicotiana tabacum (tobacco) | 15 | 1 | 4 | 20 | 6 | 26 |
| Solanum tuberosum (potato) | 146 | 1 | 9 | 156 | 45 | 201 |
| Solanum lycopersicum (tomato) | 87 | 9 | 16 | 112 | 11 | 123 |
| Mimulus guttatus (monkey flower) | 16 | 0 | 2 | 18 | 22 | 40 |
| Musa acuminata (banana) | 18 | 0 | 5 | 23 | 6 | 29 |
| Oryza sativa ssp. japonica (rice) | 25 | 4 | 4 | 33 | 23 | 56 |
| Oryza sativa ssp. indica (rice) | 6 | 2 | 2 | 10 | 6 | 16 |
| Oryza rufipogon (wild rice) | 5 | 3 | 1 | 9 | 4 | 13 |
| Panicum virgatum (switchgrass) | 10 | 0 | 0 | 10 | 14 | 24 |
| Setaria italica (foxtail millet) | 16 | 1 | 1 | 18 | 898 | 916 |
| Zea mays (maize) | 21 | 0 | 1 | 22 | 239 | 261 |
| Sorghum bicolor (sorghum) | 40 | 2 | 6 | 48 | 19 | 67 |
| Amborella trichopoda | 19 | 2 | 2 | 23 | 42 | 65 |
| Cycas rumphii | 12 | 0 | 0 | 12 | 1 | 13 |
| Ginkgo biloba | 25 | 0 | 0 | 25 | 0 | 25 |
| Physcomitrella patens | 0 | 0 | 0 | 0 | 4 | 4 |
| Volvox carteri | 5 | 0 | 2 | 7 | 20 | 27 |
| Chlamydomonas reinhardtii | 126 | 34 | 49 | 209 | 70 | 279 |
| 24-nt phasiRNA generating loci | ||||||
| Oryza rufipogon (wild rice) | 0 | 0 | 0 | 0 | 7 | 7 |
| Setaria italica (foxtail millet) | 0 | 0 | 0 | 0 | 23 | 23 |
| Sorghum bicolor (sorghum) | 0 | 0 | 0 | 0 | 3 | 3 |
Conserved and unique PHAS loci from non-coding DNA sequences
The non-coding TAS loci (TAS1, TAS2, TAS3, and TAS4) were first identified in Arabidopsis thaliana. miR173 is responsible for triggering TAS1 and TAS2, which seems to be unique in A. thaliana [11, 35]. In contrast, TAS3 and its cognate trigger miR390 were found from moss to most of the land plants examined [12]. Our searches in 23 plant species confirmed that TAS1 was unique to A. thaliana, TAS2 could only be found in A. thaliana and A. lyrata, and TAS3 was present in many land plant species.
The taxonomic distribution of TAS4 and its cognate trigger miR828 was enigmatic. Previous studies postulated that the miR828–TAS4 regulatory circuit might exist only in Arabidopsis thaliana and a few closely related species, but the possibility of a wider distribution was proposed based on the presence of miR828 and/or TAS4 homologs in several other plant genomes [35]. Our searches identified bona fide TAS4 loci based on the production of phasiRNAs in several dicot species, including poplar (Populus trichocarpa), sweet orange (Citrus sinensis), grape (Vitis vinifera), tomato (Solanum lycopersicum), tobacco (Nicotiana tabacum), potato (S. tuberosum), and monkey flower (Mimulus guttatus) (Additional file 6). The miR828–TAS4 regulatory circuit was suggested to play a role in trichome development in A. thaliana [36] and apple [20], fiber growth in cotton [36], flavonoid biogenesis in A. thaliana [37], and perhaps fruit development in tomato [38]. In line with previous reports [35], we did not detect the presence of miR828 or TAS4-derived phasiRNAs in any of the tested monocot species. Interestingly, although the MIR828 gene could be found in the genomes of all test species, its reads were only found in poplar and grape sRNA libraries (both have miR828 reads below 5 RPM) (Additional file 6), in contrast to the detection of highly abundant miR828 in cotton [36]. It is possible that the miR828 level was too low to be detected in sRNA libraries from many plant species investigated in this study.
Intriguingly, we found a miR828 homolog in the basal angiosperm Amborella trichopoda (Additional file 4: Figure S2A) based on the homology of mature miRNA sequences. This miRNA was predicted to trigger phasiRNA production from an intergenic region. It is noteworthy that phasiRNA production triggered by Atr-miR828 in Amborella trichopoda did not yield the conserved D4(−) sequence (Additional file 4: Figure S2B) that was found in Arabidopsis thaliana and many other species [35]. Our current data are insufficient to draw a conclusion about the phylogenetic relationship between the miR828–PHAS regulatory circuit in Amborella trichopoda and that in dicots, but raise the interesting possibility that the Amborella trichopoda circuit was a prototype that has stabilized and been refined in dicots but been lost in monocots during evolution.
Our searches uncovered many PHAS loci from noncoding sequences from various species (Additional files 3 and 5) and we did not analyze them in further detail. Our subsequent analyses were devoted to PHAS loci derived from protein-coding genes.
An expansive and diverse repertoire of PHAS loci originated from protein-coding genes
Our analyses identified approximately 1,600 protein-coding genes, belonging to 217 categories (Additional file 7) based on their functional annotations, that serve as PHAS loci in at least one species, significantly expanding the list of 119 currently identified gene categories that can produce phasiRNAs (Additional file 8). These PHAS loci cover a wide range of genes, which include those encoding disease resistance and wound-response proteins, hormone response factors, transcription factors, RNA silencing components, proteins involved in signal transduction, transporters, protein translation machinery components, photosystem components, histone and DNA methylation proteins, the cytoskeleton and associated factors, intracellular trafficking machinery, kinases, and other enzymes involved in diverse metabolic pathways. (Additional file 7). Transcription factors and kinases were also found to function as PHAS loci in soybean [16].
When the total number of PHAS loci from leaves of land plants was compared, some interesting patterns emerged (Additional file 4: Figure S3 and Additional file 9). The eudicots and basal angiosperm Amborella trichopoda had relatively higher numbers of PHAS loci than monocots. Among eudicots, sweet orange had the largest number of PHAS categories from the annotated gene loci in leaves. Grapevine and poplar also had a large number of PHAS loci from annotated gene loci in leaves. Closely related species may have different protein-coding PHAS loci enriched. For example, PPR-containing protein genes were the dominant PHAS loci in Arabidopsis thaliana leaves, whereas TIR-NBS-LRR class disease resistance genes were the most enriched in Arabidopsis lyrata leaves.
Disease resistance genes, including NBS-LRR genes, constitute the most conserved group of PHAS loci, confirming and expanding previous reports [15, 16, 23]. Our extensive search found phasiRNAs produced from NBS-LRR genes in gymnosperms, the basal angiosperm Amborella trichopoda and monocots, although much less than in dicots (Additional files 5 and 7). Thus, disease resistance genes evolved as PHAS loci in early seed plants but were strongly selected for in dicot plants. Expression of R genes constitutes a high cost for plants [39]. Infection of several viruses and a bacterium which encode silencing suppressors led to down-regulated expression of miRNAs and siRNAs that trigger phasiRNAs from R genes [18], leading to the hypothesis that repressed expression of R genes by phasiRNAs under non-infection conditions functions to minimize the costs for plants [17, 18] and that viral or bacterial suppressor-induced expression of R genes enables or enhances plant defense responses during infection [18]. The wide taxonomic distribution of disease resistance genes as PHAS loci in our analysis lends further support to this hypothesis.
While some PHAS loci, such as disease resistance genes and Myb domain-containing genes, were widespread among the plants we examined, most loci were found in only a few species or just one single species. For example, a squamosal promoter-binding protein gene served as a PHAS locus only in grapevine though this gene family is a conserved target of miR156 in many plants. The stochastic occurrence of many PHAS loci in the plant kingdom suggests their young evolutionary origins and the existence of yet-to-be identified biogenesis factors. This, together with the diversity of protein-coding genes acting as PHAS loci, suggests phasiRNAs as the fastest-evolving riboregulators to regulate the expression of continuously expanding numbers and types of protein-coding genes. As such, this regulation may be an important contributor to the diversification of certain plant phenotypes.
Broad taxonomic distribution of RNA silencing machinery components as PHAS loci
PhasiRNAs can be generated from RNA silencing machinery components, including DICER-LIKE (DCL)s and SUPPRESSOR OF GENE SILENCING 3 (SGS3) in Medicago truncatula [15], peach [23], and soybean [16], as well as ARGONAUTE2 (AGO2) in peach [23], and soybean [16]. We found SGS3 to be a PHAS locus in leaf and cone samples of the two gymnosperm species examined (Cycas rumphii and Gingko biloba) and in leaf and fruit samples of grape. Interestingly, Arabidopsis thaliana SGS3 did not produce any siRNAs. We also observed DCL-derived 21-nt phasiRNAs from papaya, sweet orange, tomato, and tobacco, AGO-derived 21-nt phasiRNAs from A. thaliana, A. lyrata, monkey flower, tomato and sorghum, and RNA-DEPENDENT RNA POLYMERASE (RDR)-derived 21-nt phasiRNAs from foxtail millet and Amborella trichopoda. These new observations, together with earlier reports [15, 16, 23], support the notion that phasiRNAs generated from RNA silencing machinery genes may function in a feedback mechanism to regulate the expression of these genes [15], and that this mechanism may be widespread in plants.
Novel biogenesis of phasiRNAs from introns or exon-intron junctions
The noncoding TAS2 locus in Arabidopsis thaliana produces siRNAs from an alternative transcript that retained an intron [40]. In the present study, we found a large number of protein-coding genes that generate phasiRNAs from annotated introns alone (Additional file 10) or intron–exon junctions (Additional file 11) in many plant species. For example, we found that phasiRNA production from the SGS3 locus in grape progressed from an exon into an adjacent intron (Additional file 4: Figure S4A). In addition, a serine/threonine protein kinase gene produced phasiRNAs only from its second intron (Additional file 4: Figure S4B). In total, we identified 107 PHAS loci producing phasiRNAs only from annotated introns and 157 loci producing phasiRNAs from intron–exon junctions. To validate intron annotations, we used GBrowser in the Phytozome [41], Solgenomics [42], and Amborella genome [43] databases to examine expressed sequence tag and/or RNA-sequencing (RNA-Seq) data for these PHAS loci. The data validated annotations for approximately one third (34 out of 107) of the intron-only and one fourth (37 out of 157) of the intron–exon junction PHAS loci (Additional files 10 and 11). Intriguingly, the majority of PHAS loci that generated phasiRNAs from introns or intron–exon junctions were disease resistance genes (Additional file 4: Figure S5, and Additional files 10 and 11).
An outstanding question is whether phasiRNAs from the intron-only PHAS loci are generated from free-standing introns or from alternatively spliced or aberrant transcripts. We addressed this question by analyzing the published tomato dataset that contains RNA-seq and sRNA data from the same samples [44]. We found three annotated intron regions that generated phasiRNAs but had no RNA-seq coverage, indicating that they are bona fide introns. An example is shown in Fig. 2a. We also found 11 annotated intron regions, which generated phasiRNAs with good RNA-seq coverage, indicating that they are retained in the alternatively spliced transcripts (an example is shown in Fig. 2b). These data provide experimental evidence that free-standing introns or introns retained in the alternatively spliced transcripts can indeed be the direct sources of phasiRNAs.
Fig. 2.
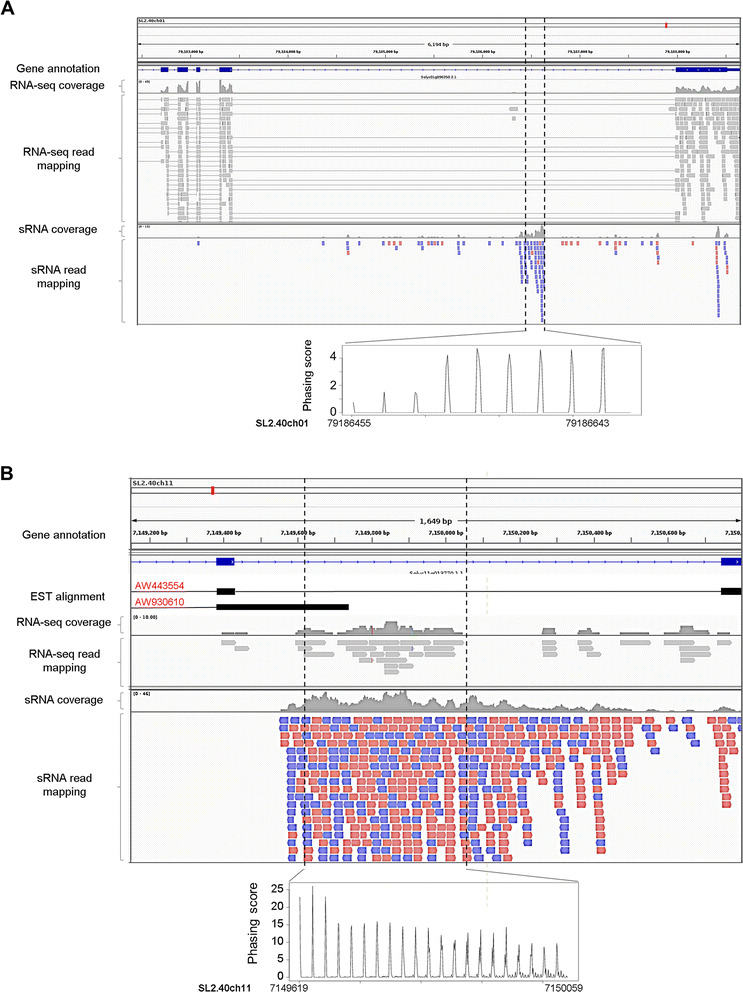
PhasiRNAs generated from introns in tomato. Examples of a PHAS locus (Sly015) that generates phasiRNAs from a bona fide intron (a) and a PHAS locus (Sly110) that generates phasiRNAs from an alternatively retained intron (b). RNA-seq and sRNA reads were derived from the same organ (fruit at the breaker stage or 42 days after pollination) and mapped to the tomato genome. For sRNA mapping, red depicts reads aligned in forward orientation and blue in reverse orientation. EST expressed sequence tag
It remains unclear whether the phasiRNAs from free-standing introns are generated via the conventional phasiRNA biogenesis pathway where a miRNA trigger functions in the cytoplasm or via a novel pathway operating in the nucleus. Regardless of the specific pathway(s), our findings revealed an unexpected link among mRNA splicing, mRNA stability, and phasiRNA regulation. This might represent a new mechanism of removing aberrant (i.e., unspliced or incorrectly spliced) RNA transcripts that escape the nucleus to accumulate in the cytoplasm. Alternatively, phasiRNA biogenesis from introns and/or intron–exon junctions of certain loci may function to regulate pre-mRNA stability or other aspects of gene expression.
Unique developmental regulation of PHAS expression in different plants
Comprehensive analysis of phasiRNAs in soybean identified a number of tissue-specific PHAS loci [16]. Most plant species we analyzed had sRNA libraries prepared from different organs and/or from various developmental stages, thereby allowing examination of the developmental significance of phasiRNA generation among different taxonomic groups. It is worth noting that sRNA libraries we used for comparative analysis for a given plant were all derived from the same genotype and generated by the same laboratory, and have been used to explore the widespread conservation and divergence of microRNAs in plants [33]. Our analyses showed that many protein-coding genes that acted as PHAS loci in one organ did not produce any siRNAs or produced only abundant non-phased siRNAs in another organ(s) of the same species (Additional file 12). For example, genes encoding NAC domain-containing transcription factors were an obvious cluster in producing phasiRNAs in fruits but much less in leaves and flowers of sweet orange (Additional file 4: Figure S6), implying their unique roles in regulating sweet orange fruit development.
All tested monocot plants possessed very few loci for generating 21-nt phasiRNAs in non-reproductive organs such as leaf, shoot, and root. For species in the grass family, there was a drastic induction of the PHAS loci in certain reproductive organs, such as the flower of foxtail millet, ear and tassel of maize (Zea mays), and panicle of sorghum. Furthermore, most of these PHAS loci were in the intergenic regions as shown in Fig. 3 and Additional file 12.
Fig. 3.
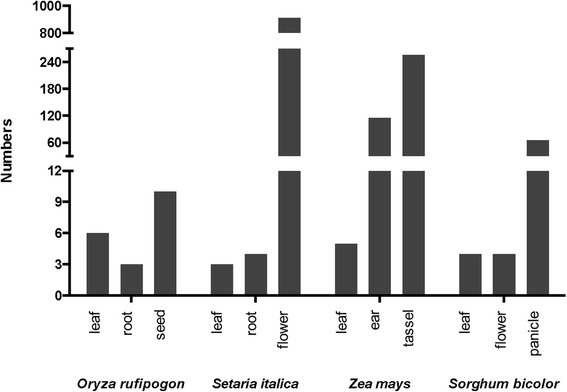
Number of PHAS loci identified in different organs of representative species in the grass family
Notably, wild rice had five PHAS loci producing phasiRNAs only in the seeds, but no siRNAs in the shoots or roots. Among these five loci, a phosphoinositide phosphatase family gene and a nuclear factor Y gene produced 20-fold more phasiRNAs than those generated by a deeply conserved noncoding PHAS (TAS3). Thus, the regulatory roles of these two loci may help us understand the function of protein-coding transcript-derived phasiRNAs in cereal development.
These observations, together with the findings from Arikit et al. [16], indicate that PHAS-based gene regulations are unique in different organs in a wide range of plants. Interestingly, organ-specific PHAS loci did not exhibit any general conservation among different plant species. Whether this may contribute to the development of distinct phenotypes in different plants remains an outstanding question.
It should be noted that the datasets we have analyzed for land plants were mostly derived from whole plant organs. A recent study reported phasiRNAs in maize anther and pollen at different developmental stages, providing deeper insights into phasiRNA biogenesis at the tissue-specific level [45]. Thus, future studies to analyze tissue- or even cell-specific expression patterns of phasiRNAs in different plants should shed new light on the functions of phasiRNAs in plant development processes.
Viral infection induces or suppresses a diverse array of PHAS loci
Some miRNAs act as master regulators to initiate the generation of phasiRNAs from mRNAs that encode NBS-LRR disease resistance proteins in legumes [15, 16] and solanaceous plants [17, 18]. Viral and bacterial infection represses the expression of miRNA triggers and consequently also phasiRNAs from some resistance gene loci [17, 18]. To gain further insight into the potential role of phasiRNAs in plant–microbe interactions, we analyzed the global expression patterns of phasiRNAs in several virus–plant systems. Specifically, we examined phasiRNA profiles in healthy and Papaya ringspot virus (PRSV)-infected leaves of papaya (Carica papaya) [33], healthy and Turnip mosaic virus-infected leaves of Arabidopsis thaliana [29], as well as healthy and Rice stripe virus (RSV)- and Rice dwarf virus (RDV)-infected shoots of rice (O. sativa L. ssp. Japonica) [31] (see Additional file 1 for the source of sRNA data).
In the papaya system, 40 and 93 PHAS loci were found in healthy and infected leaf libraries, respectively; among them, 13 were shared while many exhibited different expression patterns (Additional file 13). Six disease resistance PHAS loci showed reduced production of 21-nt siRNAs upon PRSV infection (13, 24, 33, 36, 62, and 87 %, respectively, of that in the healthy leaves). Five of them became non-PHAS loci in the infected leaves (Fig. 4b and Additional file 13). Furthermore, three out of four PHAS loci derived from auxin-signaling genes in the healthy leaves also became non-PHAS loci in the infected leaves: ARF3 had the highest degree of total siRNA reduction in infected leaves and two auxin-signaling F-box genes (AFB2 and TIR1/AFB) produced four times less siRNAs in infected leaves (Fig. 4c and Additional file 13).
Fig. 4.
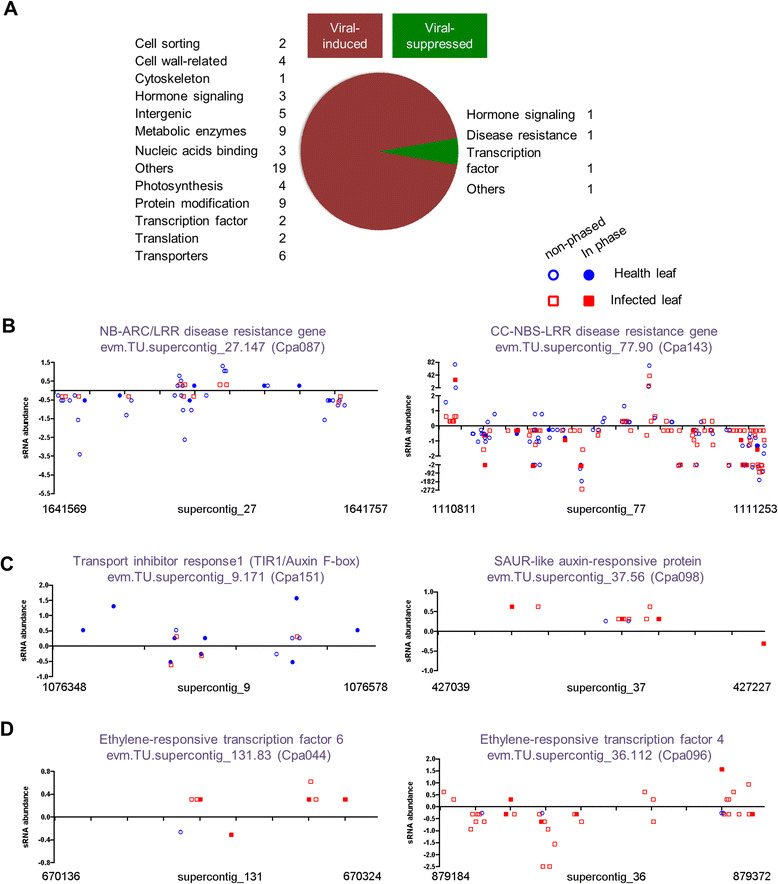
siRNA production from various PHAS loci upon viral infection. a Pie diagram showing the PHAS loci with more than five-fold changes in total 21-nt siRNA production in papaya leaves infected by Papaya ringspot virus as compared to that in healthy leaves. The number of PHAS locus or loci for each gene category showing induced or suppressed expression is indicated. b–d Specific examples of PHAS loci derived from disease-resistant genes (b), auxin-related genes (c), and ethylene-responsive transcription factor genes (d), which showed differential expression of siRNAs in healthy and infected leaves
Similarly in the rice and Arabidopsis systems, a large number of PHAS loci showed substantial changes in phasiRNA generation upon infection by different viruses (Additional file 13). It is noteworthy that an auxin response factor (ARF) gene in rice showed more than 8- and 3.5-fold changes in phasiRNA generation upon RSV and RDV infections, respectively.
While the repressed expression of some PHAS loci in the infected leaves was consistent with earlier reports in Solanaceae and Fabaceae [15–18], our analyses identified many novel PHAS loci induced by PRSV infection. These could be clustered into several groups based on their gene functions: cytoskeleton-related genes, chloroplast-related genes, cell wall-related genes, transporter genes, and hormone-signaling genes (Additional file 13). As an example, a SAUR-like ARF increased siRNA production by more than eight-fold and became a PHAS locus in infected leaves (Fig. 4c). Furthermore, two ethylene-responsive transcription factor genes were highly induced as PHAS loci in response to viral infection (Fig. 4d). Induced expression of these PHAS loci may function as part of the coordinated plant defense responses, by reducing metabolic activities to dampen viral replication. Many studies have reported up- or down-regulated expression of host genes during viral infection, with many types of genes appearing to be commonly affected in different host–virus systems [46–50]. The underlying molecular mechanisms are largely unknown. We suggest that viral infection-induced or suppressed expression of selective phasiRNAs may act as a novel mechanism to regulate the expression of selective host genes.
Nutrient stresses modulate phasiRNA biogenesis in algae
Previous reports showed the presence of phasiRNAs in unicellular alga C. reinhardtii, mainly mapped to intergenic regions [51, 52]. In the present study, we identified more than 300 PHAS loci in this alga, of which approximately 240 were mapped to protein-coding genes. Among the protein-coding PHAS loci, nine were uniquely induced by sulfate starvation, five were uniquely induced by phosphate starvation, and one was induced by both treatments. On the other hand, six protein-coding PHAS loci were suppressed by both treatments, and four were uniquely suppressed by sulfate treatment (Additional file 4: Figure S7 and Additional file 14). It is noteworthy that there was no unique PHAS locus suppressed by phosphate starvation.
The biological implications of these changes in PHAS activities remain to be understood, but the suppressed expression of transposon-polyprotein loci is of notable interest. In C. reinhardtii, a DCL1 homolog has been shown to repress retrotransposon activity [53]. However, the underlying molecular mechanism is unknown. In the case of REM1 Ty3-gypsy retrotransposon silencing, retrotransposon-derived siRNAs were postulated to be derived from double-stranded RNAs formed via a transposon polyprotein gene and its adjacent inverse transcripts [54]. However, we did not find evidence for the existence of such adjacent inverse transcripts that would contribute to the production of any siRNAs. Thus, phasiRNA-mediated retrotransposon silencing by phasiRNAs derived from the coding transcripts within the retrotransposon itself may be a novel mechanism underlying the retrotransposon silencing phenomenon in C. reinhardtii.
The multicellular alga V. carteri had much fewer PHAS loci. We found only one protein-coding PHAS locus in this alga growing under normal conditions. In contrast, five and one new protein-coding PHAS loci emerged under sulfate and phosphate starvation conditions, respectively.
Our analysis suggested that algae can respond to different abiotic stresses by differential gene expression modulated by phasiRNA biogenesis. In soybean, drought stress induced the expression of phasiRNAs from a protein-coding gene, ALLENE OXIDE SYNTHASE [16]. Future studies will determine whether dynamic changes in phasiRNA biogenesis may occur broadly in land plants in response to nutrient and other abiotic stresses.
sRNA triggers for phasiRNA biogenesis
We predicted sRNA triggers for PHAS loci mainly based on sequence complementarity (see Methods for details). We noticed that a large number of predicted triggers have a putative guided cleavage site falling out of the N × 21 bp phase windows (where N is an integer) to the positions of phasiRNAs. We discarded these out-of-phase triggers if they were not confirmed previously by other studies or not supported by the sPARTA [55] analysis described below. In total we could predict triggers for approximately 10 % of the PHAS loci we identified in this study (Additional files 15 and 16). Among these, we could find 170 miRNA triggers, among which 135 were known miRNAs (with 32 distinct sequences) and 35 putative miRNAs (with 21 distinct sequences) (Additional file 16). There are six species with available sequencing data from degradome libraries from similar sample sources/organs (Additional file 1). We used these data to test our trigger prediction with sPARTA [55]. The results are summarized in Additional file 16. For 41 triggers (4 in Arabidopsis thaliana, 4 in rice, 16 in tomato, 10 in grape, and 7 in Amborella trichopoda), the predicted cleavage sites were validated by degradome libraries. We could not validate any in P. patens, which may be attributed to the different sources for degradome and sRNA libraries. Further experimental studies are necessary to validate the remaining triggers.
sRNA triggers may work via the two-hit model (221), in which two sites of the transcript are recognized by the same sRNA [11] or two different sRNAs [13, 15, 56], or via the one-hit model (122), in which a single 22-nt sRNA directs the cleavage of the target [6, 15, 17, 18, 26]. The numbers for the predicted 21-nt and 22-nt sRNA triggers at 5′- and 3′-ends are summarized in Additional file 15. There were many cases in which we only found one 21-nt trigger at either 5′- or 3′-end, which may have been due to the limit of the algorithm in identifying imperfect alignments of sRNAs and targets.
Disease resistance genes are targeted by the miR482/miR2118 superfamily in generating phasiRNAs [18]. Other miRNA triggers, such as miR6024, miR6025, miR6027, and miR472, have also been reported [17, 19]. We found these miRNAs as triggers in various species. In addition, we predicted that miR3623 in grape was also involved in regulating phasiRNA production from disease resistance genes. Whether this suggests parallel evolution of species-specific regulators or suggests miR3623 as a relative or member of the miR482/miR2118 superfamily awaits further analysis. For NAC domain transcription factors, the predicted trigger was miR7122 in sweet orange but miR6445 in poplar. The evolutionary relationship between the MIR7122 gene and MIR6445 gene also remains unknown.
These observations show that an orthologous protein-coding gene in different plant species may be targeted by different miRNAs to produce phasiRNAs. However, because various miRNA triggers for phasiRNA biogenesis may have their own distinct expression patterns, an orthologous protein-coding gene may serve as a PHAS locus in distinct spatial-temporal manners in various species. For example, NAC domain transcription factor genes were enriched PHAS loci in the fruit sample in sweet orange and in the xylem sample from wood trunk in poplar. These observations reveal the highly complex evolutionary and functional regulatory networks of miRNAs and phasiRNAs.
Predicted target genes of phasiRNAs
We also predicted the downstream targets for phasiRNAs present with more than 10 RPM in a given library (Additional file 17). It was evident that nearly all phasiRNAs had near-perfect matches to the genes in the same family as their originating loci. Interestingly, a recent report by Boccara et al. [19] showed that phasiRNAs derived from disease resistance genes negatively regulate a broad spectrum of disease resistance genes in Arabidopsis thaliana.
Our analyses identified some phasiRNAs that may target genes outside the gene families of their originating loci. In most species, such potentially trans-regulatory phasiRNAs accounted for only a small percentage of the total phasiRNAs, generally less than 10 % with exceptions when a stringent cut-off score (1.5) was used for target prediction. Thus, in contrast to the dominant trans-regulatory mode of miRNAs [6], our analysis suggests that phasiRNAs predominantly regulate protein-coding genes from which they are derived and genes in the same families of phasiRNA-generating genes. As discussed earlier, many PHAS loci may be of recent evolutionary origin given their stochastic occurrence in plants, superficially similar to the origin of young miRNAs. However, unlike the majority of young miRNAs that do not have predicted target genes and are therefore likely function-neutral [6], phasiRNAs are all likely functional because they may target at least the genes from which they are originated.
Evolution of the phasiRNA biogenesis machinery
The stochastic presence of many PHAS loci in the species we analyzed raised questions concerning the conservation of their biogenesis machinery. Studies on the model plant Arabidopsis thaliana demonstrated that the generation of 21-nt phasiRNAs involves the coordinated actions of protein factors DCL4, AGO7, RDR6, and SGS3, in addition to the core players for miRNA biogenesis and function such as DCL1 and AGO1 [7]. Assuming this is an essential set of proteins for phasiRNA biogenesis in any plant, we determined their presence in different plants by searching the available genome sequences of 41 species, covering algae, mosses, monocots, and dicots. Gymnosperm plants were not included for lack of full genome sequences.
Our extensive search revealed that all proteins currently known to be specifically required for phasiRNA biogenesis were present in all of the tested land plants, from moss to higher plants. None of the algae species, single cellular or multicellular, had the complete set of SGS3, RDRs, DCLs, and AGOs. Among algae, only C. reinhardtii and V. carteri had dicer-like protein factors as shown in Fig. 5. Our search results match previous reports in several plant species including algae species [57], rice [58], maize [59], and tomato [60].
Fig. 5.
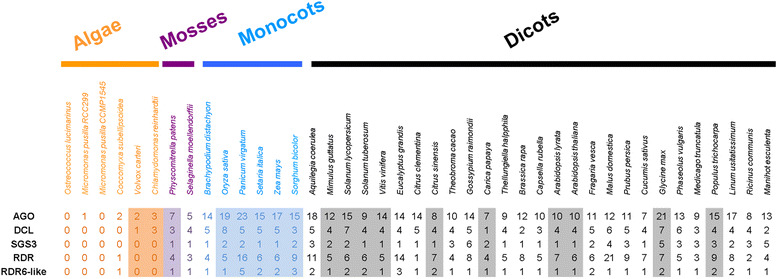
Summary of key protein factors involved in phasiRNA biogenesis in the plant kingdom. The color shades depict the species that were investigated for phasiRNA production in this study. The numbers listed indicate the copy numbers of the corresponding phasiRNA biogenesis genes
Thus, our results suggest that the currently known phasiRNA biogenesis pathway(s) has evolved only in land plants. These observations raise questions for future investigations: (1) what factors contribute to the stochastic evolution and activity of PHAS loci in different plant species, different developmental stages, and under different biotic and abiotic stress conditions; and (2) what machinery is responsible for the biogenesis of phasiRNAs in algae?
Conclusions
Our extensive search across 23 different plant species uncovered a large and surprisingly diverse repertoire of protein-coding genes as novel PHAS loci. Biogenesis of phasiRNAs from many loci is uniquely associated with speciation, development, viral infection, or nutrient stresses. Numerous PHAS loci produced phasiRNAs from intron or exon–intron junction regions, revealing an unexpected link between phasiRNA biogenesis and RNA splicing. Our analysis suggests that phasiRNAs predominantly regulate protein-coding genes from which they are derived and genes in the same families of phasiRNA-generating genes, in contrast to the dominant trans-regulatory mode of miRNAs. Interestingly, while dicot PHAS loci appear to play an overwhelming role in disease resistance, monocot PHAS loci are mostly associated with reproductive growth.
The PHAS loci derived from protein-coding genes may be considered an evolutionary novelty in that these PHAS loci possess an added regulatory function specific to some plant species rather than a replacement of the original function of protein-coding genes for these loci. The stochastic occurrence of PHAS loci in the plant kingdom suggests their young evolutionary origins. The evolutionary flux that leads to high variations for a given locus among species or taxonomic groups suggests that phasiRNA networks may contribute to plant phenotype diversity by regulating distinct developmental processes or responses to biotic or abiotic stimuli.
In summary, our new findings, together with an interactive database developed, significantly expand previous findings and will help build a critical foundation to advance further studies on how phasiRNAs may have emerged to participate in the complex regulatory networks to shape the evolution of plant phenotype diversity.
Methods
sRNA sequence processing and alignment
sRNA deep sequencing data were downloaded from the Comparative Sequencing of Plant Small RNAs website [61] and the GenBank Gene Expression Omnibus [62] (Additional file 1). All sRNA deep sequencing data were treated via standard procedures for format conversion, adaptor trimming, and read collapsing. The treated sRNA sequences were further cleaned by removing sequences that matched rRNA sequences using Bowtie [63]. The cleaned sRNA sequences were mapped to the corresponding reference genome or transcriptome sequences (Additional file 2) using Bowtie [63], allowing no mismatches and no more than six hits to the reference sequences.
Identification of candidate PHAS loci
We largely followed the methods developed by Xia et al. [23] for PHAS locus identification. Specifically, the mapped sRNA reads were denoted according to their positions in the corresponding reference genome and/or cDNA sequences. For matching sRNAs to the antisense strand of the reference sequences, a two-nucleotide positive offset was included to mimic the 3′ end overhang. A search was conducted by scanning transcriptome or genome references using a nine-phase register (21 bp as a phase register) sliding window (189 bp), each followed by a break of three-phase register (63 bp). A positive window was considered to contain no less than 10 unique reads, with more than half of the unique reads being 21 nt in length and with no less than three 21-nt unique reads falling into the phase registers. Windows were combined if they shared the same phase registers (i.e., the adjacent windows had a space of N × 21 bp, where N is an integer) and fell into the same genes.
Calculation of P-values for positive windows was based on the formula:
P-value: p(k) = ∑mX − k Pr(X)
where n was the number of total unique 21-nt sRNAs matched within a window, k was the maximum number of unique 21-nt reads falling into one of the possible phase registers, and m was the number of 21-nt phases within a window. The 21-nt PHAS loci with a P-value of less than 0.001 were identified as the positive PHAS loci.
Phasing score was calculated following the methods described in de Paoli et al. [64] and Xia et al. [23] using the following formula:
where n was the number of phase registers occupied by at least one unique 21-nt sRNA within a nine-phase register window, P was the total number of reads for all 21-nt sRNA reads falling into a given phase in a given window, and U was the total number of unique reads for all 21-nt sRNAs falling out of a given phase.
The same formulas were applied to the 24-nt PHAS windows in all tested libraries, with a change of the phase register length from 21 nt to 24 nt. We used a cut-off phasing-score of 15 to identify positive 24-nt PHAS loci according to Johnson et al. [65].
Prediction of PHAS triggers
PhasiRNAs are mainly triggered by 21-nt and/or 22-nt miRNA-mediated transcript cleavage in land plants. Occasionally, siRNAs could also serve as triggers. sPARTA [55] was used to search for the 21-nt/22-nt miRNA/siRNA triggers with relatively high abundance (>10 RPM in a given library) that potentially target 200 bp upstream and 200 bp downstream of a PHAS window with an alignment score cutoff of 4. The positive candidates were subject to Vienna RNA package [66] analysis to predict the miRNAs. The predicted miRNAs were then aligned with miRBase [67] registers (release 19) to unveil conserved miRNAs. sPARTA [55] was also employed to validate the miRNA-guided cleavage of transcripts from the cognate PHAS loci using the available degradome library data (Additional file 1). We removed the out-of-phase triggers that were not previously confirmed or were not supported by our sPARTA analysis.
Prediction of phasiRNA targets
Target prediction for phasiRNAs was performed with sPARTA [55]. Twenty-one–nucleotide phasiRNAs with an abundance of no less than 10 RPM were used to predict their downstream targets. The alignment score cutoff was set to 3.
Identification of protein factors responsible for phasiRNA biogenesis in plants
We followed the methods described in previous studies [58–60] with minor modifications to mine protein factors responsible for phasiRNA biogenesis in the genomes of 41 plant species. Briefly, the published amino acid sequences of each gene family from Arabidopsis thaliana were used to search against a plant genome database [41] using BLAST with an E-value cutoff of 1e-10. The candidates were examined for their functional domains, with those missing the essential domains removed. Specifically, for DCL homologs, we searched for candidates having high amino acid sequence homology (>50 % similarity) to the known DCLs and containing at least four of the six characteristic domains: DEAD, PAZ, Helicase-C, Duf283, RNase III, and double-stranded RNA-binding (DRB). We excluded those that do not have RNase III, PAZ, or Helicase-C domains. For AGO candidates, we looked for those having high sequence similarity (>50 %) to known AGOs and all three functional domains: DUF1785, PIWI, and PAZ. For RDRs in A. thaliana, RDR6 or RDR2 share little sequence similarity to RDR1, RDR3, RDR4, and RDR5, so we used RDR1, RDR2, and RDR6 individually as baits to search for candidates that shared high amino acid sequence homology and contained an RDR functional domain. For SGS3, we searched for those candidates having high amino acid sequence homology (>50 % similarity) to A. thaliana SGS3 and containing only XS zinc finger and XS domains.
Additional files
Summary of small RNA and degradome RNA libraries.
Reference genome and transcriptome databases used in this study.
List of 24-nt phasiRNA generating loci.
Number of PHAS loci identified from all sRNA libraries in each species. Figure S2. Identification of Atr-miR828 and TAS4-like sequences in Amborella trichopoda. Figure S3. Number of PHAS loci identified in leaf samples of each species. Figure S4. phasiRNAs mapped to intron–exon junctions or to an intron alone. Figure S5. Numbers of PHAS loci mapped to introns or intron–exon junctions in different plant species. Figure S6. Developmentally regulated phasiRNA production. Figure S7.PHAS loci in response to nutrient starvation in Chlamydomonas reinhardtii.
List of 21-nt phasiRNA generating loci.
List of phasiRNA-generating TAS4 loci.
Number of PHAS loci mapped to protein-coding genes.
List of previously reported gene families that generate phasiRNAs.
Summary of PHAS loci identified in each small RNA library.
PHAS loci containing only intron-derived sRNAs.
PHAS loci containing both intron- and exon-derived sRNAs (i.e., at intron–exon junctions).
Developmentally controlled PHAS loci.
Differential phasiRNA production in papaya, Arabidopsis , and rice between healthy and virus-infected leaf samples.
PHAS loci that were responsive to nutrient starvation.
Summary of PHAS loci with identified triggers.
Analysis for PHAS loci triggers.
Complete list of predicted target genes of phasiRNAs with RPM value no less than 10.
Acknowledgements
We thank Dr Blake Meyers for helpful discussions. We are grateful to Dr Yi Li at Peking University for making the small RNA library data from virus-infected and healthy rice plants available to us. This work was supported by grants from the Agriculture and Food Research Initiative Competitive Grant of the USDA National Institute of Food and Agriculture to BD and ZF (2014-67013-21550), National Science Foundation to BD (IOS-1051655) and to ZF (IOS-0923312 and IOS-1313887), and Ohio Plant Biotechnology Consortium to BD.
Footnotes
Yi Zheng and Ying Wang contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BD, YW, YZ, and ZF designed the project. YZ, YW, and JW performed data analyses. YZ, YW, JW, BD, and ZF wrote the paper. All authors read and approved the manuscript.
Contributor Information
Yi Zheng, Email: yz357@cornell.edu.
Ying Wang, wang.974@buckeyemail.osu.edu.
Jian Wu, wu.1495@buckeyemail.osu.edu.
Biao Ding, Email: ding.35@osu.edu.
Zhangjun Fei, Email: zf25@cornell.edu.
References
- 1.Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–87. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- 2.Simon SA, Meyers BC. Small RNA-mediated epigenetic modifications in plants. Curr Opin Plant Biol. 2011;14:148–55. doi: 10.1016/j.pbi.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Rogers K, Chen X. miRNA biogenesis and turnover in plants. Cold Spring Harb Symp Quant Biol. 2012;77:183–94. doi: 10.1101/sqb.2013.77.014530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axtell MJ, Westholm JO, Lai EC. Vive la difference: biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 2011;12:221. doi: 10.1186/gb-2011-12-4-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 2011;12:846–60. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 6.Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. Plant Cell. 2011;23:431–42. doi: 10.1105/tpc.110.082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fei Q, Xia R, Meyers BC. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell. 2013;25:2400–15. doi: 10.1105/tpc.113.114652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Axtell MJ. Classification and comparison of small RNAs from plants. Annu Rev Plant Biol. 2013;64:137–59. doi: 10.1146/annurev-arplant-050312-120043. [DOI] [PubMed] [Google Scholar]
- 9.Peragine A, Yoshikawa M, Wu G, Albrecht HL, Poethig RS. SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev. 2004;18:2368–79. doi: 10.1101/gad.1231804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vazquez F, Vaucheret H, Rajagopalan R, Lepers C, Gasciolli V, Mallory AC, et al. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell. 2004;16:69–79. doi: 10.1016/j.molcel.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 11.Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121:207–21. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Axtell MJ, Jan C, Rajagopalan R, Bartel DP. A two-hit trigger for siRNA biogenesis in plants. Cell. 2006;127:565–77. doi: 10.1016/j.cell.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 13.Cho SH, Coruh C, Axtell MJ. miR156 and miR390 regulate tasiRNA accumulation and developmental timing in Physcomitrella patens. Plant Cell. 2012;24:4837–49. doi: 10.1105/tpc.112.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dotto MC, Petsch KA, Aukerman MJ, Beatty M, Hammell M, Timmermans MC. Genome-wide analysis of leafbladeless1-regulated and phased small RNAs underscores the importance of the TAS3 ta-siRNA pathway to maize development. PLoS Genet. 2014;10:e1004826. doi: 10.1371/journal.pgen.1004826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhai J, Jeong DH, De Paoli E, Park S, Rosen BD, Li Y, et al. microRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011;25:2540–53. doi: 10.1101/gad.177527.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arikit S, Xia R, Kakrana A, Huang K, Zhai J, Yan Z, et al. An atlas of soybean small RNAs identifies phased siRNAs from hundreds of coding genes. Plant Cell. 2014;26:4584–601. doi: 10.1105/tpc.114.131847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li F, Pignatta D, Bendix C, Brunkard JO, Cohn MM, Tung J, et al. microRNA regulation of plant innate immune receptors. Proc Natl Acad Sci. 2012;109:1790–5. doi: 10.1073/pnas.1118282109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shivaprasad PV, Chen HM, Patel K, Bond DM, Santos BA, Baulcombe DC. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell. 2012;24:859–74. doi: 10.1105/tpc.111.095380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boccara M, Sarazin A, Thiebeauld O, Jay F, Voinnet O, Navarro L, et al. The Arabidopsis miR472-RDR6 silencing pathway modulates PAMP- and effector-triggered immunity through the post-transcriptional control of disease resistance genes. PLoS Pathog. 2014;10:e1003883. doi: 10.1371/journal.ppat.1003883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia R, Zhu H, An YQ, Beers EP, Liu Z. Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 2012;13:R47. doi: 10.1186/gb-2012-13-6-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu H, Xia R, Zhao B, An YQ, Dardick CD, Callahan AM, et al. Unique expression, processing regulation, and regulatory network of peach (Prunus persica) miRNAs. BMC Plant Biol. 2012;12:149. doi: 10.1186/1471-2229-12-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howell MD, Fahlgren N, Chapman EJ, Cumbie JS, Sullivan CM, Givan SA, et al. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell. 2007;19:926–42. doi: 10.1105/tpc.107.050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia R, Meyers BC, Liu Z, Beers EP, Ye S, Liu Z. microRNA superfamilies descended from miR390 and their roles in secondary small interfering RNA biogenesis in eudicots. Plant Cell. 2013;25:1555–72. doi: 10.1105/tpc.113.110957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen HM, Li YH, Wu SH. Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc Natl Acad Sci U S A. 2007;104:3318–23. doi: 10.1073/pnas.0611119104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Si-Ammour A, Windels D, Arn-bouldoires E, Kutter C, Ailhas J, Meins F, et al. miR393 and secondary siRNAs regulate expression of the TIR1/AFB2 auxin receptor clade and auxin-related development of Arabidopsis leaves. Plant Physiol. 2011;157:683–91. doi: 10.1104/pp.111.180083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Itaya A, Zhong X, Wu Y, Zhang J, van der Knaap E, et al. Function and evolution of a microRNA that regulates a Ca2+−ATPase and triggers the formation of phased small interfering RNAs in tomato reproductive growth. Plant Cell. 2011;23:3185–203. doi: 10.1105/tpc.111.088013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Axtell MJ, Snyder JA, Bartel DP. Common functions for diverse small RNAs of land plants. Plant Cell. 2007;19:1750–69. doi: 10.1105/tpc.107.051706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahlgren N, Jogdeo S, Kasschau KD, Sullivan CM, Chapman EJ, Laubinger S, et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell. 2010;22:1074–89. doi: 10.1105/tpc.110.073999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Ruiz H, Takeda A, Chapman EJ, Sullivan EM, Fahlgren N, Brempelis KJ, et al. Arabidopsis RNA-dependent RNA polymerases and dicer-like proteins in antiviral defense and small interfering RNA biogenesis during Turnip mosaic virus infection. Plant Cell. 2010;22:481–96. doi: 10.1105/tpc.109.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He G, Zhu X, Elling AA, Chen L, Wang X, Guo L, et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22:17–33. doi: 10.1105/tpc.109.072041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du P, Wu J, Zhang J, Zhao S, Zheng H, Gao G, et al. Viral infection induces expression of novel phased microRNAs from conserved cellular microRNA precursors. PLoS Pathog. 2011;7:e1002176. doi: 10.1371/journal.ppat.1002176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Bai X, Yan C, Gui Y, Wei X, Zhu QH, et al. Genomic dissection of small RNAs in wild rice (Oryza rufipogon): lessons for rice domestication. New Phytol. 2012;196:914–25. doi: 10.1111/j.1469-8137.2012.04304.x. [DOI] [PubMed] [Google Scholar]
- 33.Montes RA, de Fatima R-CF, De Paoli E, Accerbi M, Rymarquis LA, Mahalingam G, et al. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat Commun. 2014;5:3722. doi: 10.1038/ncomms4722. [DOI] [PubMed] [Google Scholar]
- 34.Plant Phased Secondary siRNA Database. [http://bioinfo.bti.cornell.edu/tool/phasiRNA].
- 35.Rock CD. Trans-acting small interfering RNA4: key to nutraceutical synthesis in grape development? Trends Plant Sci. 2013;18:601–10. doi: 10.1016/j.tplants.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan X, Pang M, Nah G, Shi X, Ye W, Stelly DM, et al. miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat Commun. 2014;5:3050. doi: 10.1038/ncomms4050. [DOI] [PubMed] [Google Scholar]
- 37.Luo QJ, Mittal A, Jia F, Rock CD. An autoregulatory feedback loop involving PAP1 and TAS4 in response to sugars in Arabidopsis. Plant Mol Biol. 2012;80:117–29. doi: 10.1007/s11103-011-9778-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuo J, Zhu B, Fu D, Zhu Y, Ma Y, Chi L, et al. Sculpting the maturation, softening and ethylene pathway: the influences of microRNAs on tomato fruits. BMC Genomics. 2012;13:7. doi: 10.1186/1471-2164-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian D, Traw MB, Chen JQ, Kreitman M, Bergelson J. Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature. 2003;423:74–7. doi: 10.1038/nature01588. [DOI] [PubMed] [Google Scholar]
- 40.Yoshikawa M, Peragine A, Park MY, Poethig RS. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev. 2005;19:2164–75. doi: 10.1101/gad.1352605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phytozome. [http://www.phytozome.net].
- 42.Sol Genomics Network. [http://solgenomics.net].
- 43.Amborella Genome Database. [http://www.amborella.org].
- 44.Zhong S, Fei Z, Chen YR, Zheng Y, Huang M, Vrebalov J, et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol. 2013;31:154–9. doi: 10.1038/nbt.2462. [DOI] [PubMed] [Google Scholar]
- 45.Zhai J, Zhang H, Arikit S, Huang K, Nan GL, Walbot V, et al. Spatiotemporally dynamic, cell-type-dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc Natl Acad Sci U S A. 2015;112:3146–51. doi: 10.1073/pnas.1418918112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babu M, Gagarinova AG, Brandle JE, Wang A. Association of the transcriptional response of soybean plants with soybean mosaic virus systemic infection. J Gen Virol. 2008;89:1069–80. doi: 10.1099/vir.0.83531-0. [DOI] [PubMed] [Google Scholar]
- 47.Catoni M, Miozzi L, Fiorilli V, Lanfranco L, Accotto GP. Comparative analysis of expression profiles in shoots and roots of tomato systemically infected by Tomato spotted wilt virus reveals organ-specific transcriptional responses. Mol Plant Microbe Interact. 2009;22:1504–13. doi: 10.1094/MPMI-22-12-1504. [DOI] [PubMed] [Google Scholar]
- 48.Hanssen IM, van Esse HP, Ballester AR, Hogewoning SW, Parra NO, Paeleman A, et al. Differential tomato transcriptomic responses induced by Pepino mosaic virus isolates with differential aggressiveness. Plant Physiol. 2011;156:301–18. doi: 10.1104/pp.111.173906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu J, Du ZX, Kong J, Chen LN, Qiu YH, Li GF, et al. Transcriptome analysis of Nicotiana tabacum infected by Cucumber mosaic virus during systemic symptom development. PLoS One. 2012;7:e43447. doi: 10.1371/journal.pone.0043447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen T, Lv Y, Zhao T, Li N, Yang Y, Yu W, et al. Comparative transcriptome profiling of a resistant vs. susceptible tomato (Solanum lycopersicum) cultivar in response to infection by Tomato yellow leaf curl virus. PLoS One. 2013;8:e80816. doi: 10.1371/journal.pone.0080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molnar A, Schwach F, Studholme DJ, Thuenemann EC, Baulcombe DC. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature. 2007;447:1126–9. doi: 10.1038/nature05903. [DOI] [PubMed] [Google Scholar]
- 52.Zhao T, Li G, Mi S, Li S, Hannon GJ, Wang XJ, et al. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 2007;21:1190–203. doi: 10.1101/gad.1543507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Casas-Mollano JA, Rohr J, Kim EJ, Balassa E, van Dijk K, Cerutti H. Diversification of the core RNA interference machinery in Chlamydomonas reinhardtii and the role of DCL1 in transposon silencing. Genetics. 2008;179:69–81. doi: 10.1534/genetics.107.086546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez-Alegre M, Dubus A, Fernandez E. REM1, a new type of long terminal repeat retrotransposon in Chlamydomonas reinhardtii. Mol Cell Biol. 2005;25:10628–38. doi: 10.1128/MCB.25.23.10628-10638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kakrana A, Hammond R, Patel P, Nakano M, Meyers BC. sPARTA: a parallelized pipeline for integrated analysis of plant miRNA and cleaved mRNA data sets, including new miRNA target-identification software. Nucleic Acids Res. 2015;42:e139. doi: 10.1093/nar/gku693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arif MA, Fattash I, Ma Z, Cho SH, Beike AK, Reski R, et al. Dicer-like2 activity in Physcomitrella patens Dicer-like4 mutants causes severe developmental dysfunction and sterility. Mol Plant. 2012;5:1281–94. doi: 10.1093/mp/sss036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cerutti H, Ma X, Msanne J, Repas T. RNA-mediated silencing in Algae: biological roles and tools for analysis of gene function. Eukaryot Cell. 2011;10:1164–72. doi: 10.1128/EC.05106-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, et al. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics. 2008;9:451. doi: 10.1186/1471-2164-9-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qian Y, Cheng Y, Cheng X, Jiang H, Zhu S, Cheng B. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 2011;30:1347–63. doi: 10.1007/s00299-011-1046-6. [DOI] [PubMed] [Google Scholar]
- 60.Bai M, Yang GS, Chen WT, Mao ZC, Kang HX, Chen GH, et al. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene. 2012;501:52–62. doi: 10.1016/j.gene.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 61.The Comparative Sequencing of Plant Small RNAs website. [http://smallRNA.udel.edu].
- 62.National Center for Biotechnology Information, Gene Expression Omnibus. [http://www.ncbi.nlm.nih.gov/geo].
- 63.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Paoli E, Dorantes-Acosta A, Zhai J, Accerbi M, Jeong DH, Park S, et al. Distinct extremely abundant siRNAs associated with cosuppression in petunia. RNA. 2009;15:1965–70. doi: 10.1261/rna.1706109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson C, Kasprzewska A, Tennessen K, Fernandes J, Nan GL, Walbot V, et al. Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res. 2009;19:1429–40. doi: 10.1101/gr.089854.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hofacker IL. Vienna RNA, secondary structure server. Nucleic Acids Res. 2003;31:3429–31. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.miRBase. [http://mirbase.org].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of small RNA and degradome RNA libraries.
Reference genome and transcriptome databases used in this study.
List of 24-nt phasiRNA generating loci.
Number of PHAS loci identified from all sRNA libraries in each species. Figure S2. Identification of Atr-miR828 and TAS4-like sequences in Amborella trichopoda. Figure S3. Number of PHAS loci identified in leaf samples of each species. Figure S4. phasiRNAs mapped to intron–exon junctions or to an intron alone. Figure S5. Numbers of PHAS loci mapped to introns or intron–exon junctions in different plant species. Figure S6. Developmentally regulated phasiRNA production. Figure S7.PHAS loci in response to nutrient starvation in Chlamydomonas reinhardtii.
List of 21-nt phasiRNA generating loci.
List of phasiRNA-generating TAS4 loci.
Number of PHAS loci mapped to protein-coding genes.
List of previously reported gene families that generate phasiRNAs.
Summary of PHAS loci identified in each small RNA library.
PHAS loci containing only intron-derived sRNAs.
PHAS loci containing both intron- and exon-derived sRNAs (i.e., at intron–exon junctions).
Developmentally controlled PHAS loci.
Differential phasiRNA production in papaya, Arabidopsis , and rice between healthy and virus-infected leaf samples.
PHAS loci that were responsive to nutrient starvation.
Summary of PHAS loci with identified triggers.
Analysis for PHAS loci triggers.
Complete list of predicted target genes of phasiRNAs with RPM value no less than 10.