

Abstract
Cortical inputs to the dorsolateral striatum (DLS) are dynamically regulated during skill learning and habit formation, and are dysregulated in disorders characterized by impaired action control. Therefore, a mechanistic investigation of the processes regulating corticostriatal transmission is key to understanding DLS-associated circuit function, behaviour and pathology. Presynaptic GABAB and group II metabotropic glutamate (mGlu2/3) receptors exert marked inhibitory control over corticostriatal glutamate release in the DLS, yet the signalling pathways through which they do so are unclear. We developed a novel approach using the genetically encoded calcium (Ca2+) indicator GCaMP6 to assess presynaptic Ca2+ in corticostriatal projections to the DLS. Using simultaneous photometric presynaptic Ca2+ and striatal field potential recordings, we report that relative to P/Q-type Ca2+ channels, N-type channels preferentially contributed to evoked presynaptic Ca2+ influx in motor cortex projections to, and excitatory transmission in, the DLS. Activation of GABAB or mGlu2/3 receptors inhibited both evoked presynaptic Ca2+ transients and striatal field potentials. mGlu2/3 receptor-mediated depression did not require functional N-type Ca2+ channels, but was attenuated by blockade of P/Q-type channels. These findings reveal presynaptic mechanisms of inhibitory modulation of corticostriatal function that probably contribute to the selection and shaping of behavioural repertoires.
Key points
Plastic changes at cortical inputs to the dorsolateral striatum (DLS) underlie skill learning and habit formation, so characterizing the mechanisms by which these inputs are regulated is important for understanding the neural basis of action control.
We developed a novel approach using the genetically encoded calcium (Ca2+) indicator GCaMP6 and brain slice photometry to assess evoked presynaptic Ca2+ transients in cortical inputs to the DLS and study their regulation by GABAB and mGlu2/3 receptors.
GABAB and mGlu2/3 receptor activation caused clear reductions in electrical stimulus-evoked presynaptic Ca2+ transients in corticostriatal inputs to the DLS.
Functional P/Q-type voltage-gated Ca2+ channels were required for the normal inhibitory action of corticostriatal mGlu2/3 receptors.
We provide direct evidence of presynaptic Ca2+ inhibition by G protein-coupled receptors at corticostriatal projections.
Introduction
The sensorimotor or dorsolateral striatum (DLS) is a critical node in cortico-basal ganglia loops that mediate action selection, skill learning and habit formation (Costa, 2007; Graybiel, 2008). Cortical inputs to the DLS drive activity in the intrinsically quiescent striatal output neurons, the medium spiny neurons (Wilson, 1995). These cortical inputs are dynamically regulated during skill learning (Costa et al. 2004; Yin et al. 2009; Koralek et al. 2012) and habit formation (Nazzaro et al. 2012; Smith & Graybiel, 2013; Gremel & Costa, 2013), and are dysregulated in disorders characterized by impaired action control, including substance misuse and obsessive-compulsive disorders, Huntington's disease and Parkinson's disease (Costa, 2007; Graybiel, 2008). Therefore, cellular processes that regulate the precise timing and efficacy of corticostriatal transmission are key determinants of DLS-associated circuit function, behaviour and pathology.
Two important regulators of corticostriatal transmission in the DLS are the presynaptic inhibitory Gi/o G protein-coupled receptors (GPCRs), GABAB and group II metabotropic glutamate (mGlu2/3) receptors. GABA released from recurrent collaterals of medium spiny neurons and striatal interneurons (particularly neuroglioform interneurons) activates GABAB receptors on corticostriatal terminals to suppress glutamate release (Calabresi et al. 1990, 1991; Nisenbaun et al. 1992; 1993; Lacey et al. 2005; Logie et al. 2013). mGlu2/3 receptors, expressed perisynaptically (Yokoi et al. 1996; Scanziani et al. 1997; Ohishi et al. 1998; Tamaru et al. 2001) on corticostriatal terminals (Kahn et al. 2001; Robbe et al. 2002), serve as autoreceptors that negatively regulate glutamate release in the DLS following periods of pronounced activity (Lovinger, 1991; Lombardi et al. 1993; Lovinger & McCool, 1995; Kahn et al. 2001).
GABAB and mGlu2/3 receptors can reduce synaptic transmission by suppressing adenylyl cyclase activity and cAMP accumulation (Travagli et al. 1991; Schoepp et al. 1995; Tzounopoulos et al. 1998), inhibiting voltage-gated calcium (Ca2+) channels (VGCCs; Stefani et al. 1994; Wu & Saggau, 1995; Takahashi et al. 1996, 1998), and/or inhibiting component(s) of the vesicle release machinery downstream of presynaptic Ca2+ influx (Scanziani et al. 1992, 1995; Tyler & Lovinger, 1995; Dittman & Regehr, 1996). The pathways through which these receptors regulate glutamate release from corticostriatal terminals in the DLS, however, remain unclear. Functional presynaptic Q-type VGCCs were found to be necessary for the increase in paired-pulse ratios (PPRs) of dorsal striatal population spikes seen following GABAB receptor activation (Barral et al. 2000). However, direct evidence for GABAB receptor-mediated effects on presynaptic Ca2+ in corticostriatal inputs is lacking. Similarly, despite evidence of a mitogen-activated protein kinase-dependent, cAMP/protein kinase A-independent mechanism for mGlu2/3 receptor-mediated depression in the dorsal striatum (Kahn et al. 2001), a clear role for presynaptic Ca2+ in this depression has yet to be established. In fact, early work using a corticostriatal co-culture preparation showed that mGlu2/3 receptors can inhibit glutamate release downstream of VGCCs, as mGlu2/3 receptor activation reduced the frequency of miniature excitatory postsynaptic currents that are independent of VGCC function (Tyler & Lovinger, 1995).
To clarify the role of presynaptic Ca2+ in the modulatory effects of GABAB and mGlu2/3 receptors on corticostriatal transmission, we developed a novel approach using the genetically encoded Ca2+ indicator GCaMP6s and brain slice photometry to assess presynaptic Ca2+ in a population of corticostriatal inputs to the DLS. We provide direct evidence that both GABAB and mGlu2/3 receptors inhibit stimulus-evoked presynaptic Ca2+ transients (PreCaTs) in corticostriatal afferents, and that functional P/Q-type VGCCs are required for the normal inhibitory action of corticostriatal mGlu2/3 receptors. In most instances, changes in evoked PreCaTs paralleled changes in DLS output as measured by simultaneous field potential recordings, indicating that inhibition of presynaptic Ca2+ probably contributes to inhibition of synaptic transmission by these receptors. However, some inconsistencies between these two measures were observed, and in this context we discuss the strengths and limitations of the photometry technique.
Methods
Ethical approval
All procedures conformed to the US National Institutes of Health Guide to the Care and Use of Laboratory Animals and were approved by the National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committee.
Animals
Male and female Emx1Cre mice (Jackson Laboratory, Bar Harbor, ME, USA; B6.129S2-Emx1tm1(cre)Krj/J) expressing an internal ribosomal entry site Cre recombinase gene under control of the Emx1 promoter (Gorski et al. 2002) were used. Emx1Cre mice were backcrossed to C57BL/6 J for more than eight generations and bred to heterozygosity for the Emx1Cre allele. To assess central Cre expression, Emx1Cre mice were crossed with mice expressing a loxP-flanked STOP sequence upstream of the enhanced green fluorescent protein (EGFP) variant, ZsGreen1 (Jackson Laboratory; B6.Cg-Gt(ROSA)26Sortm9(CAG-ZsGreen1)Hze/J). All mice were housed in groups of two to four on a 12 h light cycle (lights on at 06:30 h) with ad libitum access to food and water.
Viral vectors
Adeno-associated viral (AAV) vectors encoding double-floxed inverted open reading frame constructs of GCaMP6s (AAV1.CAG.FLEX.GCAMP6s.WPRE.bGH; ∼213 GC ml−1) and EGFP (AAV2/9.CAG.FLEX.EGFP.WPRE.bGH; ∼213 GC ml−1) were purchased from Penn Vector Core (Philadelphia, PA, USA).
Surgery
Under isoflurane anaesthesia, Emx1Cre mice (postnatal days (PD)30–40) were stereotaxically injected with 300 nl of either AAV-FLEX-GCaMP6s or AAV-FLEX-EGFP bilaterally into M1 motor cortex. M1 coordinates relative to bregma were (in mm): A/P: +1.1; M/L: ± 1.7; D/V: −0.9. Ketoprofen (5 mg kg−1, s.c.) was injected 30 min prior to completion of the surgery. Five to 8 weeks following surgery, brains were removed for slice recordings.
Brain slice preparation
PD70–90 mice were anaesthetized with isoflurane, brains were removed and 350 μm thick coronal sections containing the striatum were prepared using a vibratome in carbogen-bubbled, 4°C cutting solution containing (in mM): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4 and 10 d-glucose. Sections were equilibrated for 30 min at 33°C in carbogen-bubbled artificial cerebrospinal fluid (aCSF) containing (in mm): 124 NaCl, 4.5 KCl, 1 MgCl2, 2 CaCl2, 26 NaHCO3, 1.2 NaH2PO4 and 10 d-glucose. Sections were then incubated in aCSF at 23–24°C until transferred as hemisections to the recording chamber.
Photometry and extracellular field recordings
Photometry and extracellular field recordings were acquired at 23–24°C from GCaMP6s- or EGFP-expressing brain slices perfused with aCSF at 2 ml min−1 in a submersion chamber. Similar to the approach recently used by Sgobio et al. (2014a,2014b), fluorophores were excited using 470 ± 20 nm light from a mercury lamp (HBO 100 with Zeiss FluoArc Variable Intensity Lamp Control; 100 W Osram Mercury Short Arc, BulbDirect, 35% intensity) delivered through a 40 × 0.8 NA water immersion objective on an upright microscope (Axioskop 2 FS Plus, Zeiss, Oberkochen, Germany). Epifluorescence from a 180 μm2 area within the DLS (in a 350 μm thick slice) was filtered at 525 ± 25 nm and directed to a photomultiplier tube (PMT; Model D-104, Photon Technology International, Edison, NJ, USA). PMT voltage output (time constant: 5 ms; gain: 400 × 10–1 μA V–1) was low-pass filtered at 1 kHz and digitized at 6.67 kHz using a Digidata 1322A and Clampex 9.2 software (Axon Instruments, Molecular Devices LLC, Sunnyvale, CA, USA). The magnitude of the PMT output represents the total photon count at each time point. Control of a shutter driver (VCM-D1, Uniblitz, Vincent Associates, Rochester, NY, USA) and shutter (Model V25, Uniblitz) by Clampex 9.2 software (Axon Instruments, Union City, CA, USA) limited slice illumination to 4 s intervals every 30 s. An electrical pulse (0.5–0.8 mA, 40–100 μs) was delivered 2 s into each 4 s illumination interval though a concentric bipolar tungsten stimulating electrode (WE3CEA5, Microprobes, Gaithersburg, MD, USA) placed at the border of the DLS and the overlying white matter, at the edge of the selected 180 μm2 area. This stimulation elicited both evoked PreCaTs, which were proportional to the evoked increase in photons detected by the PMT, and evoked extracellular field population spikes (PSs), which were detected through thin-wall glass recording electrodes (2–5 MΩ) containing 0.9% NaCl solution placed within the DLS. Extracellular field potentials were amplified (×1000; DP-301 digital amplifier, Warner Instrument Corp., Hamden, CT, USA; Axopatch 200B Integrated Patch Amplifier, Axon Instruments), high- and low-pass filtered at 1 and 3 kHz, respectively, and digitized at 6.67 kHz (Digidata 1322A and Clampex 9.2 software, Axon Instruments). PreCaTs and extracellular field potentials were recorded simultaneously or separately (≥25% simultaneous recordings in each experiment).
GCaMP6s was chosen over other variants (GCaMP6m or 6f) to optimize the signal-to-noise ratio (SNR) of the evoked PreCaTs (Chen et al. 2013). The 180 μm2 optical recording area in the DLS was determined on the basis of previous studies from our laboratory (Sgobio et al. 2014a) and pilot studies aimed at optimizing PreCaT SNR. PreCaTs were strongest when the optical recording area contained a dense field of tortuous GCaMP6s-expressing presynaptic elements (Fig.1E). Bundles of GCaMP6s-expressing fibres passing through striatum (as shown in dorsomedial striatum in Fig.1D) were avoided because they were unresponsive to electrical stimulation and probably lack VGCCs that enable activity-dependent Ca2+ influx (Robitaille et al. 1990; Schneggenburger et al. 2012). Baseline fluorescence and evoked PreCaT amplitude stabilized after a 20–30 min period of 4 s bouts of 470 nm illumination and single-pulse electrical stimulation (both occurring every 30 s). PreCaT and field potential recordings began after this stabilization period.
Figure 1.
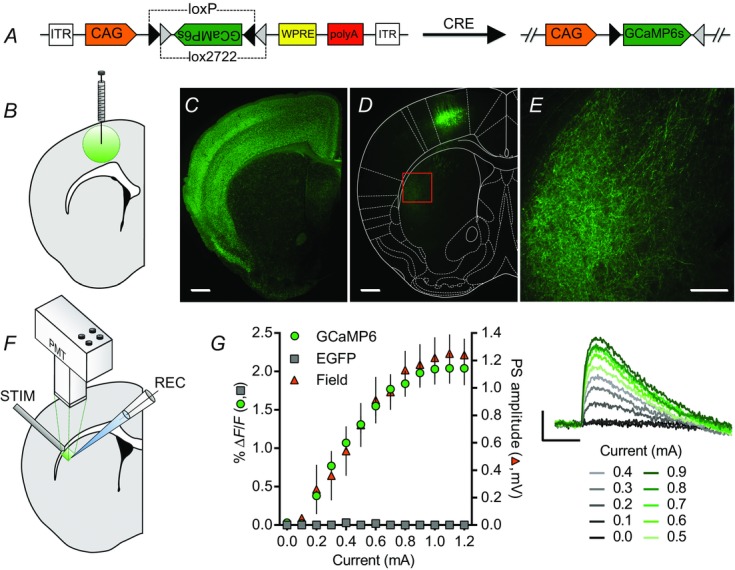
GCaMP6s expression in corticostriatal inputs to dorsolateral striatum
A, schematic diagram of the AAV-FLEX-GCaMP6s construct. In the presence of Cre recombinase, permanent recombination of two pairs of loxP and lox2272 sites changes the orientation of the GCaMP6 sequence from anti-sense to sense with respect to its promoter, CAG. B, schematic diagram of an AAV-FLEX-GCaMP6s injection into M1. C, cre-mediated expression of EGFP preferentially in cortical pyramidal neurons (and cortical glia; Gorski et al. 2002) in a striatal-containing coronal brain slice from an Emx1Cre;CAG-floxedSTOP-ZsGreen mouse. D and E, cre-mediated GCaMP6s expression in M1 cortical pyramidal cells (D) and presynaptic corticostriatal elements in the DLS (D, E) of an Emx1Cre mouse injected with AAV-FLEX-GCaMP6s into M1. The area shown in E is indicated by the box in D. F, schematic diagram of brain slice photometry and electrophysiology recordings. A concentric bipolar stimulating electrode placed at the border of the DLS and overlying white matter evoked striatal fluorescent transients detected by a photomultiplier tube (PMT) and striatal field potentials detected by a glass recording electrode. G, current–response curves of 100 μs electrical pulse-evoked fluorescence and field potentials recorded in the DLS of Emx1Cre mice injected with AAV-FLEX-GCaMP6s or AAV-FLEX-EGFP. GCaMP6s- (green circles, n = 6) but not EGFP-injected mice (grey squares, n = 6) showed evoked fluorescent transients that scaled with stimulation current amplitude and population spike (PS) amplitude (orange triangles, n = 6) (traces on right; average of four evoked responses per current amplitude). Scale bars of fluorescence images: 500 μm (C, D), 200 μm (E). Scale bars of evoked PreCaTs from AAV-FLEX-GCaMP6s-injected Emx1Cre mice: 1% ΔF/F, 0.5 s (G).
Analysis
Photometric traces were averaged in 2 min bins (four traces) in Clampfit 9.2 and exported to GraphPad Prism 6 (GraphPad Software) and Microsoft Excel for processing. An exponential decay was fitted to each averaged trace (excluding the period corresponding to the evoked PreCaT) and used to linearize and subtract the underlying baseline photometric recording (comparable to Sgobio et al. 2014a). PreCaT amplitude was measured as the ratio of the average peak amplitude of the stimulus-evoked fluorescent transient and the average fluorescence measured immediately before the stimulus, and then converted to a percentage (% ΔF/F). In experiments examining the time course of PreCaT amplitudes during experimental manipulations, amplitudes were expressed as the percentage of the average amplitude obtained during baseline recording within each slice.
Single pulse-evoked PreCaTs with amplitudes < 0.6% ΔF/F (after stabilization) were omitted to avoid inadequate SNRs following drug application. Approximately 85% of GCaMP6s-injected Emx1Cre mice yielded at least one brain slice with adequate corticostriatal GCaMP6s expression in DLS to evoke PreCaT amplitudes of ≥0.6% ΔF/F.
To calculate PPRs of PreCaT amplitudes, groups of four single-pulse traces and four paired-pulse traces were evoked for each inter-pulse interval (IPI). Each four-trace group was averaged, and the underlying baselines of the averages were linearized and subtracted using exponential decays, as described above. Each averaged single-pulse response was subtracted from its associated PPR to isolate the response to the second of the paired pulses. The ratio of the peak ΔF/F of the second pulse to the peak ΔF/F of the associated single pulse was considered the PreCaT PPR.
Mean PS amplitude, defined as the voltage difference between the peak post-artifact positivity and subsequent peak negativity, was measured for individual traces in Clampfit 9.2 and analysed in GraphPad Prism. In experiments examining the time course of PS amplitudes during experimental manipulations, PS amplitudes were averaged in 2 min bins and expressed as the percentage of the average amplitude obtained during baseline recording within each slice.
All comparisons of both single pulse-evoked PreCaT and PS amplitudes were conducted using two-tailed, paired or unpaired Student's t tests, as appropriate. Unless otherwise specified, 2 min bins of data at specific time points were used for these comparisons, as indicated above. Two minute bins of data at t = 6 min were used for baseline comparisons. PPRs were analysed with two-way repeated measures ANOVAs with Sidak's post hoc comparisons using the within-subjects factor of IPI and between-subjects factor of drug treatment.
Drugs
TTX, 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo(f)-quinoxaline (NBQX), dl-2-amino-5-phosphonopentanoic acid (AP5), ω-conotoxin GVIA, ω-agatoxin IVA and LY379268 were prepared in distilled H2O. Baclofen was prepared in 0.5% acetic acid. CGP55845, LY341495, nifedipine and ryanodine were prepared in DMSO. All drugs were diluted to their final concentrations in aCSF (final DMSO concentrations were ≤0.1%). TTX and AP5 were purchased from Abcam (Cambridge, MA, USA). ω-Conotoxin GVIA and ω-agatoxin IVA were purchased from Sigma-Aldrich (St Louis, MO, USA). All other drugs were purchased from Tocris Bioscience (Ellisville, MO, USA).
Immunohistochemistry
Emx1Cre;ZsGreen and GCaMP6-injected Emx1Cre mice were transcardially perfused under deep isoflurane anaesthesia with 0.1 m PBS and 4% formaldehyde in PBS (pH 7.4). Brains were collected and post-fixed in 4% formaldehyde at 4°C for 24 h. Coronal sections 50 μm thick were made using a vibratome through frontal cortex and striatum. On a revolving platform, sections were incubated for 2 h in PBST (PBS with 0.2% Triton X-100), blocked in 5% BSA in PBST for 4 h and incubated in chicken polyclonal anti-GFP antibody (Abcam, ab13970, 1:2000) in PBST for 12 h at 4°C. Following three 1 h washes in PBST, sections were incubated in Alexa Fluor 488 goat anti-chicken antibody (Life Technologies, Carlsbad, CA, USA; A-11039, 1:2000) in PBST for 12 h at 4°C. Following a 1 h wash in PBST and two 1 h washes in PBS, sections were mounted on Superfrost Plus slides (Daigger, Vernon Hills, IL, USA; EF15978Z) using Fluoromount Aqueous Mounting Medium (Sigma, F4680), coverslipped and imaged using Zeiss AxioVision LE 4.3 software with a Zeiss Axiocam on a Zeiss SteREO Lumar microscope. Brightness was adjusted uniformly in each image for presentation.
Results
Characterization of corticostriatal PreCaTs in DLS
To detect Ca2+ in presynaptic elements of corticostriatal projections, we injected AAV-FLEX-GCaMP6s (Fig.1A) bilaterally into M1 of Emx1Cre mice (Fig.1B) and assessed, using fluorescence photometry, the PreCaTs evoked in the DLS by local electrical stimulation (Fig.1F). Crossing Emx1Cre mice with reporter mice that express EGFP following Cre-mediated recombination (floxed-STOP-CAG-ZsGreen1) revealed dense expression of the fluorophore throughout cortex (Fig.1C). Accordingly, M1 injection of AAV-FLEX-GCaMP6s into Emx1Cre mice resulted in expression of the genetically encoded calcium indicator (GECI) in M1 (Fig.1D) and M1 projection neuron targets, including the DLS, assessed by direct visualization of GCaMP6s or immunostaining for its GFP moiety (Fig.1E). Importantly, no GCaMP6s-expressing somata were detected in striatum in any slice analysed in this study. Evoked PreCaTs that scaled with both stimulation current amplitude and field PS amplitude were detected in the DLS of Emx1Cre mice injected with AAV-FLEX-GCaMP6s (n = 6 GCaMP6s, Field), but not AAV-FLEX-EGFP (n = 3; Fig.1G), indicating that PreCaTs represent activity-dependent increases in cytosolic Ca2+ in corticostriatal processes.
We next assessed whether corticostriatal PreCaTs were action potential- and Ca2+-dependent. PreCaTs were abolished by bath application of the sodium channel blocker, TTX (1 μm, n = 4; Fig.2A), and Ca2+-free aCSF (n = 4; Fig.2B), and dramatically reduced by application of the non-specific VGCC blocker cadmium (100 μm; 10.6 ± 1.9% of baseline at t = 60 min, n = 5, P < 0.0001; Fig.2C). As expected, all three treatments also abolished the PSs measured in field potential recordings at rates comparable to their effects on PreCaTs (n = 3–5; Fig.2A–C). Consistent with a presynaptic locus of expression, PreCaTs, but not PSs, were insensitive to co-application of ionotropic glutamate receptor antagonists, NBQX (10 μm) and AP5 (50 μm; 95.2 ± 5.5% of baseline at t = 40 min, n = 7, P = 0.44) (Fig.2D). These results indicate that PreCaTs reflect action potential-dependent Ca2+ entry into presynaptic corticostriatal elements in the DLS.
Figure 2.
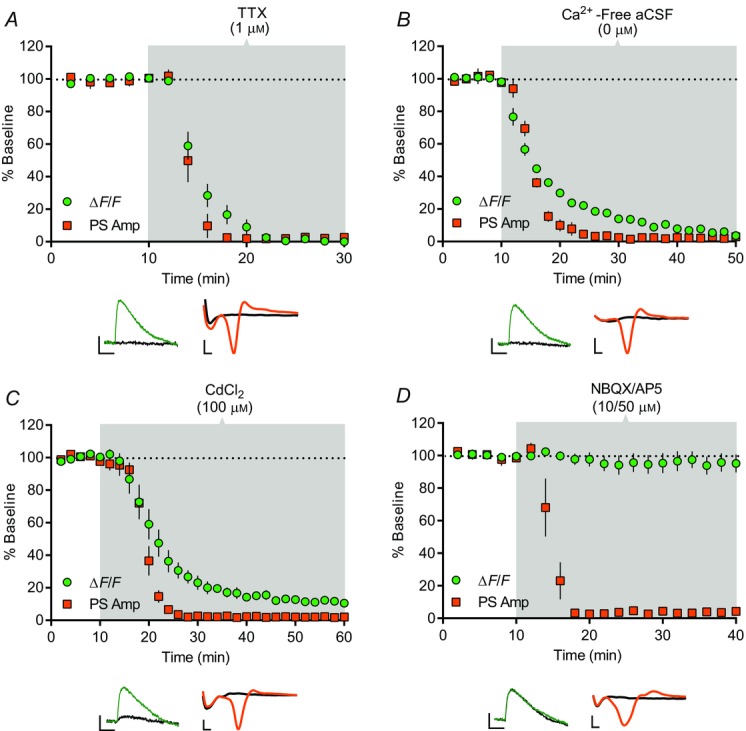
Characterization of corticostriatal PreCaTs in dorsolateral striatum
Time course of the effects of TTX, Ca2+-free aCSF, CdCl2 and NBQX/AP5 on electrical pulse-evoked photometric PreCaT and electrophysiological field potential recordings in the DLS. PreCaT amplitude is expressed as % baseline ΔF/F (green circles) and population spike amplitude is expressed as % baseline PS Amp (orange squares). Shaded areas correspond to periods of drug application. Representative traces from PreCaT (left, average of four evoked responses) and field potential recordings (right, average of eight evoked responses) are shown below each graph. A, bath application of the sodium channel blocker, TTX (1 μm), for 20 min abolished PreCaTs and PSs. B, bath application of Ca2+-free aCSF for 40 min abolished PreCaTs and PSs. C, bath application of the non-selective Ca2+ channel blocker, CdCl2 (100 μm), for 50 min dramatically reduced PreCaT amplitude and abolished PSs. D, bath application of the AMPA and NMDA receptor antagonists, NBQX (10 μm) and AP5 (50 μm), for 30 min had no effect on PreCaT amplitude, but abolished PSs. Trace time points: green/orange, 10 min; black, 70 min (A), 50 min (B), 60 min (C), 40 min (D). Trace scale bars: PreCaTs, 1% ΔF/F, 0.5 s; field potentials, 0.5 mV, 1 ms.
VGCC contribution to corticostriatal PreCaTs and transmission
Using combined PreCaT and field potential recordings, we next tested the contribution of specific VGCCs to corticostriatal transmission in the DLS. Application of the N-type VGCC blocker, ω-conotoxin GVIA (1 μm), strongly reduced PreCaT (67.6 ± 2.2% of baseline at t = 70 min, n = 6, P < 0.0001) and PS amplitude (49.7 ± 7.9% of baseline at t = 70 min, n = 8, P < 0.005) (Fig.3A). The P/Q-type VGCC blocker, ω-agatoxin IVA (200 nm), did not induce a significant reduction in PreCaT amplitude (86.4 ± 5.7% of baseline at t = 70 min, n = 6, P = 0.052), but a significant reduction in PS amplitude (80.8 ± 8.2% of baseline at t = 70 min, n = 6, P < 0.05) (Fig.3B) was observed. Co-application of ω-conotoxin GVIA (1 μm) and ω-agatoxin IVA (200 nm) resulted in an additive suppression of PreCaT (60.2 ± 8.6% of baseline at t = 70 min, n = 6, data not shown) and PS amplitude (28.6 ± 11.6% of baseline at t = 70 min, n = 5, data not shown). In contrast, nifedipine, an L-type VGCC blocker (1 μm), had no effect on either PreCaTs (95.6 ± 3.7% of baseline at t = 50 min, n = 6, P = 0.4) or PSs (102.1 ± 2.3 of baseline at t = 50 min, n = 3, P = 0.84) (Fig.3C). Together, these data support roles for N-, and to a lesser extent, P/Q-type VGCCs, in evoked presynaptic Ca2+ influx in motor corticostriatal projections and excitatory transmission in the DLS.
Figure 3.
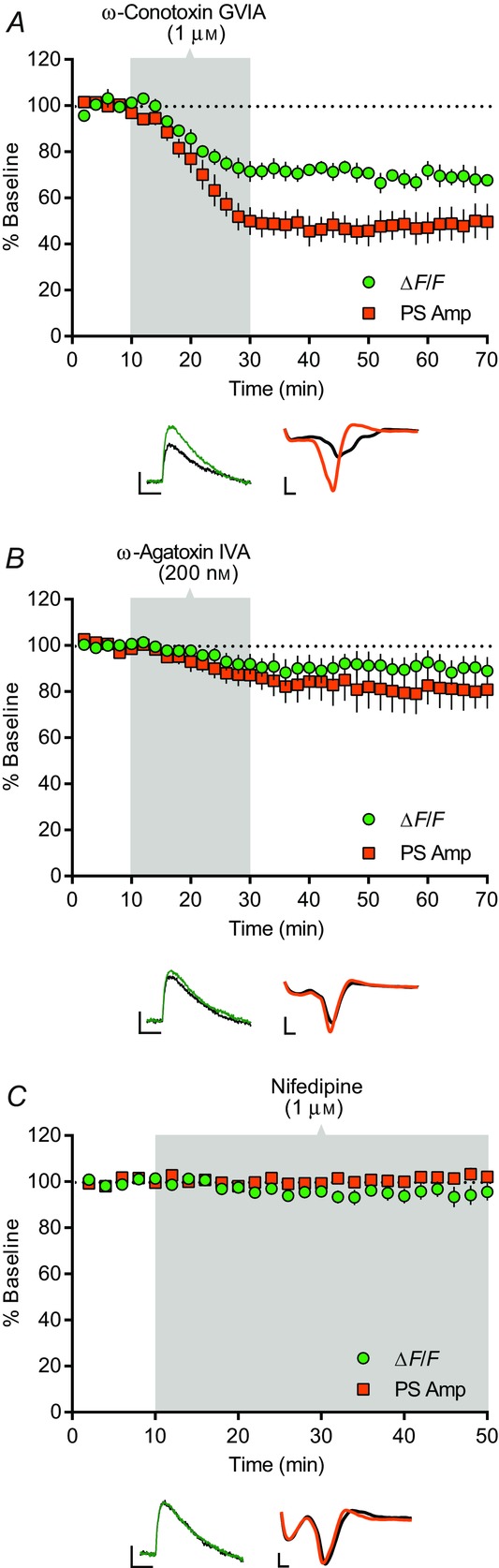
VGCC contribution to corticostriatal PreCaTs and transmission
Time course of the effects of ω-conotoxin GVIA, ω-agatoxin IVA and nifedipine on electrical pulse-evoked PreCaT (ΔF/F) and field potential (PS Amp) recordings in the DLS. Shaded areas correspond to periods of drug application. Representative traces from PreCaT (left, average of four evoked responses) and field potential recordings (right, average of eight evoked responses) are shown below each graph. A, bath application of the N-type VGCC blocker, ω-conotoxin GVIA (1 μm), for 20 min suppressed PS amplitude. B, bath application of the P/Q-type VGCC blocker, ω-agatoxin IVA (200 nm), for 20 min modestly suppressed PS amplitude. C, bath application of the L-type VGCC blocker, nifedipine (1 μm), for 40 min had no effect on PreCaT or PS amplitude. Trace time points: green/orange, 10 min; black, 70 min (A, B), 50 min (C). Trace scale bars: PreCaTs, 1% ΔF/F, 0.5 s; field potentials, 0.5 mV, 1 ms.
PPRs of corticostriatal PreCaTs and transmission
Corticostriatal synapses can exhibit short-term depression in response to pairs of afferent stimuli (paired-pulse depression (PPD)) (Niittykosko et al. 1999; Akopian et al. 2000), but residual Ca2+ in the presynaptic terminal at the time of the second stimulus drives these synapses towards paired-pulse facilitation (PPF) at short IPIs (Choi & Lovinger, 1997; Calabresi et al. 1997; Zucker & Regehr, 2002). Without means to directly assess presynaptic Ca2+ at these synapses, the mechanisms by which presynaptic Ca2+ contributes to this short-term plasticity could not be studied. We therefore measured corticostriatal PreCaTs (and PSs) in response to pairs of electrical pulses separated by intervals ranging from 10 ms to 6 s.
We observed pronounced PPF of PreCaT amplitude that peaked at IPIs of 20 ms (2.7 ± 0.2 PreCaT2/PreCaT1, n = 7) and was no longer present at IPIs of 6 s (1.1 ± 0.0) (Fig.4A). This facilitation contrasted with the overall PPD seen in PS amplitude during field recordings using identical stimulation parameters (Control: n = 7; Fig.4B). PreCaT PPF was insensitive to pharmacological manipulations that alter PPRs of transmission (Zucker & Regehr, 2002). Indeed, application of high [Ca2+] aCSF (4 mm), which itself enhanced single pulse-evoked PreCaT amplitude (177.8 ± 7.9%, 20 min after application, n = 4, data not shown), had no effect on PreCaT PPF relative to control conditions (2.6 ± 0.1 PreCaT2/PreCaT1 at 20 ms IPI, n = 4; IPI × Treatment interaction: F10,90 = 1.15, P = 0.34; Fig.4A). In contrast, PS amplitude was unaltered by application of 4 mm Ca2+ (97.2 ± 5.1 mV, 20 min following application, n = 5, data not shown), but PPD of PS amplitude was enhanced at several short IPIs in 4 mm Ca2+ (n = 5; IPI × Treatment interaction: F10,100 = 17.18, P < 0.0001; IPI 20, 40, 80 and 120 ms: P < 0.05; Fig.4B). PreCaT PPF following bath application of a cocktail of ω-conotoxin GVIA (1 μm) and ω-agatoxin IVA (200 nm) was also indistinguishable from controls at all IPIs tested (2.8 ± 0.1 PreCaT2/PreCaT1 at 20 ms IPI, n = 4; IPI × Treatment interaction: F10,90 = 0.49, P = 0.89; Fig.4A). Prevention of Ca2+-induced Ca2+ release (CICR) from internal Ca2+ stores by ryanodine (20 μm) did not alter corticostriatal PreCaTs evoked by single pulses (97.5 ± 13.4%, 20 min following application, n = 4, data not shown) or PreCaT PPF at all IPIs tested (2.5 ± 0.1 PreCaT2/PreCaT1 at 20 ms IPI, n = 4; IPI × Treatment interaction: F10,90 = 0.98, P = 0.47; Fig.4A).
Figure 4.
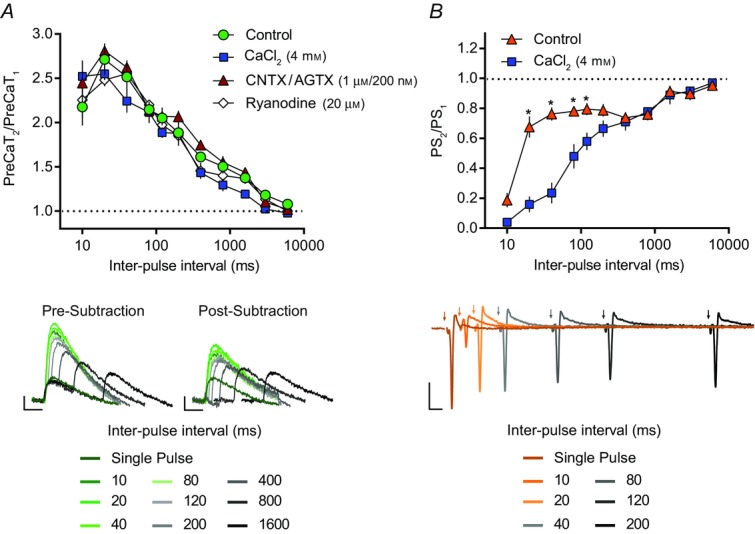
Paired-pulse ratios of corticostriatal PreCaTs and transmission
The ratio of two PreCaTs (PreCaT2/PreCaT1) (A) or two population spikes (PS2/PS1) (B) evoked by two electrical pulses separated by IPIs of 10, 20, 40, 80, 120, 200, 400, 800, 3000 and 6000 ms (IPIs presented on a log scale). A, the effects of CaCl2, ω-conotoxin GVIA (CNTX) with ω-agatoxin IVA (AGTX), and ryanodine on PreCaT PPRs were assessed relative to control conditions. The evoked PreCaT amplitude observed in response to the second of two paired electrical pulses was robustly enhanced relative to the PreCaT amplitude evoked by a single pulse at the majority of IPIs tested (i.e. PPF of PreCaT amplitude, green circles, n = 7). PPF in the presence of high [Ca2+] aCSF (4 mm CaCl2; blue squares, n = 4), following co-application of ω-conotoxin GVIA and ω-agatoxin IVA (1 μm and 200 nm; red triangles, n = 4), or following ryanodine (20 μm; white diamonds, n = 4) was indistinguishable from control conditions (green circles, n = 7). Representative traces of control PreCaTs in response to single- and paired-pulse stimulations, before (left) and following (right) subtraction of the corresponding single-pulse response, are shown below (average of four evoked responses per IPI). Trace scale bars, 1% ΔF/F, 0.5 s. B, the effect of CaCl2 on PS PPRs was assessed relative to control conditions. The evoked PS amplitude observed in response to the second of two paired electrical pulses was depressed relative to the PS amplitude evoked by the first pulse at the majority of IPIs tested (i.e. PPD of PS amplitude, orange triangles, n = 7). PPD in the presence of high [Ca2+] aCSF (4 mm CaCl2; blue squares, n = 5) was enhanced at 20, 40, 80 and 120 ms IPIs relative to control conditions (orange triangles, n = 7). Representative traces of control field recordings in response to single- and paired-pulse stimulations are shown below (average of two evoked responses per IPI). Trace scale bars, 0.5 mV, 10 ms.
GABAB- and mGlu2/3 receptor-mediated depression of corticostriatal PreCaTs and transmission
We next tested whether presynaptic Gi/o protein-coupled GABAB and mGlu2/3 receptors suppress corticostriatal transmission by inhibiting presynaptic Ca2+ influx. Bath application of the GABAB receptor agonist, baclofen (10 μm), for 5 min reduced PreCaT amplitudes to 79.9 ± 2.4% of baseline (n = 5, P < 0.05) and PS amplitudes to 43.7 ± 11.1% of baseline (n = 6, P < 0.005) 5 min following application (Fig.5A). This depression reversed shortly after removal of baclofen from the bath; PreCaT and PS amplitudes were indistinguishable from baseline 45 min following the cessation of baclofen application (PreCaT: 96.2 ± 3.3% of baseline, n = 5, P = 0.50 vs. baseline; PS: 94.7 ± 2.4% of baseline, n = 6, P = 0.07 vs. baseline) (Fig.5A). Pre-application of the selective GABAB receptor antagonist, CGP55845 (2 μm), blocked baclofen-induced short-term depression of both PreCaTs (98.6 ± 2.1% of baseline at t = 20 min, n = 5, P = 0.74 vs. baseline, P < 0.005 vs. baclofen) and PSs (103.6 ± 4.1% of baseline at t = 20 min, n = 5, P = 0.51 vs. baseline, P < 0.005 vs. baclofen) (Fig.5B). Application of CGP55845 (2 μm) alone had no effect on PS amplitude (100.8 ± 4.2% of baseline at 45 min following application, n = 3, P = 0.87; data not shown). These results indicate that GABAB receptor-mediated short-term depression of corticostriatal transmission involves inhibition of presynaptic Ca2+ influx.
Figure 5.
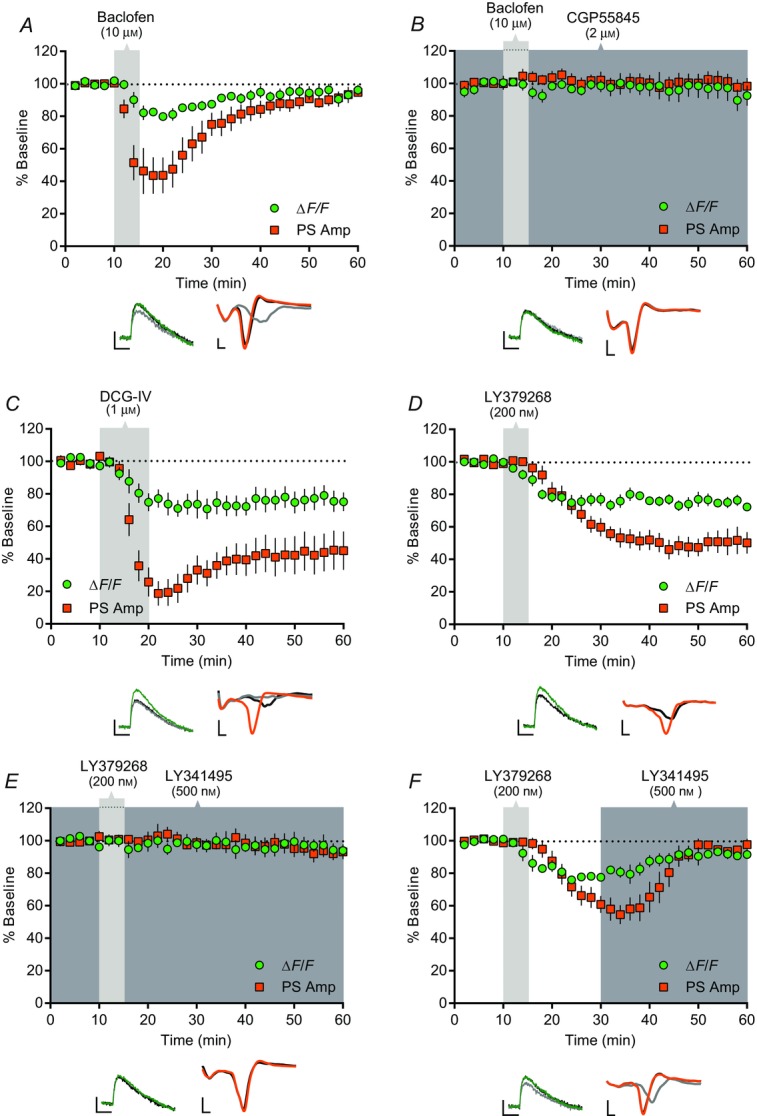
GABAB- and mGlu2/3 receptor-mediated depression of corticostriatal PreCaTs and transmission
Time course of the effects of baclofen, DCG-IV and LY379268 on electrical stimulation-evoked PreCaT (ΔF/F) and field potential (PS Amp) recordings in the DLS. Shaded areas correspond to periods of drug application. Representative traces from PreCaT (left, average of four evoked responses) and field potential recordings (right, average of eight evoked responses) are shown below each graph. A, bath application of the GABAB receptor agonist, baclofen (10 μm), for 5 min transiently suppressed both PreCaT and PS amplitude. B, pre-application of the GABAB receptor antagonist, CGP55845 (2 μm), prevented baclofen- (10 μm) induced suppression of PreCaT and PS amplitude. C, bath application of the mGlu2/3 receptor agonist, DCG-IV (1 μm), for 10 min suppressed both PreCaT and PS amplitude. D, bath application of the mGlu2/3 receptor agonist, LY379268 (200 nm), for 5 min suppressed both PreCaT and PS amplitude. E, pre-application of the mGlu2/3 receptor antagonist, LY341495 (500 nm), prevented LY379268- (200 nm) induced suppression of PreCaT and PS amplitude. F, bath application of LY341495 (500 nm) for 30 min reversed LY379268- (200 nm) induced suppression of PreCaT and PS amplitude. Trace time points: green/orange, 10 min; grey, 20 min (A), 22 min (C), 30 min (F); black, 60 min. Trace scale bars: PreCaTs, 1% ΔF/F, 0.5 s; field potentials: 0.5 mV, 1 ms.
Bath application of the group II mGlu receptor agonist, DCG-IV (1 μm), for 10 min strongly depressed PreCaT amplitude (75.1 ± 5.8% of baseline at t = 60 min, n = 5, P < 0.05) (Fig.5C). DCG-IV-induced depression of PS amplitude was most pronounced immediately at the end of the 10 min application, but remained strongly inhibited at least 40 min after cessation of DCG-IV application (45.1 ± 11.8% of baseline at t = 60 min, n = 6, P < 0.01) (Fig.5C). Similarly, a 5 min application of the more potent and selective mGlu2/3 receptor agonist, LY379268 (200 nM), strongly depressed PreCaTs (72.3 ± 3% of baseline at t = 60 min, n = 7, P < 0.0005) and PSs (50.2 ± 6.7% of baseline at t = 60 min, n = 8, P < 0.0001) (Fig.5D).
Pre-application of the selective mGlu2/3 antagonist, LY341495 (500 nm), blocked LY379268-induced depression of PreCaTs (94.6 ± 2.4% of baseline at t = 60 min, n = 4, P = 0.13 vs. baseline, P < 0.001 vs. LY379268) and PSs (93.1 ± 4.5% of baseline at t = 60 min, n = 7, P = 0.26 vs. baseline, P < 0.0005 vs. LY379268) (Fig.5E). Interestingly, LY341495 also reversed the long-lasting LY379268-induced depression. Specifically, PreCaT amplitude, which was reduced to 79.8 ± 2.6% of baseline at 30 min following agonist application, returned to near baseline levels following a subsequent 30 min application of LY341495 (94.5 ± 3.4% of baseline at t = 60 min, n = 4, P = 0.21 vs. baseline, P < 0.005 vs. LY379268). Similarly, LY379268-induced depression of PS amplitude to 60.9 ± 5.3% of baseline at 30 min was fully reversed by LY341495 (97.7 ± 3.6% of baseline at t = 60 min, n = 7, P = 0.23 vs. baseline, P < 0.0001 vs. LY379268) (Fig.5F). Application of LY341495 alone had no effect on PS amplitude (97.3 ± 5.6% of baseline at 45 min following application, n = 3, P = 0.88; data not shown). These results indicate that activation of mGlu2/3 receptors induces a labile form of long-term depression (LTD; Atwood et al. 2014) at corticostriatal synapses that involves a reduction in presynaptic Ca2+ influx.
P/Q-type VGCCs contribute to mGlu2/3 receptor-mediated depression
Given the clear effects of mGlu2/3 receptor activation on presynaptic Ca2+, we next performed a series of occlusion experiments to test whether blockade of select VGCCs can interfere with mGlu2/3 receptor function. We first followed bath application of the N-type VGCC blocker, ω-conotoxin GVIA, with application of LY379268 (Fig.6A). Twenty minutes of bath application of ω-conotoxin reduced PreCaT amplitude to 73.1 ± 5.5% of baseline (n = 4). Subsequent application of LY379268 further inhibited PreCaTs to 54.8 ± 5.5% of baseline after 40 min. This inhibition by LY379268 following ω-conotoxin was not significantly different from the inhibition observed when LY379268 was applied alone (74.2 ± 3.3% of 25–30 min ω-conotoxin baseline at 65–70 min, P < 0.005 vs. ω-conotoxin baseline, P = 0.93 vs. LY379268) (Fig.6C). We observed a similar lack of effect of ω-conotoxin on LY379268-induced depression of PS amplitude (Fig.6A). Following ω-conotoxin pre-application, which reduced PS amplitude to 59.4 ± 9.7% of baseline (n = 6), LY379268 further inhibited PS amplitude to 28.7 ± 10.5% of baseline after 40 min. Again, this inhibition by LY379268 after N-type VGCC blockade did not differ from that observed after LY379268 alone (39.5 ± 10.9% of 25–30 min ω-conotoxin baseline, P < 0.0005 vs. ω-conotoxin baseline, P = 0.46 vs. LY379268) (Fig.6C). Together, these results indicate that mGlu2/3 receptor-mediated depression at corticostriatal synapses can occur independently of N-type VGCC function.
Figure 6.
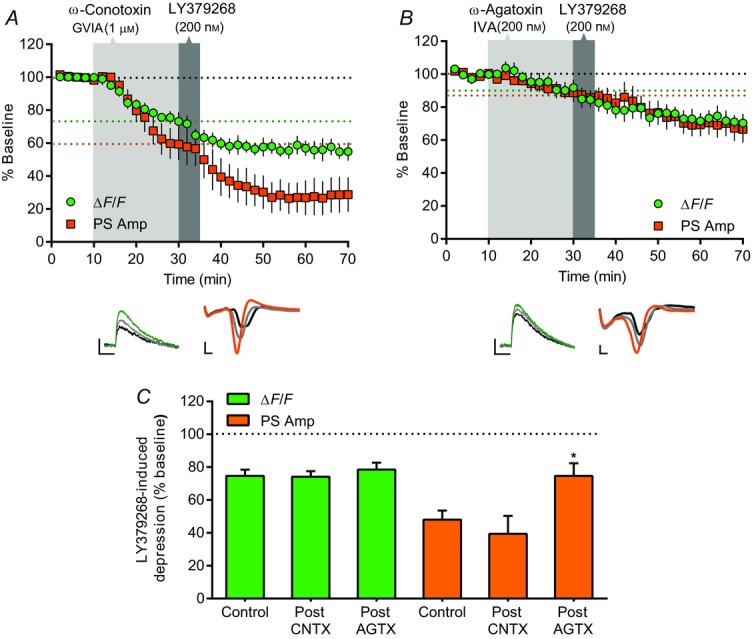
P/Q-type VGCCs contribute to mGlu2/3 receptor-mediated depression
A and B, time course of the effects of ω-conotoxin GVIA (CNTX) or ω-agatoxin IVA (AGTX) on LY379268-induced depression of electrical stimulation-evoked PreCaT (ΔF/F) and field potential (PS Amp) recordings in the DLS. Shaded areas correspond to periods of drug application. The dotted lines denote the average % baseline ΔF/F (green line) and PS Amp (orange line) achieved at termination of VGCC blocker application; these values served as the baselines to which the effects of subsequent LY379268 application were compared. Representative traces from PreCaT (left, average of four evoked responses) and field potential recordings (right, average of eight evoked responses) are shown below each graph. A, pre-application of the N-type VGCC blocker, ω-conotoxin GVIA (1 μm), for 20 min had no effect on LY379268- (200 nm) induced suppression of PreCaT and PS amplitude. B, pre-application of the P/Q-type VGCC blocker, ω-agatoxin IVA (200 nm), for 20 min attenuated LY379268- (200 nm) induced suppression of PS amplitude. LY379268-induced suppression of PreCaT amplitude was unaltered by pre-application of ω-agatoxin IVA. Trace time points: green/orange, 10 min; grey, 30 min; black, 70 min. Trace scale bars: PreCaTs, 1% ΔF/F, 0.5 s; field potentials: 0.5 mV, 1 ms. C, summary of LY379268-induced inhibition of PreCaT and PS amplitude with or without pre-application of VGCC blockers. Inhibition of PS amplitude by LY379268 after P/Q-type VGCC blockade (Post AGTX) was significantly less than that observed after LY379268 alone (Control); *P < 0.05.
In contrast, a parallel series of occlusion experiments revealed a role for P/Q-type VGCCs in mGlu2/3 receptor function in the DLS. Bath application of the P/Q-type VGCC blocker, ω-agatoxin IVA, non-significantly reduced PreCaT amplitude to 91.9 ± 4.8% of baseline (n = 6, P = 0.49), which was further reduced by subsequent LY379268 application to 70.3 ± 5.5% (P < 0.01, Fig.6B). This inhibition by LY379268 following ω-agatoxin was not significantly different from the inhibition observed when LY379268 was applied alone (78.5 ± 4.2% of 25–30 min ω-agatoxin baseline, P < 0.01 vs. ω-agatoxin baseline, P = 0.51 vs. LY379268) (Fig.6C). However, pre-application of ω-agatoxin did modify LY379268-induced depression of PS amplitude (Fig.6B). Following application of ω-agatoxin, which reduced PS amplitude to 84.8 ± 4.4% of baseline (n = 8), LY379268 further inhibited PS amplitude to only 65.5 ± 8.9% of baseline after 40 min. This inhibition by LY379268 after P/Q-type VGCC blockade was significantly blunted relative to that observed after LY379268 alone (75.3 ± 8.7% of 25–30 min ω-agatoxin baseline, P < 0.05 vs. ω-agatoxin baseline, P < 0.05 vs. LY379268) (Fig.6C). Together, these results indicate that full mGlu2/3 receptor-mediated depression at corticostriatal synapses requires functional P/Q-type VGCCs.
Discussion
Here we developed and validated a novel approach using the genetically encoded calcium indicator, GCaMP6, to assess presynaptic Ca2+ in cortical inputs to the DLS. Using simultaneous photometric PreCaT and electrophysiological field recordings, we found that: (1) evoked presynaptic Ca2+ influx into motor cortex inputs to the DLS, like excitatory transmission in the DLS (Lovinger et al. 1994; Bargas et al. 1998), is preferentially controlled by N-type VGCCs, relative to P/Q-type VGCCs; (2) GABAB and mGlu2/3 receptor activation inhibits presynaptic Ca2+ influx in corticostriatal inputs to the DLS; and (3) mGlu2/3 receptor-mediated depression involves P/Q-type VGCCs. These findings reveal novel presynaptic mechanisms of autoreceptor and heteroreceptor control over corticostriatal function that may contribute to striatal-dependent learning and action control.
Several factors have slowed the development of viable corticostriatal presynaptic Ca2+ measurements in brain slices. Unlike many hippocampal and cerebellar projections, corticostriatal projections are ill suited to presynaptic loading of conventional Ca2+ indicator dyes because striatal-projecting pyramidal neurons are distributed widely in cortex, local cortical regions innervate many striatal subregions (Kincaid et al. 1998) and corticostriatal axons are multiplanar (Kawaguchi et al. 1989; Gorelova & Yang, 1997). Corticostriatal boutons are also smaller than many terminals canonically used to study presynaptic Ca2+ function (Amaral & Dent, 1981; Hámori & Somogyi, 1983; Liu et al. 2011), making high-resolution Ca2+ imaging of individual corticostriatal terminals technically challenging (Park et al. 2014). By combining modern viral, optogenetic and transgenic tools with conventional photomultiplier-based fluorescence detection, we developed an approach that allows sensitive and highly temporally resolved assessment of presynaptic Ca2+ dynamics in genetically and regionally specified corticostriatal inputs.
Our work provides direct evidence that GABAB and mGlu2/3 receptors suppress evoked corticostriatal transmission by inhibiting presynaptic Ca2+ influx in corticostriatal projections. These findings are consistent with reports of GABAB and mGlu2/3 receptor-mediated inhibition of presynaptic Ca2+ at other central synapses (Travagli et al. 1991; Swartz et al. 1993; Doze et al. 1995; Scanziani et al. 1995; Wu & Saggau, 1995; Yoshino & Kamiya, 1995; Choi & Lovinger, 1996; Dittman & Regehr, 1996; Takahashi et al. 1996), and extend previous studies at corticostriatal synapses that relied on VGCC blockers to probe GABAB and mGlu2/3 receptor mechanisms. For example, Barral et al. (2000) showed that blockade of Q-type VGCCs by ω-agatoxin TK occluded the ability of the GABAB agonist baclofen to enhance PPRs of striatal field potentials. Similarly, mGlu2/3 receptor-mediated LTD of excitatory transmission in the nucleus accumbens (NAc) was shown to occlude, and be occluded by, the inhibitory effects of ω-agatoxin IVA (Robbe et al. 2002). We show for the first time that these same channels are necessary for normal mGlu2/3 receptor-mediated plasticity in the DLS, a striatal subregion with inputs, outputs and functions that differ from those of the NAc (Groenewegen et al. 1999; Gerfen et al. 1992). Together, these data strongly support a role for P/Q-type VGCCs in the metabotropic glutamate and GABA receptor modulation of corticostriatal synapses.
P/Q-type VGCC blockade reduced, but did not abolish, mGlu2/3 receptor-mediated depression of transmission. The P/Q-insensitive action of mGlu2/3 receptors is consistent with reports of Ca2+-independent mechanisms of mGlu2/3 (and GABAB) receptor-mediated inhibition of neurotransmitter release within and beyond the dorsal striatum (Tyler & Lovinger, 1995; Dittman & Regehr, 1996; Wu & Saggau, 1997). Notably, the magnitude of depression induced by ω-agatoxin IVA alone closely mimicked the magnitude by which prior application of ω-agatoxin blunted mGlu2/3 receptor-mediated depression (∼20% of baseline).
Unlike transmission, mGlu2/3 receptor-mediated inhibition of PreCaT amplitude was unaltered by prior P/Q-type VGCC blockade. Several explanations could account for this observation. For example, it is well established that small changes in presynaptic Ca2+ current exert large effects on transmission, given the approximate fourth-power relationship between transmitter release and presynaptic Ca2+ influx (Katz & Miledi, 1970; Wu & Saggau, 1994). Accordingly, drug-induced reductions in percentage baseline PreCaT amplitude in the present study were consistently smaller than the associated reductions in PS amplitude. Therefore, small changes in PS amplitude may accompany PreCaT changes that are too small to detect. In fact, application of ω-agatoxin IVA alone produced a 15–20% reduction in PS amplitude and no significant reduction in PreCaT amplitude (Fig.3B, 6B). In the same manner, pretreatment with ω-agatoxin, which blunted the LY379268-induced reduction in PS amplitude by ∼20%, was unlikely to yield a significant change in the LY379268-induced reduction in PreCaT amplitude.
Field photometry offers many advantages for studying the modulation of presynaptic Ca2+, including high detection sensitivity and frequency response with real-time measurement, low noise, affordability and adaptability to optically record from a wide variety of neuronal inputs. Its major shortcoming, however, is its limited spatial resolution. The strongest modulatory effects on presynaptic Ca2+ are likely to occur within or near presynaptic terminals, where the majority of presynaptic VGCCs and modulatory receptors are located. With population-level field photometry, however, these effects are measured against a background of presynaptic elements (i.e. axons) that express high levels the Ca2+ indicator but low levels of VGCCs and receptors. As a result, field photometry may not reveal subtle or spatially restricted effects of presynaptic modulators.
Many factors, including limited spatial resolution of optical recordings, can contribute to discrepancies between measures of presynaptic Ca2+ and postsynaptic activity. Although highly related, evoked presynaptic Ca2+ influx and postsynaptic cell firing are notably distinct events that can be independently regulated. Presynaptic terminals and postsynaptic cells receive distinct inputs, express unique receptors and respond differently to the various transmitters and neuromodulators evoked by local electrical stimulation (GABA, dopamine, etc.). Also, inputs that express the Ca2+ indicator and therefore contribute to the PreCaT signal (e.g. M1-derived inputs) are possibly distinct from (and probably a subset of) those that contribute to the evoked PSs. These different populations may exhibit unique transmission properties or mechanisms of modulation. Furthermore, optical recordings can be affected by factors (e.g. indicator kinetics and affinities) that have no bearing on electrical recordings.
These and other factors may help explain not only the subtle differences in time course of some of the drug effects on PreCaT and PS amplitude, but also the stark contrast between the robust PPF seen in our PreCaT recordings and the PPD seen in our PS recordings. Given that manipulations of extracellular Ca2+ and VGCC function that alter PPR of transmission (Zucker & Regehr, 2002) had no effect on PreCaT PPF, and that the exact stimulation parameters that produced PreCaT PPF yielded PPD of PS amplitudes, it is unlikely that the PreCaT PPF represents an enhanced conductance of presynaptic VGCCs (and thus Ca2+ influx) during the second of two closely paired stimuli. Instead, the PreCaT PPF is more probably attributable to Ca2+ buffering within corticostriatal presynaptic elements expressing high levels of GCaMP6s, a GECI with a relatively high Ca2+ affinity (Kd ≈ 144 nm) (Chen et al. 2013). Notably, the PS is not a direct measure of synaptic transmission, and is subject to factors that affect neuronal excitability. Indeed, the large PPD observed in PSs at short IPIs is not observed when excitatory postsynaptic currents are measured in striatal neurons (Choi & Lovinger, 1997; Calabresi et al. 1997). Overall, our findings indicate that there are limitations to measuring PPRs with either of these field-based measurements, and it is better to rely on direct measurement of synaptic responses in this context.
In conclusion, cortical inputs to the dorsal striatum are essential for the control of voluntary actions, and the precise regulation of these inputs underlies our ability to select and shape adaptive behavioural repertoires. Our work provides mechanistic insight into the actions of two prominent sources of regulatory control over corticostriatal function. Our approach to assessing presynaptic Ca2+ in corticostriatal projections innervating the DLS can be readily applied to the study of other GPCR neuromodulators and activity-induced forms of plasticity at these synapses (Huang et al. 2001; Gerdeman et al. 2002; Mathur et al. 2011; Atwood et al. 2014), and adapted to probe related cortico-basal ganglia circuits (e.g. prefrontal cortex inputs to the dorsomedial striatum or NAc). As new fluorescent sensors for other signalling molecules continue to become available (e.g. Ponsioen et al. 2004; Allen & Zhang, 2006), comparable fluorescence photometry approaches will prove invaluable to achieve a more sophisticated understanding of plasticity mechanisms. Given emerging evidence for GABAB and mGlu2/3 receptors in basal ganglia pathologies such as substance misuse and Huntington's disease (Moussawi & Kalivas, 2010; Beveridge et al. 2011; Reiner et al. 2012; Kim & Seo, 2014), our work also provides a foundation for studies using disease models in which corticostriatal signalling processes may be affected.
Acknowledgments
We thank Dr Carmelo Sgobio for his valuable advice and technical expertise, and all members of the Lovinger laboratory for their helpful discussions, particularly Drs Guohong Cui, Matthew J. Pava and Brian N. Mathur. We also thank Dr Margaret I. Davis and Austin Feng for assistance in the early characterization of Cre and GCaMP6 expression in Emx1Cre mice, and Guoxiang Luo for help with mouse genotyping. We are also grateful to the Fisher's Lane Animal Care staff for their excellent animal husbandry and veterinary care.
Glossary
- AAV
adeno-associated virus
- AP5
dl-2-amino-5-phosphonopentanoic acid
- CICR
calcium-induced calcium release
- DLS
dorsolateral striatum
- EGFP
enhanced green fluorescent protein
- GPCR
G protein-coupled receptor
- GECI
genetically encoded calcium indicator
- GCaMP6
calmodulin-based genetically encoded fluorescent calcium indicator 6
- IPI
inter-pulse interval
- LTD
long-term depression
- mGlu2/3
group II metabotropic glutamate receptors
- NBQX
2,3-Dihydroxy-6-nitro-7-sulfamoylbenzo(f)-quinoxaline
- NAc
nucleus accumbens
- PMT
photomultiplier tube
- PPD
paired-pulse depression
- PPF
paired-pulse facilitation
- PPR
paired-pulse ratio
- PreCaT
presynaptic calcium transient
- PS
population spike
- PS Amp
population spike amplitude
- SNR
signal-to-noise ratio
- VGCC
voltage-gated calcium channel
Additional information
Competing interests
None.
Author contributions
D.A.K. and D.M.L. designed the experiments. D.A.K. performed the experiments and analysed the data. D.A.K. and D.M.L. wrote the manuscript.
Funding
This work was supported by the Division of Intramural Clinical and Biological Research of the NIAAA, and the Natural Sciences and Engineering Research Council (NSERC) of Canada (PDF 438487-13).
References
- Allen MD. Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 2006;348:716–721. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- Akopian G, Musleh W, Smith R. Walsh JP. Functional state of corticostriatal synapses determines their expression of short- and long-term plasticity. Synapse. 2000;38:271–280. doi: 10.1002/1098-2396(20001201)38:3<271::AID-SYN6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Amaral DG. Dent JA. Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J Comp Neurol. 1981;195:51–86. doi: 10.1002/cne.901950106. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA. Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci. 2014;17:540–548. doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lovinger DM. Mathur BN. Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 2014;37:663–673. doi: 10.1016/j.tins.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargas J, Ayala GX, Hernández E. Galarraga E. Ca2+-channels involved in neostriatal glutamatergic transmission. Brain Res Bull. 1998;45:521–524. doi: 10.1016/s0361-9230(97)00439-5. [DOI] [PubMed] [Google Scholar]
- Barral J, Toro S, Galarraga E. Bargas J. GABAergic presynaptic inhibition of rat neostriatal afferents is mediated by Q-type Ca2+ channels. Neurosci Lett. 2000;283:33–36. doi: 10.1016/s0304-3940(00)00909-5. [DOI] [PubMed] [Google Scholar]
- Beveridge TJ, Smith HR, Nader MA. Porrino LJ. Group II metabotropic glutamate receptors in the striatum of non-human primates: dysregulation following chronic cocaine self-administration. Neurosci Lett. 2011;496:15–19. doi: 10.1016/j.neulet.2011.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Mercuri NB, De Murtas M. Bernardi G. Endogenous GABA mediates presynaptic inhibition of spontaneous and evoked excitatory synaptic potentials in the rat neostriatum. Neurosci Lett. 1990;188:99–102. doi: 10.1016/0304-3940(90)90258-b. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Mercuri NB, De Murtas M. Bernardi G. Involvement of GABA systems in feedback regulation of glutamate-and GABA-mediated synaptic potentials in rat neostriatum. J Physiol. 1991;440:581–599. doi: 10.1113/jphysiol.1991.sp018726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A. Bernardi G. Endogenous adenosine mediates the presynaptic inhibition induced by aglycemia at corticostriatal synapses. J Neurosci. 1997;15:4509–4516. doi: 10.1523/JNEUROSCI.17-12-04509.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K. Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. Lovinger DM. Metabotropic glutamate receptor modulation of voltage-gated Ca2+ channels involves multiple receptor subtypes in cortical neurons. J Neurosci. 1996;16:36–45. doi: 10.1523/JNEUROSCI.16-01-00036.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. Lovinger DM. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci U S A. 1997;94:2665–2670. doi: 10.1073/pnas.94.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Cohen D. Nicolelis M. Differential corticostriatal plasticity during fast and slow motor skill learning. Curr Biol. 2004;14:1124–1134. doi: 10.1016/j.cub.2004.06.053. [DOI] [PubMed] [Google Scholar]
- Costa RM. Plastic corticostriatal circuits for action learning: what's dopamine got to do with it? Ann N Y Acad Sci. 2007;1104:172–191. doi: 10.1196/annals.1390.015. [DOI] [PubMed] [Google Scholar]
- Dittman JS. Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doze VA, Cohen GA. Madison DV. Calcium channel involvement in GABAB receptor-mediated inhibition of GABA release in area CA1 of the rat hippocampus. J Neurophysiol. 1995;74:43–53. doi: 10.1152/jn.1995.74.1.43. [DOI] [PubMed] [Google Scholar]
- Dreosti E, Odermatt B, Dorostkar MM. Lagnado L. A genetically encoded reporter of synaptic activity in vivo. Nat Methods. 2009;6:883–889. doi: 10.1038/nmeth.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J. Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL. Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelova N. Yang CR. The course of neural projection from the prefrontal cortex to the nucleus accumbens in the rat. Neuroscience. 1997;76:689–706. doi: 10.1016/s0306-4522(96)00380-6. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. Habits, rituals, and the evaluative brain. Annu Rev Neurosci. 2008;31:359–387. doi: 10.1146/annurev.neuro.29.051605.112851. [DOI] [PubMed] [Google Scholar]
- Gremel CM. Costa RM. Orbitofrontal and striatal circuits dynamically encode the shift between goal-directed and habitual actions. Nat Commun. 2013;4:2264. doi: 10.1038/ncomms3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenewegen HJ, Wright CI, Beijer AV. Voorn P. Convergence and segregation of ventral striatal inputs and outputs. Ann N Y Acad Sci. 1999;877:49–63. doi: 10.1111/j.1749-6632.1999.tb09260.x. [DOI] [PubMed] [Google Scholar]
- Hámori J. Somogyi J. Differentiation of cerebellar mossy fiber synapses in the rat: a quantitative electron microscope study. J Comp Neurol. 1983;220:365–377. doi: 10.1002/cne.902200402. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lo SW. Hsu KS. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol. 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn L, Alonso G, Robbe D, Bockaert J. Manzoni OJ. Group 2 metabotropic glutamate receptors induced long-term depression in mouse striatal slices. Neurosci Lett. 2001;316:178–182. doi: 10.1016/s0304-3940(01)02397-7. [DOI] [PubMed] [Google Scholar]
- Katz B. Miledi R. Further study of the role of calcium in synaptic transmission. J Physiol. 1970;207:789–801. doi: 10.1113/jphysiol.1970.sp009095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ. Emson PC. Intracellular recording of identified neostriatal patch and matrix spiny cells in a slice preparation preserving cortical inputs. J Neurophysiol. 1989;62:1052–1068. doi: 10.1152/jn.1989.62.5.1052. [DOI] [PubMed] [Google Scholar]
- Kim W. Seo H. Baclofen, a GABAB receptor agonist, enhances ubiquitin–proteasome system functioning and neuronal survival in Huntington's disease model mice. Biochem Biophys Res Commun. 2014;443:706–711. doi: 10.1016/j.bbrc.2013.12.034. [DOI] [PubMed] [Google Scholar]
- Kincaid AE, Zheng T. Wilson CJ. Connectivity and convergence of single corticostriatal axons. J Neurosci. 1998;18:4722–4731. doi: 10.1523/JNEUROSCI.18-12-04722.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koralek AC, Jin X, Long JD, 2nd, Costa RM. Carmena JM. Corticostriatal plasticity is necessary for learning intentional neuroprosthetic skills. Nature. 2012;483:331–335. doi: 10.1038/nature10845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey CJ, Boyes J, Gerlach O, Chen L, Magill PJ. Bolam JP. GABAB receptors at glutamatergic synapses in the rat striatum. Neuroscience. 2005;136:1083–1095. doi: 10.1016/j.neuroscience.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Liu B, Ouyang L, Mu S, Zhu Y, Li K, Zhan M, Liu Z, Jia Y. Lei W. The morphological characteristics of corticostriatal and thalamostriatal neurons and their intrastriatal terminals in rats. Surg Radiol Anat. 2011;33:807–817. doi: 10.1007/s00276-011-0823-9. [DOI] [PubMed] [Google Scholar]
- Logie C, Bagetta V. Bracci E. Presynaptic control of corticostriatal synapses by endogenous GABA. J Neurosci. 2013;33:15425–15431. doi: 10.1523/JNEUROSCI.2304-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi G, Alesiani M, Leonardi P, Cherici G, Pellicciari R. Moroni F. Pharmacological characterization of the metabotropic glutamate receptor inhibiting D-[3H]-aspartate output in rat striatum. Br J Pharmacol. 1993;110:1407–1412. doi: 10.1111/j.1476-5381.1993.tb13977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. Trans-1-aminocyclopentane-1,3- dicarboxylic acid (t-ACPD) decreases synaptic excitation in rat striatal slices through a presynaptic action. Neurosci Lett. 1991;129:17–21. doi: 10.1016/0304-3940(91)90710-b. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Merritt A. Reyes D. Involvement of N- and non-N-type calcium channels in synaptic transmission at corticostriatal synapses. Neuroscience. 1994;62:31–40. doi: 10.1016/0306-4522(94)90312-3. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. McCool BA. Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGluR2 or 3. J Neurophysiol. 1995;73:1076–1083. doi: 10.1152/jn.1995.73.3.1076. [DOI] [PubMed] [Google Scholar]
- Mathur BN, Capik NA, Alvarez VA. Lovinger DM. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–7411. doi: 10.1523/JNEUROSCI.6250-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K. Kalivas PW. Group II metabotropic glutamate receptors (mGlu2/3) in drug addiction. Eur J Pharmacol. 2010;639:115–122. doi: 10.1016/j.ejphar.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazzaro C, Greco B, Cerovic M, Baxter P, Rubino P, Trusel M, Parolaro D, Tkatch T, Benfenati F, Pedarzani P. Tonini R. SK channel modulation rescues striatal plasticity and control over habit in cannabinoid tolerance. Nat Neurosci. 2012;15:285–295. doi: 10.1038/nn.3022. [DOI] [PubMed] [Google Scholar]
- Niittykoski M, Ruotsalainen S, Haapalinna A, Larson J. Sirviö J. Activation of muscarinic M3-like receptors and β-adrenoceptors, but not M2-like muscarinic receptors or α-adrenoceptors, directly modulates corticostriatal neurotransmission in vitro. Neuroscience. 1999;90:95–105. doi: 10.1016/s0306-4522(98)00447-3. [DOI] [PubMed] [Google Scholar]
- Nisenbaun ES, Berger TW. Grace AA. Presynaptic modulation by GABAB receptors of glutamatergic excitation and GABAergic inhibition of neostriatal neurons. J Neurophysiol. 1992;67:477–481. doi: 10.1152/jn.1992.67.2.477. [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Berger TW. Grace AA. Depression of glutamatergic and GABAergic synaptic responses in striatal spiny neurons by stimulation of presynaptic GABAB receptors. Synapse. 1993;14:221–242. doi: 10.1002/syn.890140306. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Neki A. Mizuno N. Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res. 1998;30:65–82. doi: 10.1016/s0168-0102(97)00120-x. [DOI] [PubMed] [Google Scholar]
- Park H, Popescu A. Poo MM. Essential role of presynaptic NMDA receptors in activity-dependent BDNF secretion and corticostriatal LTP. Neuron. 2014;84:1009–1022. doi: 10.1016/j.neuron.2014.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL. Jalink K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5:1176–1180. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR. Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Lafferty DC, Wang HB, Del Mar N. Deng YP. The group 2 metabotropic glutamate receptor agonist LY379268 rescues neuronal, neurochemical and motor abnormalities in R6/2 Huntington's disease mice. Neurobiol Dis. 2012;47:75–91. doi: 10.1016/j.nbd.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Chaumont S, Bockaert J. Manzoni OJ. Role of p/q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci. 2002;22:4346–4356. doi: 10.1523/JNEUROSCI.22-11-04346.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Adler EM. Charlton MP. Strategic location of calcium channels at transmitter sites of frog neuro-muscular synapses. Neuron. 1990;5:773–779. doi: 10.1016/0896-6273(90)90336-e. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gähwiler BH. Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH. Thompson SM. Presynaptic inhibition of excitatory synaptic transmission by muscarinic and metabotropic glutamate receptor activation in the hippocampus: are Ca2+ channels involved? Neuropharmacology. 1995;34:1549–1557. doi: 10.1016/0028-3908(95)00119-q. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC. Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Han Y. Kochubey O. Ca2+ channels and transmitter release at the active zone. Cell Calcium. 2012;52:199–207. doi: 10.1016/j.ceca.2012.04.011. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Johnson BG, Salhoff CR, Valli MJ, Desai MA, Burnett JP, Mayne NG. Monn JA. Selective inhibition of forskolin-stimulated cyclic AMP formation in rat hippocampus by a novel mGluR agonist, 2R,4R-4-aminopyrrolidine-2,4-dicarboxylate. Neuropharmacology. 1995;34:843–850. doi: 10.1016/0028-3908(95)00061-a. [DOI] [PubMed] [Google Scholar]
- Sgobio C, Kupferschmidt DA, Cui G, Sun L, Li Z, Cai H. Lovinger DM. a Optogenetic measurement of presynaptic calcium transients using conditional genetically encoded calcium indicator expression in dopaminergic neurons. PLoS One. 2014;9:e111749. doi: 10.1371/journal.pone.0111749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgobio C, Cai H. Lovinger DM. b Dual recording of real-time dopamine release and presynaptic calcium transients reveals distinct properties of dopaminergic afferents in dorsal and ventral striatum. Soc Neurosci Abstract. 2014 [Google Scholar]
- Smith KS. Graybiel AM. A dual operator view of habitual behavior reflecting cortical and striatal dynamics. Neuron. 2013;79:361–374. doi: 10.1016/j.neuron.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani A, Pisani A, Mercuri NB, Bernardi G. Calabresi P. Activation of metabotropic glutamate receptors inhibits calcium currents and GABA-mediated synaptic potentials in striatal neurons. J Neurosci. 1994;14:6734–6743. doi: 10.1523/JNEUROSCI.14-11-06734.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz KJ, Merritt A, Bean BP. Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y. Tsujimoto T. G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J Neurosci. 1998;18:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travagli RA, Ulivi M. Wojcik WJ. γ-Aminobutyric acid-B receptors inhibit glutamate release from cerebellar granule cells: consequences of inhibiting cyclic AMP formation and calcium influx. J Pharmacol Exp Ther. 1991;258:903–909. [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M. Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N. Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503. doi: 10.1016/s0306-4522(01)00305-0. [DOI] [PubMed] [Google Scholar]
- Tyler EC. Lovinger DM. Metabotropic glutamate receptor modulation of synaptic transmission in corticostriatal co-cultures: role of calcium influx. Neuropharmacology. 1995;34:939–952. doi: 10.1016/0028-3908(95)00066-f. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Janz R, Südhof TC, Nicoll RA. Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Wilson CJ. The contribution of cortical neurons to the firing pattern of striatal spiny neurons. In: Houk JC, Davis JL, Beiser DG, editors. Models of Information Processing in the Basal Ganglia. Cambridge, MA: MIT Press; 1995. pp. 29–50. [Google Scholar]
- Wu LG. Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG. Saggau P. GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J Physiol. 1995;485:649–657. doi: 10.1113/jphysiol.1995.sp020759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG. Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilario MR, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM. Costa RM. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakaguchi I, Katsuura G, Shigemoto R, Ohishi H, Nomura S, Nakamura K, Nakao K, Katsuki M. Nakanishi S. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science. 1996;273:645–647. doi: 10.1126/science.273.5275.645. [DOI] [PubMed] [Google Scholar]
- Yoshino M. Kamiya H. Suppression of presynaptic calcium influx by metabotropic glutamate receptor agonists in neonatal rat hippocampus. Brain Res. 1995;695:179–185. doi: 10.1016/0006-8993(95)00743-a. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]