

Abstract
Key points
Anoxia induces hyper-excitability and cell death in mammalian brain but in the western painted turtle (Chrysemys picta bellii) enhanced GABA transmission prevents injury.
The mechanism responsible for increased GABA transmission is unknown; however, reactive oxygen species (ROS) generated by mitochondria may play a role because this is an oxygen-sensitive process.
In this study, we show that inhibition of mitochondrial ROS production is sufficient to initiate a redox-sensitive GABA signalling cascade that suppresses pyramidal neuron action potential frequency.
These results further our understanding of the turtle's unique strategy for reducing ATP consumption during anoxia and highlights a natural mechanism in which to explore therapies to protect mammalian brain from low-oxygen insults (e.g. cerebral stroke).
Abstract
Anoxia induces hyper-excitability and cell death in mammalian brain but in the anoxia-tolerant western painted turtle (Chrysemys picta bellii) neuronal electrical activity is suppressed (i.e. spike arrest), adenosine triphosphate (ATP) consumption is reduced, and cell death does not occur. Electrical suppression is primarily the result of enhanced γ-aminobutyric acid (GABA) transmission; however, the underlying mechanism responsible for initiating oxygen-sensitive GABAergic spike arrest is unknown. In turtle cortical pyramidal neurons there are three types of GABAA receptor-mediated currents: spontaneous inhibitory postsynaptic currents (IPSCs), giant IPSCs and tonic currents. The aim of this study was to assess the effects of reactive oxygen species (ROS) scavenging on these three currents since ROS levels naturally decrease with anoxia and may serve as a redox signal to initiate spike arrest. We found that anoxia, pharmacological ROS scavenging, or inhibition of mitochondrial ROS generation enhanced all three types of GABA currents, with tonic currents comprising ∼50% of the total current. Application of hydrogen peroxide inhibited all three GABA currents, demonstrating a reversible redox-sensitive signalling mechanism. We conclude that anoxia-mediated decreases in mitochondrial ROS production are sufficient to initiate a redox-sensitive inhibitory GABA signalling cascade that suppresses electrical activity when oxygen is limited. This unique strategy for reducing neuronal ATP consumption during anoxia represents a natural mechanism in which to explore therapies to protect mammalian brain from low-oxygen insults.
Introduction
Maintaining neuronal ion homeostasis and membrane potential (Vm) are critical to the survival and proper function of a healthy brain. These properties are sustained by the activity of ion pumps such as the Na+/K+ ATPase, which consumes ∼50% of the total neuronal adenosine triphosphate (ATP) turnover to regulate intracellular Na+ following the generation of action potentials (APs) (Howarth et al. 2012). Under normoxic conditions, neurons rely on aerobic energy production to generate the ATP required to drive these pumps and restore ion gradients after an AP. Conversely, during periods of low oxygen stress, aerobic metabolism ceases and anaerobic ATP production is insufficient to support ATP-driven ion transport. As a result, ion gradients and Vm run down and neurons undergo excitotoxic cell death (Choi, 1992). However, this does not occur in the brain of the anoxia-tolerant turtle, Chrysemys picta bellii, which can survive days (22°C) to months (3°C) of complete anoxia (Ultsch, 1985).
In C. picta brain, the catastrophic cascade of events that is characteristic of mammalian neuronal responses to low oxygen stress is avoided; instead, a suite of natural anoxic defence mechanisms is activated. These responses combine to down-regulate neuronal ATP consumption sufficiently to enable anoxic turtle neurons to subsist on anaerobically derived ATP (Bickler & Buck, 2007). As a result, the brain of this species is remarkably tolerant of complete anoxia and also of global or focal ischaemia and high K+ insults (Pamenter et al. 2012). To reduce cellular energy consumption during anoxia, AP firing frequency decreases due to a neuroprotective strategy termed spike arrest (SA) (Perez-Pinzon et al. 1992; Pamenter et al. 2011). The primary mechanism of SA is a potentiation of GABA transmission that induces an inhibitory shunting current and clamps Vm near the GABA reversal potential (EGABA, ∼−70 mV). This shunt impedes AP generation and thereby reduces the need for compensatory ATP-driven ion pumping (Pamenter et al. 2011). SA is critical to tolerance of anoxia and ischaemia; however, the mechanism through which decreases in oxygen availability are sensed and how this modulates GABA transmission has yet to be elucidated.
Reactive oxygen species (ROS) generation is a consequence of aerobic respiration through the mitochondrial electron transport chain (ETC) and thus inherently sensitive to changes in oxygen availability, making it a potentially important component of ROS-mediated signalling cascades. Indeed, sustained anoxia results in the elimination of intracellular ROS ([ROS]i) in turtle dorsal cortex (Pamenter et al. 2007; Dukoff et al. 2014). ROS have long been associated with pathological disease states, but more recently, evidence has emerged that ROS can regulate a variety of redox-sensitive proteins and signalling pathways (Veal et al. 2007). Of particular interest is superoxide (O2•−), which is converted to hydrogen peroxide (H2O2) in the cytosol (Adam-Vizi, 2005). A number of neurotransmitter systems have been found to be redox sensitive. For example, H2O2 suppresses dopamine release in striatal brain slices (Rice, 2011) and Ca2+-dependent exocytosis of glutamate in cortical synaptosomes (Zoccarato et al. 1995). GABA release is also sensitive to ROS with O2•− preventing GABA release in hypothalamus (Chen & Pan, 2007) and oxidants preventing GABA release in spinal cord (Yowtak et al. 2011). In addition, GABAA receptors are potentiated by reducing agents and inhibited by oxidizing agents (Amato et al. 1999; Calero et al. 2011).
Given the important inhibitory role of GABA during anoxia in turtle brain and the potential for redox modulation of GABA release and GABAA receptors we investigated the effects of redox-modulation on GABAergic transmission in cortical pyramidal neurons located in the dorsal cortex. Mitochondria are the major producers of ROS in the cell and are potentially important oxygen sensors; therefore, we asked whether modulation of mitochondrial ROS (mROS) generation is sufficient to initiate GABA-mediated SA.
Methods
Animals
This study was approved by the University of Toronto Animal Care Committee and conforms to the relevant guidelines issued by the Canadian Council on Animal Care regarding the care and use of experimental animals, and conforms to the principles of UK regulations, as described in Drummond (2009). Fifty adult male and female turtles (carapace diameter >15 cm, 300–600 g) were purchased from Niles Biological Inc. (Sacramento, CA, USA). Turtles were housed as described previously (Shin & Buck, 2003). All experiments were conducted at room temperature (22°C).
Whole-cell electrophysiology
Cortical sheet dissection and anoxic whole-cell patch-clamp recording methods are described elsewhere (Pamenter & Buck, 2008). Briefly, animals were decapitated and cortical sheets were dissected free and stored in artificial cerebrospinal fluid (aCSF) containing (in mm): 107 NaCl, 2.6 KCl, 1 MgCl2, 2 NaH2PO4 (2H2O), 26.5 NaHCO3, 10 glucose, 5 imidazole, 1.2 CaCl2 (2H2O) (pH 7.4, adjusted with 12 n HCl; osmolarity 285–290 mosmol (l solution)−1 at 4°C. For experiments, tissue was perfused with aCSF at 22°C. Patch-clamp recordings were made using fire-polished 4–8 MΩ borosilicate glass pipettes (Harvard Apparatus Ltd, Holliston, MA, USA). Physiological pipette solution contained (in mm): 130 potassium gluconate, 3 NaCl, 5 sodium gluconate, 1 MgCl2, 10 NaHepes, 0.3 NaGTP, 2 NaATP, 0.0001 CaCl2 (adjusted to pH 7.4 with methanesulfonic acid; osmolarity 295–300 mosmol (l solution)−1). For perforated-patch experiments the pipette solution included 25 μg ml−1 gramicidin. Whole-cell capacitance was compensated and typical whole-cell access resistances (Ra) were 20–30 MΩ. Ra was monitored periodically and recordings were discarded if it changed by more than 20% or whole-cell leak currents changed > ±30 pA. Electrophysiological recordings were performed on pyramidal neurons located in the dorsal cortex and dorsomedial cortex of the turtle cerebrocortex. These pyramidal neurons are glutamatergic and account for ∼80–90% of all the neurons in the cerebrocortex (Ulinski, 2007; Shepherd, 2011). The remaining 10–20% are non-pyramidal with GABAergic stellate interneurons as the primary inhibitory cells which provide feedforward and feedback inhibition to pyramidal cells (Connors & Kriegstein, 1986; Shepherd, 2011). Pyramidal neurons were differentiated from stellate cells by their responses to somatic current injections which produce trains of APs that accommodate in pyramidal cells but do not in stellate cells (Connors & Kriegstein, 1986; Shin & Buck, 2003).
Whole-cell conductance (Gw) was assessed by clamping neurons at voltage steps from −100 to −30 mV in 10 mV increments lasting 250 ms each. Current amplitudes were measured between 200 and 220 ms to avoid any capacitance effects, and a slope conductance was determined from the resultant current–voltage (I–V) relationship (Ghai & Buck, 1999; Pamenter et al. 2011). The reversal potential of the GABA-induced current (EGABA) was measured using a similar protocol (voltage steps from −100 to −50 mV in 10 mV increments) applied before and after a 15 s GABA (2 mm) application. Note: 15 s of 2 mm GABA application is sufficient to depolarize Vm to EGABA, and Vm recovers rapidly following cessation of GABA drip perfusion. I–V relationships were analysed by linear regression in the presence and absence of GABA, and EGABA was estimated by the intersection point of these two curves, as described elsewhere (Watanabe et al. 2009). This intersection point is the voltage at which there is no difference in current between the indicated treatment and GABA treatment and is a measure of the cell's background current indicating no effect of GABA perfusion and therefore the GABA reversal potential. In this study there was no significant difference between whole-cell and gramicidin-perforated patch measurements of Gw or EGABA; therefore, the data sets were grouped together. Redox agents react with Ag electrodes to produce voltage offsets (Berman & Awayda, 2013). To reduce potential artefacts in electrophysiological recordings molten AgCl was re-applied often on bath and reference electrodes. With turtle aCSF in the bath and physiological pipette solution in the recording pipette the liquid junction potential (LJP) was experimentally assessed as 14 mV (protocol based on Neher, 1992), and this value is supported by LJP calculations using a generalized version of the Henderson equation (Clampex junction potential calculator; Molecular Devices, Sunnyvale, CA, USA). The LJPs associated with ROS scavenger N-(2-mercaptopropionyl) glycine (MPG) and the oxidant hydrogen peroxide (H2O2) were 19 and −1 mV, respectively. All data have been corrected for these values offline. Experiments were performed in a repeated measures paired design starting with a 10 min normoxic pre-treatment, followed by a 30 min treatment and then a 30 min normoxic recovery period unless otherwise stated. H2O2, gabazine (GZ) and bicuculline methiodide (BIC) were applied for 5 min. Analysis of electrophysiological data was performed using Clampfit 10 software (Molecular Devices).
Measurement of GABAA receptor currents and charge transfer
To aid in the detection of GABAA receptor currents, neurons were voltage clamped at a holding potential of −100 mV and pipette [Cl−] was increased to 130 mm by equimolar substitution of CsCl for potassium gluconate. After establishing the whole-cell configuration, a 10 min equilibration period was provided for the exchange of ions, stabilization of the patch and for antagonism of non-GABAergic receptors and ion channels. Turtle pyramidal neurons exhibit two types of phasic GABAA receptor-mediated currents: fast spontaneous IPSCs (sIPSCs) and slower giant IPSCs (gIPSCs). Spontaneous IPSCS recorded in the presence of TTX are referred to as miniature IPSCs (mIPSCs). Both sIPSCs and mIPSCs were detected using a threshold search protocol with a trigger level set to 3 times the baseline noise (∼3 pA). All recordings were visually inspected and only events with a rapid onset (10–90% rise time <5 ms) were included in the analysis. Fast sIPSC and mIPSC data were assessed for 2 min at the end of each treatment period. Giant IPSCs are unique to turtle pyramidal neurons and have previously been identified as phasic currents (Pamenter et al. 2011). To differentiate them from sIPSCs they are referred to as gIPSCs throughout this manuscript. Due to the lower incidence of gIPSCs they were assessed for 5 min at the end of each treatment period. If required, mIPSCs or sIPSCs were normalized to the normoxic 5 min time point (>15 min after patch formation).
The amplitude of the tonic current was calculated as the difference between the holding current measured before and after BIC or H2O2 application. Two changes in the baseline holding current were calculated and statistically compared. The first measurement was between two 10 s periods 1 min apart immediately prior to inhibition of tonic GABA current; this was the average change in baseline current with no treatment. The second measurement was taken 1 min after BIC or H2O2 application; this was the average change in baseline current due to GABAA receptor inhibition. To ensure an accurate measurement of holding current, baseline was sampled at 5 ms epochs every 100 ms over a 10 s period, and any baseline points falling on the decay phase of an IPSC were omitted (Nusser & Mody, 2002; Bright & Smart, 2013). Tonic currents were measured following a 10 min normoxic control period and after 30 min of treatment.
Charge transfer (Q (pA × ms); the integrated area under an IPSC) associated with sIPSCs and gIPSCs was calculated according to the equation: QIPSC = (ftreatment × Qtreatment) × Δt, where ftreatment is the mean frequency (Hz) of the IPSCs, Qtreatment is the mean charge transfer (pC) per IPSC, and Δt is time. The charge transfer associated with the tonic current was calculated according to the equation QTC = ITC × Δt, where ITC is the tonic current (pA) (Bai et al. 2001).
Fluorescence imaging
Methods for dye loading and fluorescence imaging of intracellular ROS ([ROS]i) with the membrane-permeable ROS-sensitive fluorescent indicator 5-(and-6)-chloromethyl-2',7'-dichlorodihydro-fluorescein diacetate, acetyl ester (CM-H2DCFDA; Invitrogen, Burlington, ON, Canada) are described elsewhere (Dukoff et al. 2014). Briefly, cortical sheets were incubated in aCSF containing 5 μm CM-H2DCFDA (from a 1 mm stock solution in dimethylsulfoxide (DMSO)) for 30 min (4°C) followed by a 30 min wash in aCSF (22°C). Once in the cell the acetate groups are cleaved by intracellular esterases yielding the non-fluorescent 5-(and-6)-chloromethyl-2',7'-dichlorodihydro-fluorescein (CM-H2DCF) (Koopman et al. 2006). Steady state normoxic generation of ROS leads to oxidation of the CM-H2DCF to fluorescent 5-(and-6)-chloromethyl-2',7'-dichloro-fluorescein (CM-DCF) and a subsequent increase in fluorescence. Cessation of ROS generation results in no change in CM-DCF fluorescence.
Mitochondrial O2•− generation was assessed using the cell-permeant fluorescent probe MitoSOX Red (excitation/emission: 510 nm/580 nm; Invitrogen) (Robinson et al. 2008). Cortical sheets were incubated in 5 μm MitoSOX Red (dissolved in DMSO) for 30 min at 4°C, then washed in aCSF for 15 min at 22°C. For both CM-DCF and MitoSOX experiments cortical sheets were exposed to normoxic control aCSF for 10 min, followed by treatment aCSF for 20–30 min until steady state, and in some experiments reperfused with normoxic control aCSF for 20 min or treated with 50 μm H2O2 for 5 min. In both sets of experiments, the average change in regions of interest from the centre of the cell body of 10 neurons per cortical sheet were chosen and used as a single replicate. To assess treatment effects on ROS generation the fluorescence value at the onset of treatment steady state was compared to a linear regression line fitted to the 10 min normoxic portion of the trace. Data are presented as percentage change expressed relative to that fitted normoxic regression line.
Pharmacology
Elimination of intracellular ROS was achieved using the ROS scavenger N-(2-mercaptopropionyl) glycine (MPG; 0.5 mm) (Leroy et al. 1991). Elimination of mitochondrial O2•− was achieved using the mitochondrial O2•− scavenger MitoTEMPO (20 μm). Mitochondrial ROS generation was decreased with a complex IV (cytochrome c oxidase) inhibitor sodium cyanide (CN; 0.5 mm) (Turrens, 2003). Modulators of [ROS]i were dissolved in aCSF and drip and bath applied. The following drugs were drip perfused: H2O2 (50 μm), GABA (2 mm), GZ (SR-95531; 25 μm), BIC (100 μm), strychnine (2 μm), (2R)-amino-5-phosphonopentanoate (AP5; 25 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 25 μm) and tetrodotoxin (TTX; 1 μm). All chemicals were obtained from Sigma-Aldrich Canada Ltd (Oakville, ON, Canada), except TTX, which was obtained from Tocris Cookson (Bristol, UK).
Statistics
Electrophysiological and fluorescence data were analysed using SigmaPlot software package version 11.0 (Systat Software, Inc., San Jose, CA, USA). All data were tested for normality and equal variance, and either an analysis of variance or a Kruskal–Wallis analysis was used for between-group comparisons where appropriate. Post hoc analyses consisted of Holm-Sidak or Tukey's. Significance was assessed relative to 30 min normoxia unless otherwise stated. Data are expressed as means ± SEM; P < 0.05 was considered statistically significant. For Vm and GABA current data sets pre-treatment data were not different from normoxic control data; therefore, treatments are compared to 30 min normoxic control data.
Results
Fluorescent assessment of ROS generation in cortical pyramidal neurons
To investigate a role for ROS in modulating GABA receptor currents we first confirmed that [ROS]i were eliminated by our various treatment protocols. We have previously shown that in cortical brain sheets anoxia and general ROS scavengers (MPG and N-acetylcysteine) decrease CM-DCF fluorescence indicating that under these conditions cellular ROS production is inhibited (Pamenter et al. 2007; Dukoff et al. 2014). Compared to the 10 min normoxic baseline, the rate of increase in CM-DCF fluorescence did not change over 1 h of normoxic perfusion (0.9 ± 0.2%; n = 4; Fig.1A and Ba), indicating maintenance of cellular redox homeostasis. However, anoxia significantly decreased fluorescence compared to normoxic controls (−8.0 ± 1.2%; n = 4; P < 0.001; Fig.1A and Bb). Mitochondrial ROS generation is arrested in anoxic cells because there is no oxygen available to remove electrons from the ETC. This could be an important signal to initiate ROS-mediated signalling cascades. To assess if decreases in mROS generation mimic anoxia we treated cortical sheets with CN, a complex IV (cytochrome c oxidase) inhibitor. Consistent with Pamenter et al. (2007), perfusion of normoxic or anoxic aCSF plus CN decreased CM-DCF fluorescence compared to normoxic control (−8.6 ± 1.5 and −8.4 ± 0.7%, respectively; n = 5 each; P < 0.001 for both; Fig.1A and Bc). To confirm that ROS produced by the mitochondria has the capacity to modulate intracellular redox state we next assessed the effect of scavenging mitochondrial O2•− with the mitochondrial-specific O2•− scavenger MitoTEMPO. Compared to normoxic control, MitoTEMPO decreased CM-DCF fluorescence by −10.7 ± 0.8% (n = 4; P < 0.001 for both; Fig.1A and Bd). Application of H2O2 during any of the treatments increased CM-DCF fluorescence confirming sensitivity of the dye to exogenous H2O2.
Figure 1.
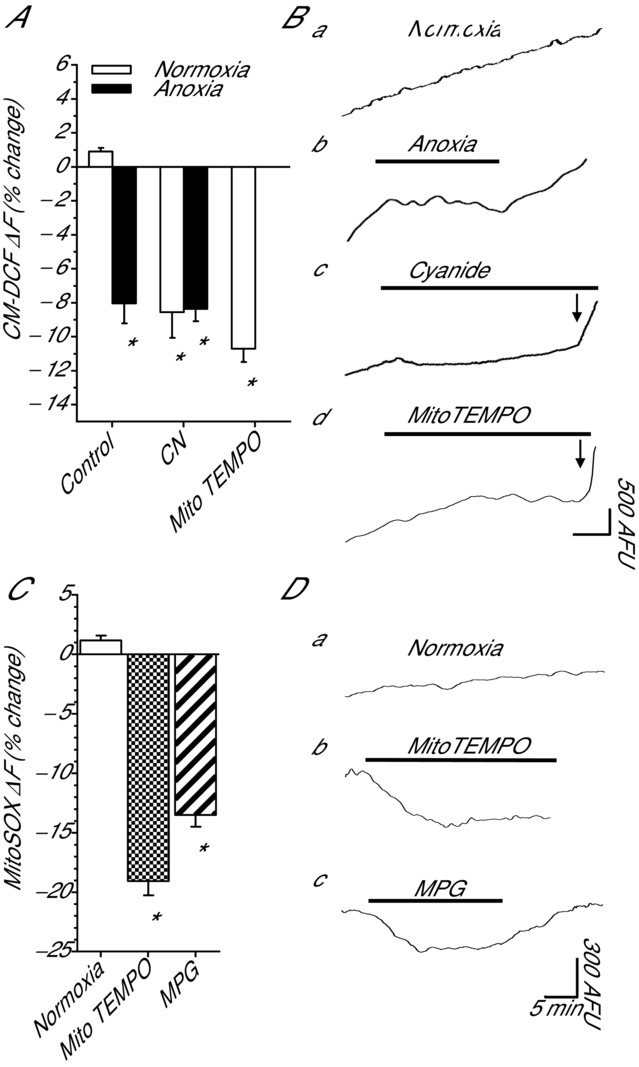
Fluorescent assessment of mitochondrial ROS generation and intracellular redox state following ROS-modulating treatments in cortical neurons
A, summary of changes in CM-DCF fluorescence. B, sample CM-DCF fluorescence recordings from A. Note: when trace is horizontal there is zero [ROS]i generation. Horizontal scale bar: 5 min in a–c, and 10 min in d. C, summary of changes in MitoSOX fluorescence. D, sample MitoSOX fluorescence recordings from C. Arrow indicates onset of H2O2 application. Bars represent duration of treatment. Arbitrary fluorescence units (AFU). Treatments: normoxia (95% O2/5% CO2-bubbled aCSF), anoxia (95% N2/5% CO2-bubbled aCSF), CN (0.5 mm), MPG (0.5 mm), MitoTEMPO (20 μm) and H2O2 (50 μm). Data are means ± SEM, n = 4–5 replicates per treatment. *Significant difference of treatment from normoxic controls (P < 0.05).
To specifically assess mROS generation we next used the mitochondrial targeted O2•− indicator MitoSOX. Compared to the 10 min normoxic baseline, the rate of change in MitoSOX fluorescence did not change over a 1 h normoxic treatment (1.17 ± 0.1%; n = 4; Fig.1C and Da), indicating a steady state rate of mitochondrial O2•− generation which agrees with the CM-DCF measurements. Compared to normoxic control, treatment with MitoTEMPO or MPG decreased O2•− generation by −19.1 ± 1.3% and −13.5 ± 1.0% (n = 4 for each; P < 0.001 for both; Fig.1C and Dbc). These data indicate that treatment with MitoTEMPO and MPG decrease mitochondrial ROS generation and mimic the anoxia-mediated decreases in [ROS]i. MitoSOX is positively charged and accumulates preferentially in mitochondria due to the strong negative membrane potential (Robinson et al. 2008); therefore, meaningful measurements using MitoSOX require a stable mitochondrial membrane potential. In turtle cortical neurons, anoxia (Hawrysh & Buck, 2013), and CN (data not shown) depolarize mitochondrial membrane potential and this reduces the driving force on MitoSOX uptake and could result in underestimation of superoxide generation. Therefore, under these conditions it is not possible to properly assess the effects of anoxia and cyanide on mitochondrial superoxide generation.
Pharmacological or anoxia-mediated decreases in [ROS]i shift Vm to EGABA by activating GABAA receptors
Cortical pyramidal neurons are the primary excitatory neurons in the turtle dorsal cortex; therefore, under anoxic conditions electrical suppression of these neurons is of particular interest because this will reduce ATP consumption and prolong survival. To investigate the effects of ROS modulation on GABAergic transmission in these neurons, we recorded in the perforated-patch and whole-cell patch clamp configuration. In passive current clamp recordings, normoxic pyramidal neurons had a Vm of −87.3 ± 1.9 mV (n = 10), and this was hyperpolarized relative to EGABA (−77.6 ± 1.6 mV; n = 10; P < 0.001; Fig.2A). EGABA was not affected by oxygen availability or any pharmacological treatment (n = 4–10 each; P = 0.920; not shown); therefore, data are compared to normoxic EGABA. Anoxia depolarized Vm to −77.6 ± 1.5 mV (n = 9; P < 0.001) and it was not different from EGABA (P = 0.986; Fig.2A and Bb). To determine if decreases in [ROS]i alone could mimic the anoxic shift in Vm we treated cortical sheets with MPG. Compared to normoxic control, MPG treatment depolarized Vm (−77.2 ± 1.4 mV; n = 11; P < 0.001) to EGABA (P = 0.863; Fig.2A and Bc). Co-treatment with anoxia plus MPG depolarized Vm to −75.3 ± 1.9 mV (n = 11; P < 0.001) and Vm was not different from anoxic Vm (P = 0.375) or EGABA (P = 0.347) (Fig.2A). A subsequent 5 min application of GZ hyperpolarized Vm under both normoxic (−83.5 ± 2.8 mV; n = 6) and anoxic (−85.3 ± 2.1 mV; n = 4) conditions, and Vm was different from MPG treatment alone (P = 0.040 and P = 0.013, respectively; Fig.2A), and from EGABA (P = 0.042 and P = 0.022, respectively; Fig.2A).
Figure 2.
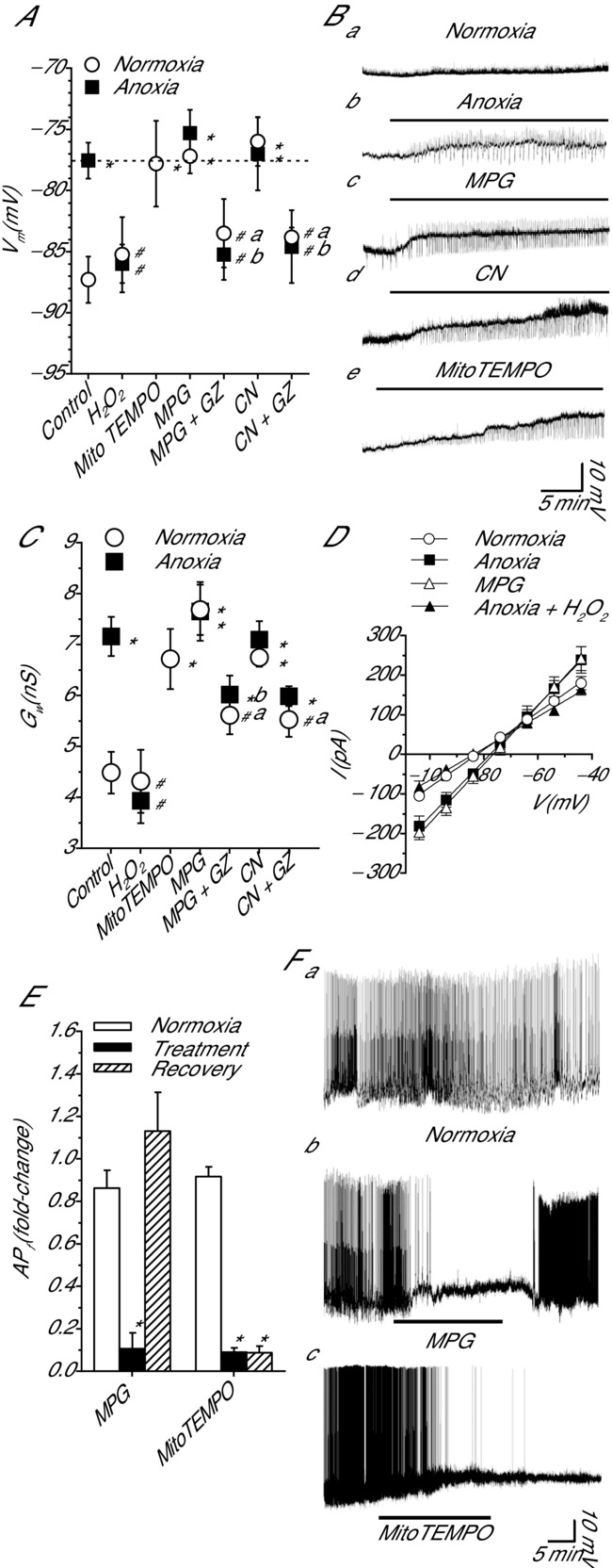
Pharmacological or anoxia-mediated decreases in [ROS]i shift pyramidal neuron Vm to EGABA by activating a GZ-sensitive increase in Gw; ROS scavenging decreases APf in a subset of spontaneously active cortical pyramidal neurons
A, summary graph showing Vm depolarization following a 30 min ROS-depleting treatment, anoxia, ROS scavenger MPG and mitochondrial cytochrome c oxidase inhibitor CN. Addition of the GABAA receptor inhibitor GZ prevents Vm changes, and the oxidant H2O2 reverses Vm changes. Note: the dashed line represents EGABA. B, sample raw traces showing treatment-induced changes in Vm. C, summary of changes in Gw following treatments as outlined in A. D, sample I–V relationships used to determine Gw in C. E, summary of change in APf following treatment with MPG or MitoTEMPO. F, sample raw traces used to generate E. Horizontal bars represent duration of treatment. Treatments: normoxia (95% O2/5% CO2-bubbled aCSF), anoxia (95% N2/5% CO2-bubbled aCSF), MPG (0.5 mm), MitoTEMPO (20 μm), GZ (25 μm), CN (0.5 mm) and H2O2 (50 μm). Data are means ± SEM, n = 4–11 replicates per treatment. *Significant difference from normoxic controls. #Significant difference from EGABA in A or anoxia in C. The letters ‘a’ and ‘b’ indicate a significant within-treatment different due to GZ (normoxia and anoxia, respectively) (P < 0.05).
Since CN-mediated inhibition of complex IV decreased [ROS]i to an extent similar to the effect of anoxia we next tested the effects of CN on Vm. CN treatment depolarized Vm (−76.0 ± 2.0 mV; n = 6; P < 0.001) and it was not different from EGABA (P = 0.577; Fig.2A and Bd). Co-application of CN and anoxia depolarized Vm compared to normoxic control (−77.0 ± 3.0 mV; n = 5; P < 0.001) but not more than anoxia alone (P = 0.859; Fig2A). Application of GZ reversed the CN-mediated Vm shift under both normoxic (−83.8 ± 0.9 mV; n = 6) and anoxic conditions (−84.6 ± 3.0 mV; n = 5), and Vm was different from CN treatment alone (P = 0.003 and P = 0.006, respectively; Fig.2A) and compared to EGABA (P = 0.032 and P = 0.023, respectively; Fig.2A). Scavenging mitochondrial O2•− with MitoTEMPO depolarized Vm to −77.8 ± 3.5 mV compared to normoxic control (n = 5; P = 0.003), and this value was not different from EGABA (P = 0.947; Fig.2A and Bd). Finally, we tested whether this effect could be reversed by the addition of an oxidant. Application of H2O2 did not change Vm under normoxic conditions (−85.3 ± 3.1 mV; n = 4; P = 0.525). However, in anoxic neurons H2O2 application hyperpolarized Vm back to normoxic values (−77.5 ± 1.3 to −86.0 ± 1.6 mV; n = 4; P = 0.687; Fig.2A).
An interesting observation is the appearance of depolarizing excitatory postsynaptic potentials under the treatment conditions outlined in Fig.2B. Preliminary studies indicate that these are a combination of glutamatergic and GABAergic currents (data not shown). Future studies will investigate their origin and contribution to changes in Vm.
ROS scavengers increase Gw by activating GABAA receptors
A GZ-sensitive shift in Vm to EGABA indicates that a decrease in cortical ROS activate a GABAA receptor-mediated current in pyramidal neurons. To confirm this we assessed Gw in pyramidal neurons following perfusion of anoxic aCSF, ROS scavengers, CN or H2O2. Compared to normoxia, anoxic treatment caused an increase in Gw from 4.5 ± 0.4 to 7.2 ± 0.4 nS, respectively (n = 10 and 9, respectively; P < 0.001; Fig.2C and D). Normoxic MPG treatment increased Gw to 7.7 ± 0.5 nS (n = 8), and this was not different from anoxic values (P = 0.352). Subsequent application of GZ to normoxic MPG-treated neurons decreased Gw to a level not different from normoxic control (5.6 ± 0.4 nS; n = 6; P = 0.069; Fig.2C). MPG plus anoxia also increased Gw (7.7 ± 0.6 nS; n = 8) but this was not different from anoxia alone indicating activation of a similar pathway (P = 0.385; Fig.2C). Subsequent application of GZ to anoxic neurons decreased Gw compared to anoxic values (6.0 ± 0.3 nS; n = 6); however, this was not statistically significant (P = 0.066). When comparing MPG-treated neurons, GZ induced a significant decrease in Gw under both normoxic and anoxic conditions (P = 0.008 and P = 0.001, respectively). Compared to normoxic control, CN application increased Gw in normoxic and anoxic neurons (6.8 ± 0.2 and 7.1 ± 0.4 nS; n = 9 each; P < 0.001 for both; Fig.2C) and these values were not different from anoxic controls (P = 0.452 and P = 0.604, respectively). Subsequent treatment with GZ decreased Gw under both normoxic and anoxic conditions (5.5 ± 0.3 and 6.0 ± 0.2 nS, respectively; n = 6 for both); however, only under normoxic conditions was Gw not different from normoxic values (P = 0.097). When comparing CN-treated neurons, GZ induced a decrease in Gw under both normoxic and anoxia conditions; however, only in the normoxic CN + GZ-treated neurons did GZ treatment significantly decrease Gw values compared to CN treatment alone (P = 0.004; Fig.2C). Scavenging mitochondrial O2•− with MitoTEMPO increased Gw to 6.7 ± 0.6 nS compared to normoxic control (n = 6; P < 0.001; Fig.2C), and this was not different from anoxic control (P = 0.471). To test if pyramidal neuron Gw changes could be reversed by an oxidant, H2O2 was applied. Normoxic H2O2 application did not change Gw from normoxic values (4.3 ± 0.6; n = 6; P = 0.683; Fig.2C). However, compared to the anoxic control H2O2 decreased Gw under anoxic conditions (3.9 ± 0.4 nS; n = 6; P < 0.001; Fig.2C and D).
ROS scavenging decreases AP frequency
A subset of pyramidal neurons located in the medial aspect of the dorsal cortex are spontaneously active (Shen & Kriegstein, 1986). These neurons are well suited to use as a direct measure of SA since decreases in AP frequency (APf) indicate electrical suppression. Anoxic perfusion decreases APf in these neurons (Pamenter et al. 2011); therefore, the effect of ROS scavenging with MPG and MitoTEMPO on APf was assessed. Normoxic MPG treatment decreased APf from 0.86 ± 0.1 to 0.1 ± 0.1 Hz (n = 6; P < 0.001; Fig.2E and Fb), and following recovery APf returned to baseline levels (P = 0.146). Normoxic application of MitoTEMPO decreased APf from 0.92 ± 0.05 to 0.09 ± 0.04 Hz (n = 4; P < 0.001; Fig.2E and Fc). APs did not recover within the 30 min normoxic recovery period.
Decreases in ROS enhance mIPSC frequency
Enhanced GABA transmission can occur through either direct modulation of postsynaptic receptors or by a presynaptic mechanism. To determine which aspects of GABA transmission ROS scavenging regulates we assessed mIPSC amplitude and frequency. Compared to normoxia (−8.2 ± 1.7 pA; n = 6) mIPSC amplitude did not change with anoxia (−5.7 ± 0.9 pA; n = 6) or MPG treatment (−7.8 ± 1.1 pA; n = 6) (P = 0.369; Fig.3A and C). The frequency of mIPSCs was stable within a recording but variable between cells; therefore, this data set was normalized (see Methods for details). Compared to normoxia (1.0 ± 0.1; n = 6), anoxia and MPG treatment more than doubled mIPSC frequency (2.4 ± 0.2 and 3.0 ± 0.6, respectively; n = 6 for both; P = 0.029 and P = 0.002, respectively; Fig.3B and C).
Figure 3.
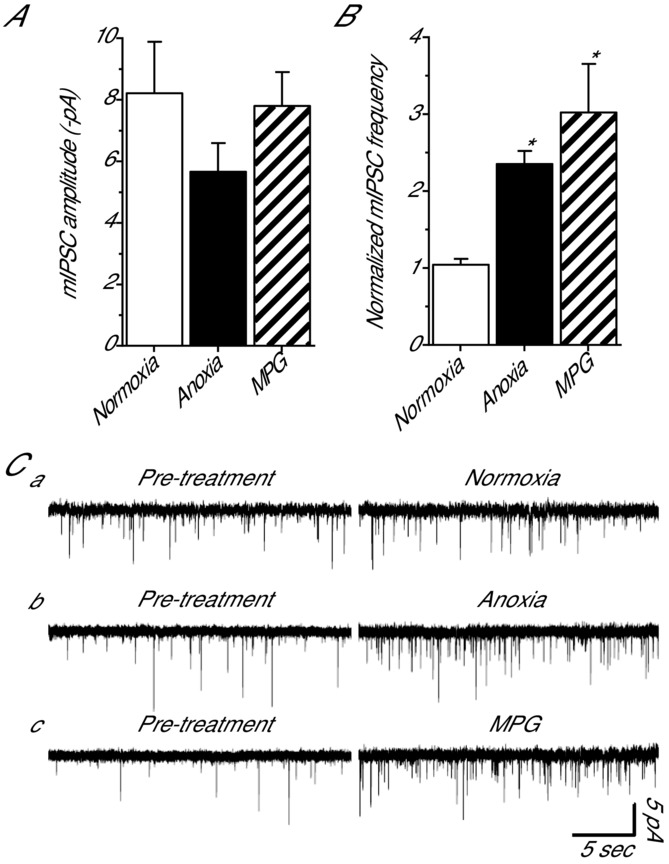
Pharmacological and anoxia-mediated decreases in [ROS]i increase mIPSC frequency but not amplitude in cortical pyramidal neurons
A, summary of mIPSC amplitude following a 30 min treatment with anoxia or the ROS scavenger MPG. B, summary of normalized mIPSC frequency under the same conditions as A. C, sample raw traces used to generate A and B. Note: normoxic pre-treatment (10 min) and treatment (30 min). GABAA receptor-mediated mIPSCs were enhanced with high [Cl−] pipette solution (130 mm) and isolated with NMDA and AMPA receptor antagonists (AP5 and CNQX, respectively; 25 μm each), a voltage-gated Na+ channel inhibitor (TTX; 1 μm), and a glycine receptor antagonist (strychnine; 2 μm). Treatments: normoxia (95% O2/5% CO2-bubbled aCSF), anoxia (95% N2/5% CO2-bubbled aCSF), MPG (0.5 mm). Data are means ± SEM, n = 6 replicates per treatment. *Significant difference from normoxic controls (P < 0.05).
Decreases in ROS enhance GABAA receptor sIPSC amplitude
To determine if sIPSCs are sensitive to ROS scavenging and to quantify their roles in GABAergic SA we assessed the effects of ROS depletion on their amplitude and frequency. Spontaneous IPSC frequency did not change following any treatment compared to normoxia (11.1 ± 0.71; n = 5–7; P = 0.498; not shown). The amplitude of sIPSCs was stable within a recording but variable between cells; therefore, this data set was normalized (see Methods for details). Anoxia increased sIPSC amplitude (1.1 ± 0.05 to 1.9 ± 0.08; n = 5 each; P = 0.009; Fig.4A, Dd and De) relative to the normoxic control measurements. MPG treatment increased sIPSC amplitude compared to normoxic control (2.0 ± 0.2; n = 7; P = 0.004; Fig.4A). Co-treatment with anoxia plus MPG was not different from anoxic conditions (2.1 ± 0.3; n = 7 each; P = 0.974; Fig.4A). CN treatment increased sIPSC amplitude (1.6 ± 0.05; n = 7; P = 0.046; Fig.4A) relative to normoxic control. Co-treatment with anoxia plus CN was not different from anoxic treatment (1.8 ± 0.04; n = 7; and P = 0.539; Fig.4A). Compared to normoxic control, treatment with MitoTEMPO doubled sIPSC amplitude to 2.0 ± 0.3 (n = 5; P = 0.004; Fig.4A and B). Treatment with GZ eliminated sIPSCs under all treatment conditions (Fig.4Df, E and F).
Figure 4.

GABAA receptor-mediated spontaneous IPSCs and giant IPSCs are sensitive to pharmacological and anoxia-mediated decreases in [ROS]i in cortical pyramidal neurons
A, summary of changes in sIPSC amplitude in response to a 30 min normoxic or anoxic treatment with the ROS scavenger MPG, mitochondrial cytochrome c oxidase inhibitor CN, or mitochondrial O2•− scavenger MitoTEMPO. Note: data were normalized to a preceding 2 min normoxic time point. B, sample raw spontaneous voltage-clamp recording demonstrating the effect of MitoTEMPO on GABAergic IPSCs. C, summary of gIPSC amplitude in response to the same 30 min treatment as in A. D, sample raw spontaneous voltage-clamp recording demonstrating the change in GABA currents during a transition to anoxia. a–c expanded trace showing the effect of anoxia and GZ on gIPSCs. d–f, expanded trace showing the effect of anoxia and GZ on sIPSCs. The scale bars correspond, from top to bottom, to: 200 pA/75 s, 200 pA/7.5 s, 40 pA/750 ms. E, sample raw spontaneous voltage-clamp recording demonstrating MPG-mediated changes in GABA currents. Note: GZ application inhibits sIPSCs and gIPSCs demonstrating ROS scavenger-mediated increase in GABA release and activation of GABAA receptors. F, superimposed spontaneous gIPSC recordings demonstrating that gIPSC amplitude increases following application of CN and these are GABAA receptor mediated because GZ inhibits them. Note: traces are averaged from 4 recordings each, 10 gIPSC events per recording. Neurons were voltage clamped at −100 mV and GABAA receptor currents were enhanced with high [Cl−] pipette solution (130 mm) and isolated with NMDA and AMPA receptor antagonists (AP5 and CNQX, respectively; 25 μm each). Note: due to high pipette chloride, anoxia and ROS scavenging occasionally caused action currents. Bars indicate treatment duration. Treatments: normoxia (95% O2/5% CO2-bubbled aCSF), anoxia (95% N2/5% CO2-bubbled aCSF), MPG (0.5 mm), GZ (25 μm), CN (0.5 mm). Data are means ± SEM, n = 5–7 replicates per treatment. *Significant difference from normoxic controls (P < 0.05).
GABA-mediated gIPSC amplitude increases with ROS depletion
Across all treatments the frequency of gIPSCs did not change relative to normoxic frequencies (0.09 ± 0.01 Hz; n = 7; P = 0.370; not shown). Relative to normoxic control (−228.1 ± 11.8 pA; n = 7), anoxia and MPG more than doubled gIPSC peak amplitude (−574.6 ± 93.2 and −515.0 ± 64.0 pA, respectively; n = 7 each; P = 0.003 and P = 0.013, respectively; Fig.4C, Da, Db and E). Co-application of MPG and anoxia also increased gIPSC amplitude over normoxic control (−675.9 ± 109.4 pA; n = 7; P < 0.001), but not more than anoxia alone (P = 0.366; Fig.4C). CN application increased gIPSC amplitude under normoxic (−470.4 ± 52.02 pA; n = 7; P = 0.034) and anoxic conditions (−541.0 ± 119.08 pA; n = 7; P = 0.007) compared to normoxic control (Fig.4C and F). Treatment with MitoTEMPO also increased gIPSC amplitude relative to normoxic control values (480.0 ± 35.0; n = 5; P = 0.029; Fig.4C). Treatment with GZ eliminated gIPSCs under all treatment conditions (Fig.4Dc, E and F).
Decreases in ROS enhance tonic GABAA receptor currents
To investigate the role of tonic GABAergic inhibition in anoxia-mediated SA we applied BIC following ROS scavenging treatments. BIC was used to measure tonic GABAA receptor-mediated currents because the extrasynaptic GABAA receptors likely to be responsible for tonic currents are more sensitive to BIC then to GZ (Bai et al. 2001). Under normoxic conditions BIC induced an outward shift in the holding current relative to baseline (19.9 ± 1.9 pA; n = 5; P = 0.034; Fig.5A and Ca) indicating inhibition of a GABAA receptor-mediated tonic current. The BIC-induced shift in holding current doubled with anoxia and was larger than the normoxic current (40.6 ± 8.2 pA; n = 5; P < 0.001; Fig.5A and Cb). Application of BIC to cortical neurons treated with MPG or CN revealed a tonic current that was significantly larger than the normoxic current (43.8 ± 7.3 and 39.0 ± 4.2 pA; n = 5 each; P < 0.001 for both) but not different from anoxia (P = 0.667 and P = 0.838, respectively; Fig.5A, Cc and Cd). Application of BIC to MitoTEMPO-treated neurons also induced an increase in holding current that was larger than normoxic control (33.9 ± 5.6 pA; n = 5; P < 0.001; Fig.5A and Ce).
Figure 5.
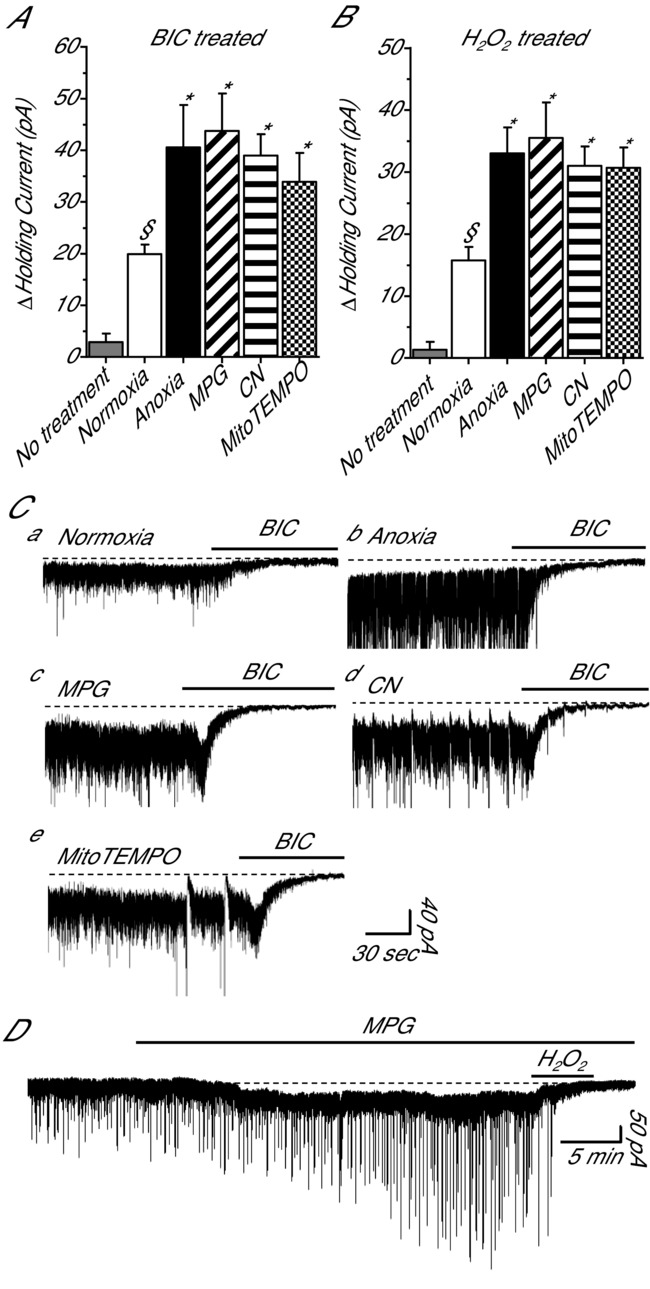
Tonic GABAA receptor currents in cortical pyramidal neurons are inhibited by H2O2 A, summary of the effects of bicuculline on tonic GABAA receptor currents following normoxia, anoxia, MPG or CN. B, summary of the effects of H2O2 on tonic GABAA receptor currents following the same treatments as in A. Note: no treatment (grey bars) in A and B represent the average change in the holding current prior to application of BIC or H2O2, respectively. C, sample raw traces used to generate A. D, sample raw trace used to generate B. Bars denote treatment duration. Dashed lines highlight the GABA-mediated tonic current. Pyramidal neurons were voltage clamped at −100 mV and GABAA receptor currents were enhanced with high [Cl−] pipette solution (130 mm) and isolated with NMDA and AMPA receptor antagonists (AP5 and CNQX, respectively; 25 μm each). Treatments: normoxia (95% O2/5% CO2-bubbled aCSF), anoxia (95% N2/5% CO2-bubbled aCSF), MPG (0.5 mm), CN (0.5 mm), BIC (100 μm) and H2O2 (50 μm). Data are means ± SEM, n = 5–7 replicates per treatment. §Significant difference from baseline. *Significant difference from normoxic controls (P < 0.05).
H2O2 inhibits GABA transmission
Since ROS scavenging increases GABA transmission we asked if this effect is reversed by the presence of the oxidant H2O2. Following treatment with either normoxia or anoxia, a 5 min application of H2O2 completely inhibited sIPSCs and gIPSCs (data not shown). Relative to baseline, H2O2 induced an outward shift in the holding current under normoxic and anoxic conditions (15.8 ± 2.2 and 33.0 ± 4.2 pA, respectively; n = 4 and 5; P = 0.022 and P < 0.001; Fig.5B). To assess if GABA transmission can be modulated exclusively by redox agents, H2O2 was applied following normoxic MPG. H2O2 application abolished MPG-mediated sIPSCs and gIPSCs (Fig.5D). Relative to baseline, H2O2 also induced an outward shift in the holding current in MPG (35.5 ± 5.7 pA; n = 4; P < 0.001; Fig.4B and D). H2O2 application to CN treatment neurons revealed a 31.0 ± 3.1 pA outward current compared to baseline (n = 4; P < 0.024; Fig.4B). Application of H2O2 to MitoTEMPO-treated neurons also induced an increase in holding current that was larger than normoxic control (30.8 ± 3.5 pA; n = 4; Fig.5B).
Decreases in ROS increase GABAA receptor-mediated charge transfer
To estimate the relative contribution of GABAergic sIPSCs, gIPSCs and tonic currents to SA we calculated the charge transfer associated with each current over a 2 min period (Fig.6A and B). Compared to pre-treatment, the charge transfer resulting from sIPSCs was stable for over 30 min (1.1 ± 0.11 and 1.2 ± 0.10 μC; n = 6; P = 0.454). Anoxia (2.0 ± 0.26 μC; n = 5; P = 0.026), MPG (1.8 ± 0.22 μC; n = 7; P = 0.039), CN (2.4 ± 0.60 μC; n = 6; P = 0.001), and MitoTEMPO (2.4 ± 0.4 μC; n = 5; P = 0.003) all doubled sIPSC-mediated charge transfer compared to normoxia, and this is consistent with their effect on current amplitude (Fig.6A). Charge transfer resulting from gIPSCs did not change over 30 min of normoxia (1.7 ± 0.62 to 1.7 ± 0.69 μC; n = 7) and conducted ∼40% more current than normoxic sIPSCs. Anoxia (3.3 ± 0.33 μC; n = 7; P = 0.06), MPG (3.6 ± 0.63 μC; n = 7; P = 0.001), CN (3.0 ± 0.4 μC; n = 7; P = 0.022), and MitoTEMPO (3.0 ± 0.5 μC; n = 5; P = 0.022) treatment doubled charge transfer compared to normoxic gIPSC values (Fig.6A). Charge transfer associated with tonic GABA currents did not change over 30 min of normoxia (2.0 ± 0.12 and 2.4 ± 0.23 μC; n = 7) and was ∼40% larger than gIPSCs and ∼200% larger than sIPSCs under normoxic conditions. Anoxia (4.4 ± 0.78 μC; n = 5; P = 0.023), MPG (5.3 ± 0.87 μC; n = 5; P = 0.006), CN (4.7 ± 0.52 μC; n = 5; P = 0.012) and MitoTEMPO (4.3 ± 0.5 μC; n = 5; P = 0.029) treatment all doubled charge transfer compared to normoxic tonic GABA currents (Fig.6A). On average charge transfer resulting from tonic currents was ∼45% larger than gIPSCs and ∼240% larger than sIPSCs within each treatment condition.
Figure 6.
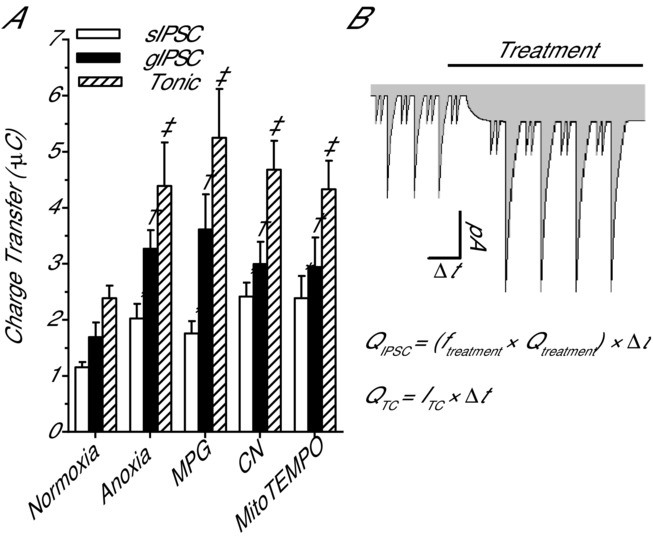
Comparison of the pharmacological and anoxia-mediated charge transfer associated with GABAA receptor-mediated sIPSCs, gIPSCs and tonic currents
A, summary of the cumulated charge transfer resulting from GABAA receptor sIPSCs, gIPSCs and tonic currents after a 30 min treatment with anoxia, MPG or CN. Note: charge transfer is calculated over a duration of 2 min at the end of the treatment period. B, schematic drawing and equations detail the methods used to calculate charge transfer. Note: grey shading indicates charge transfer associated with GABAA receptor currents in pyramidal neurons. Bar denotes treatment duration. Pyramidal neurons were voltage clamped at −100 mV and GABAA receptor currents were enhanced with high [Cl−] pipette solution (130 mm) and isolated with NMDA and AMPA receptor antagonists (AP5 and CNQX, respectively; 25 μm each). Treatments: normoxia (95% O2/5% CO2-bubbled aCSF), anoxia (95% N2/5% CO2-bubbled aCSF), MPG (0.5 mm) and CN (0.5 mm). Data are means ± SEM, n = 5–7 replicates per treatment. *Significant difference from normoxic sIPSCs. †Significant difference from normoxic gIPSCs. ‡Significant difference from normoxic gIPSCs (P < 0.05).
Discussion
In this study we explore the effects of alterations in [ROS]i on GABAergic transmission in cortical sheets of an anoxia-tolerant vertebrate. We demonstrate that in turtle dorsal cortex GABAergic transmission is enhanced by pharmacological decreases in [ROS]i (MPG), and that increases in [ROS] (H2O2) at physiological concentrations inhibits it. CN-mediated inhibition of mROS generation by inhibition of complex IV of the mitochondrial ETC or MitoTEMPO-mediated scavenging of mitochondrial O2•− mimics the effects of both anoxia and general MPG-mediated ROS scavenging, indicating that mitochondria are an in situ oxygen sensor capable of enhancing GABAergic transmission (i.e. SA). GABA-mediated SA is a critical component of the turtle's anoxia-tolerance strategy and our results identify a unique redox-sensitive inhibitory signalling pathway that enables survival during prolonged anoxic stress.
During normoxia, ROS levels are maintained by the balance between ROS-generating and natural ROS-scavenging pathways (Pitlik et al. 2009). Under anoxic conditions, however, oxygen is rapidly metabolized by mitochondria resulting in a decrease in [ROS]i which can modulate ROS-sensitive signalling pathways by shifting cellular redox state. In turtle, this is confirmed by our fluorescence measurement of [ROS]i which shows ROS generation is inhibited after ∼10 min of anoxia, and agrees with measurements of bath chamber 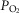 , which decreases to ∼ 0 mmHg within the same time frame (Dukoff et al. 2014). Neurons under anoxic conditions are therefore unlikely to have a significant source of ROS and intracellular proteins are expected to be in a reduced state or at least not encountering oxidative signals. The free radical scavenger MPG neutralizes ROS as they are generated and provides a general indication of the role of ROS in biological processes. In this study we used MPG to assess the sensitivity of GABAA receptors to decreases in [ROS]i. The effectiveness of MPG at scavenging intracellular ROS in turtle dorsal cortex has previously been confirmed (Dukoff et al. 2014); however, as a thiol-based ROS scavenger it may not accurately replicate the physiological effects of decreases in ROS under anoxic conditions, since MPG may additionally directly reduce cysteine residues on redox-sensitive proteins. We think this is unlikely for two reasons. First, the anoxic application of MPG did not alter the time course or the magnitude of the decrease in [ROS]i. Second, CN which does not directly modulate cysteine residues mimics both anoxia and MPG-mediated changes in CM-DCF fluorescence. An important finding from this study is that mitochondrial O2•− scavenging with MitoTEMPO reduced CM-DCF fluorescence. This is important because it indicates that mitochondrially derived O2•− is the major source of ROS in turtle cortical neurons. This is supported by the CN experiments which also prevented mROS generation under normoxic conditions.
, which decreases to ∼ 0 mmHg within the same time frame (Dukoff et al. 2014). Neurons under anoxic conditions are therefore unlikely to have a significant source of ROS and intracellular proteins are expected to be in a reduced state or at least not encountering oxidative signals. The free radical scavenger MPG neutralizes ROS as they are generated and provides a general indication of the role of ROS in biological processes. In this study we used MPG to assess the sensitivity of GABAA receptors to decreases in [ROS]i. The effectiveness of MPG at scavenging intracellular ROS in turtle dorsal cortex has previously been confirmed (Dukoff et al. 2014); however, as a thiol-based ROS scavenger it may not accurately replicate the physiological effects of decreases in ROS under anoxic conditions, since MPG may additionally directly reduce cysteine residues on redox-sensitive proteins. We think this is unlikely for two reasons. First, the anoxic application of MPG did not alter the time course or the magnitude of the decrease in [ROS]i. Second, CN which does not directly modulate cysteine residues mimics both anoxia and MPG-mediated changes in CM-DCF fluorescence. An important finding from this study is that mitochondrial O2•− scavenging with MitoTEMPO reduced CM-DCF fluorescence. This is important because it indicates that mitochondrially derived O2•− is the major source of ROS in turtle cortical neurons. This is supported by the CN experiments which also prevented mROS generation under normoxic conditions.
Rather than ROS, it is possible that the anoxic inhibition of mitochondrial ATP production might instead be the signal behind increased GABA transmission since decreases in intracellular [ATP] ([ATP]i) and [ROS]i both occur under anoxic conditions. In particular, one candidate signalling molecule that increases during anoxia is the ATP metabolite adenosine. In anoxic turtle brain, ATP breaks down into adenosine resulting in increases in extracellular [adenosine], and this adenosine signal has been identified as a protective neuromodulator in turtle brain (Nillsson & Lutz, 1992). In vertebrate brain, adenosine acts as an inhibitory neuromodulator that can decrease neuronal excitability (Bickler & Buck, 2007) and enhance GABA release through agonism of presynaptic adenosine A2A receptors (Shindou et al. 2002). However, decreases in mitochondrial ATP production and increases in extracellular adenosine probably do not initiate enhanced GABA release in anoxic turtle brain for two main reasons. First, the timeline of anoxia-induced changes in GABA transmission and ATP/adenosine do not match. In anoxic turtle brain, GABA release is enhanced by ∼10–20 min of anoxic treatment while adenosine and [ATP]i are maintained at normoxic levels for ∼60 min through an increase in anaerobic glycolysis and decreased ATP consumption (Nillsson & Lutz, 1992; Buck et al. 1998). Second, under normoxic conditions ROS scavenging with MPG or MitoTEMPO enhances GABA receptor currents even though oxidative phosphorylation and presumably ATP generation remains functioning. In turtle cortical sheets, intracellular pH decreases by 0.5 or more pH units during anoxia due to net ATP hydrolysis and anaerobic glycolysis; therefore, intracellular acidification could also be a potential signal to initiate GABA release (Buck et al. 1998). However, this is also not likely since ROS scavenger-mediated changes in GABA transmission occur during normoxia when normal pH levels are maintained. Together, these data point to decreases in ROS rather than ATP or pH as the inhibitory signal behind increased GABA transmission.
In support of a redox-sensitive GABAergic SA mechanism we demonstrate that under normoxic conditions the pharmacological elimination of ROS induces shunting inhibition by clamping Vm to EGABA. This is probably the result of a GABAA receptor-mediated increase in Gw because application of GZ decreases Gw and reverses Vm. The inability of GZ to fully reverse the MPG and CN-induced changes in Gw is possibly due to the incomplete inhibition of extrasynaptic GABAA receptors by GZ (see below for discussion). ROS scavenging did not have an effect on EGABA and therefore, GABAergic SA is not the result of modulation of Cl− regulatory machinery (e.g. potassium–chloride co-transporters (KCC2) and sodium–potassium–chloride co-transporters (NKCC1)). Anoxia plus MPG did not have an additive effect on Gw or Vm indicating that ROS scavenging and anoxia activate a similar mechanism. Importantly, ROS scavenging with MPG or with MitoTEMPO has the capacity to decrease APf ∼90%, providing direct evidence that decreases in mitochondrially derived [ROS]i can initiate SA. Application of H2O2 reversed anoxia-induced changes in Gw and Vm indicating that GABA transmission is suppressed by the presence of an oxidant. Activation of GABAA receptor-mediated shunting inhibition by decreasing mitochondrial O2•− production with MitoTEMPO indicates that the anoxic decrease in [ROS]i is probably the signal that induces GABA-mediated SA and supports the conclusion that electrical suppression results from a mitochondrially based oxygen-sensing mechanism.
To determine if GABA release is potentiated by decreases in ROS we measured changes in mIPSC frequency. We found that ROS scavenging and anoxia increase mIPSC frequency 2–3 times indicating GABA release is redox sensitive. Since vesicular GABA release is primarily Ca2+ dependent, under these experimental conditions a change in mIPSC frequency indicates an increase in presynaptic [Ca2+] (see below for potential mechanisms) (Trigo et al. 2010). Miniature IPSC frequency could also increase as a result of a redox-sensitive insertion of GABAA receptors into the postsynaptic membrane. This is known to occur through Ca2+/calmodulin-dependent protein kinase II phosphorylation of the GABAA receptor (Saliba et al. 2012). However, the amplitude of mIPSCs did not change following anoxia or ROS scavenging which indicates that postsynaptic GABA receptors are not redox modulated (Trigo et al. 2010). This is an unexpected finding since GABA transmission has been shown to be redox sensitive with reducing agents potentiating GABAA receptors and oxidizing agents inhibiting them (Amato et al. 1999; Calero et al. 2011).
The increase in sIPSC and gIPSC amplitudes could also be the result of an increase in redox-sensitive GABA release. GABAA receptors have two GABA-binding sites and elevated synaptic [GABA] results in longer more frequent receptor open times (Richter et al. 2012). Under anoxic or ROS-depleted conditions increased GABA release would lead to temporal synchronization of inhibitory events across multiple synapses and summation of IPSC amplitude. This is particularly evident with sIPSCs which occur at a high frequency (∼13–14 Hz). Giant IPSCs are a unique GABAA receptor current in turtle pyramidal neurons with characteristics similar to giant depolarizing potentials in neonatal brain (Ben-Ari et al. 2007) or GABAA,slow currents (Capogna & Pearce, 2011). Giant IPSCs are probably the result of network-driven polysynaptic events because gIPSC frequency does not change and currents last hundreds of milliseconds. This fits with a redox-sensitive GABA release mechanism because increased simultaneous release of GABA onto pyramidal neurons would result in an increase in gIPSC amplitude during anoxia or ROS depletion.
In our previous investigation of the effect of anoxia on tonic GABA currents in pyramidal neurons GZ (25 μm) was used to antagonize extrasynaptic GABAA receptors. Under these conditions a persistent GABA current was not uncovered (Pamenter et al. 2011). Extrasynaptic GABAA receptors have a different subunit composition from synaptic GABAA receptors which influences receptor inhibition and is the likely reason GZ did not reveal a tonic current (Bai et al. 2001). In this study we applied BIC and determined that there is a significant tonic GABAergic current under normoxic conditions, and it doubles in amplitude following anoxia or ROS scavenging. Importantly, inhibition of mROS generation with MitoTEMPO or CN also doubled this current indicating extrasynaptic GABAA receptor currents are regulated by mROS. This finding is consistent with a redox-sensitive increase in GABA release resulting from enhanced synaptic phasic GABA spillover or volume transmission. The analysis of the charge transfer is an important component of this study because it allows a direct comparison of the relative contribution associated with each GABAA receptor current. This analysis determined that tonic currents are responsible for 45–50% of the total GABAergic charge transfer under all treatment conditions. This highlights the important inhibitory role of extrasynaptic GABAA receptors during anoxia and shows how tonic GABAergic inhibition is an effective mechanism through which to clamp Vm at EGABA.
In turtle dorsal cortex, GABAergic stellate interneurons are the principal inhibitory neurons responsible for modulation of pyramidal neuron activity (Connors & Kriegstein, 1986; Shepherd, 2011); and therefore, the most likely site of a redox-sensitive GABA release mechanism. Enhanced GABA release from stellate interneurons likely results from activation of a local oxygen-sensing mechanism because anoxia and redox-sensitive changes in GABA transmission occur in isolated cortical sheets. Glutamate receptor antagonists do not block GABA transmission indicating that glutamatergic input or feedback excitation from pyramidal neurons is not involved in this mechanism. This is interesting because stellate interneurons receive feedback from pyramidal neurons (Colombe et al. 2004); however, it is possible that the downregulation of glutamatergic signalling during anoxia prevents this (Shin & Buck, 2003). Although the mechanism responsible for oxygen sensitivity has yet to be elucidated this evidence indicates that in turtle dorsal cortex the stellate interneuron/pyramidal neuron network is an important inhibitory control point for dampening electrical activity during anoxia.
Mitochondria are appropriately positioned to function as oxygen sensors and initiators of redox-based signalling cascades because they are the primary consumer of cellular oxygen and the most ubiquitous source of cellular ROS (Chen et al. 2003). The presynaptic localization of mitochondria at the membrane and the ROS-sensitive modulation of GABAergic transmission suggest that anoxic decreases in mROS production are a likely signal to initiate GABA-mediated SA. In addition since [ROS]i fluctuate with oxygen availability, changes in mROS production could function as an oxygen sensor. Importantly, ROS-mediated signalling would occur simultaneously throughout the brain and since this mechanism does not require energy during a time when cellular ATP is limited, it constitutes a metabolically inexpensive signal to coordinate the down-regulation of energy-consuming processes on a broad scale.
In mammals, several ion channels have been identified as redox-sensitive including voltage-gated potassium channels (Müller & Bittner, 2002) and ATP-sensitive potassium channels (Bao et al. 2009). Since K+ channel activity has a large influence on Vm, decreases in [ROS]i could inhibit potassium channels resulting in Vm depolarization and increased GABA release. For example, in pulmonary arteries, hypoxic vasoconstriction is mediated in part by the inhibition of redox-sensitive K+ channels leading to depolarization of Vm and the opening of L-type Ca2+ channels (Archer & Michelakis, 2002). In turtle stellate interneurons a similar mechanism could result in increased GABA release. Alternatively, in turtle pyramidal neurons anoxia depolarizes mitochondrial membrane potential leading to Ca2+ release and inhibition of NMDA receptors through a protein phosphatase 1- and 2A-mediated pathway (Shin et al. 2005). Since this Ca2+ signal is an integral component of the anoxia tolerance strategy in pyramidal neurons, it is possible that it also occurs in presynaptic GABAergic nerve terminals resulting in increased GABA release. A number of second messenger signalling molecules are also redox sensitive, including protein kinase C (PKC) (Chu et al. 2003), protein kinase A (Humphries et al. 2005), protein kinase G (Burgoyne et al. 2007), protein tyrosine kinase (Klann & Thiels, 1999), tyrosine phosphatase 1B (Denu & Tanner, 1998), protein phosphatase 2A and 2B (Klann & Thiels, 1999) and G proteins (Nishida et al. 2000). Activation of any of these signalling intermediates has the potential to initiate signalling cascades and modulate neuronal firing characteristics via indirect ion channel modification. This is plausible since turtle pyramidal neurons express an oxygen-sensitive Ca2+-activated potassium channel that is inhibited through a PKC-mediated mechanism that could be regulated by [ROS]i (Rodgers-Garlick et al. 2013).
In summary, we have demonstrated that in turtle dorsal cortex GABA release is redox sensitive and that decreases in [ROS]i are sufficient and necessary to induce GABAergic SA. Spontaneous IPSCs, gIPSCs and tonic GABA currents all contribute to SA; however, tonic currents are responsible for the majority of charge transfer. We have established that decreases in mitochondria ROS O2•− generation are capable of initiating GABA-mediated SA, highlighting a unique ROS-mediated signalling mechanism in a naturally anoxia-tolerant vertebrate. We propose a signalling mechanism in which anoxic decreases in mROS generation enhance stellate interneuron GABA release, resulting in inhibition of glutamatergic pyramidal neurons. Further elucidation of this mechanism will lead to a better understanding of redox-sensitive GABA transmission in an anoxia-tolerant vertebrate brain and could potentially produce medically relevant protective measures against anoxic or hypoxic insults in mammalian brain.
Acknowledgments
We thank A. Chowdhury for assistance with fibre-optic oxygen measurements and P. Hawrysh for assistance with rhodamine fluorescence measurements.
Glossary
- aCSF
artificial cerebrospinal fluid
- AP
action potential
- AP5
(2R)-amino-5-phosphonopentanoate
- BIC
bicuculline methiodide
- CM-H2DCFDA
5-(and-6)-chloromethyl-2',7'-dichlorodihydro-fluorescein diacetate acetyl ester
- CN
cyanide
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- EGABA
GABA reversal potential
- ETC
electron transport chain
- gIPSC
giant inhibitory postsynaptic current
- Gw
whole-cell conductance
- GZ
gabazine
- mIPSC
miniature inhibitory postsynaptic current
- MPG
N-(2-mercaptopropionyl) glycine
- mROS
mitochondrial reactive oxygen species
- O2•−
superoxide
- ROS
reactive oxygen species
- SA
spike arrest
- sIPSC
spontaneous inhibitory postsynaptic current
- Vm
membrane potential
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
L.T.B. and D.W.H. conceived the project and designed experiments. D.W.H., M.E.P. and D.J.D. performed the experiments, and analysed the data. D.W.H. prepared the manuscript, and L.T.B., M.E.P. and D.J.D. revised it critically for important intellectual content. All authors approved the final manuscript, qualify for authorship and are the sole authors. Experiments were performed at the University of Toronto in the laboratory of L.T.B.
Funding
This study was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant) to L.T.B.
References
- Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal. 2005;7:1140–1149. doi: 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- Amato A, Connolly CN, Moss SJ. Smart TG. Modulation of neuronal and recombinant GABAA receptors by redox reagents. J Physiol. 1999;517:35–50. doi: 10.1111/j.1469-7793.1999.0035z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S. Michelakis E. The mechanism(s) of hypoxic pulmonary vasoconstriction: potassium channels, redox O2 sensors, and controversies. News Physiol Sci. 2002;17:131–137. doi: 10.1152/nips.01388.2002. [DOI] [PubMed] [Google Scholar]
- Bai D, Zhu G, Pennefather P, Jackson MF, MacDonald JF. Orser BA. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by γ-aminobutyric acidA receptors in hippocampal neurons. Mol Pharmacol. 2001;59:814–824. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]
- Bao L, Avshalumov MV, Patel JC, Lee CR, Miller EW, Chang CJ. Rice ME. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J Neurosci. 2009;29:9002–9010. doi: 10.1523/JNEUROSCI.1706-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa J-L, Tyzio R. Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Berman JM. Awayda MS. Redox artifacts in electrophysiological recordings. Am J Physiol Cell Physiol. 2013;304:C604–C613. doi: 10.1152/ajpcell.00318.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickler PE. Buck LT. Hypoxia tolerance in reptiles, amphibians, and fishes: Life with variable oxygen availability. Annu Rev Physiol. 2007;69:145–170. doi: 10.1146/annurev.physiol.69.031905.162529. [DOI] [PubMed] [Google Scholar]
- Bright DP. Smart TG. Methods for recording and measuring tonic GABAA receptor-mediated inhibition. Front Neural Circuits. 2013;7:193. doi: 10.3389/fncir.2013.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck L, Espanol M, Litt L. Bickler P. Reversible decreases in ATP and PCr concentrations in anoxic turtle brain. Comp Biochem Physiol A Mol Integr Physiol. 1998;120:633–639. doi: 10.1016/s1095-6433(98)10079-x. [DOI] [PubMed] [Google Scholar]
- Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, Browning DD. Eaton P. Cysteine redox sensor in PKGIαenables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- Calero CI, Vickers E, Moraga Cid G, Aguayo LG, von Gersdorff H. Calvo DJ. Allosteric modulation of retinal GABA receptors by ascorbic acid. J Neurosci. 2011;31:9672–9682. doi: 10.1523/JNEUROSCI.5157-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M. Pearce RA. GABAA,slow: causes and consequences. Trends Neurosci. 2011;34:101–112. doi: 10.1016/j.tins.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Chen Q. Pan HL. Signaling mechanisms of angiotensin II-induced attenuation of GABAergic input to hypothalamic presympathetic neurons. J Neurophysiol. 2007;97:3279–3287. doi: 10.1152/jn.01329.2006. [DOI] [PubMed] [Google Scholar]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL. Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell-death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Chu F, Ward NE. O'Brian CA. PKC isozyme S-cysteinylation by cystine stimulates the pro-apoptotic isozyme PKCδ and inactivates the oncogenic isozyme PKC. Carcinogenesis. 2003;24:317–325. doi: 10.1093/carcin/24.2.317. [DOI] [PubMed] [Google Scholar]
- Colombe JB, Sylvester J, Block J. Ulinski PS. Subpial and stellate cells: two populations of interneurons in turtle visual cortex. J Comp Neurol. 2004;471:333–351. doi: 10.1002/cne.20037. [DOI] [PubMed] [Google Scholar]
- Connors BW. Kriegstein AR. Cellular physiology of the turtle visal cortex: Distinctive properties of pyramidal and stellate neurons. J Neurosci. 1986;6:164–177. doi: 10.1523/JNEUROSCI.06-01-00164.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu JM. Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukoff DJ, Hogg DW, Hawrsyh PJ. Buck LT. Scavenging ROS dramatically increases NMDA receptor whole cell currents in painted turtle cortical neurons. J Exp Biol. 2014;217:3346–3355. doi: 10.1242/jeb.105825. [DOI] [PubMed] [Google Scholar]
- Ghai HS. Buck LT. Acute reduction in whole cell conductance in anoxic turtle brain. Am J Physiol Regul Integr Comp Physiol. 1999;277:R887–R893. doi: 10.1152/ajpregu.1999.277.3.R887. [DOI] [PubMed] [Google Scholar]
- Hawrysh PJ. Buck LT. Anoxia-mediated calcium release through the mitochondrial permeability transition pore silences NMDA receptor currents in turtle neurons. J Exp Biol. 2013;216:4375–4387. doi: 10.1242/jeb.092650. [DOI] [PubMed] [Google Scholar]
- Howarth C, Gleeson P. Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab. 2012;32:1222–1232. doi: 10.1038/jcbfm.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries KM, Deal MS. Taylor SS. Enhanced dephosphorylation of cAMP-dependent protein kinase by oxidation and thiol modification. J Biol Chem. 2005;280:2750–2758. doi: 10.1074/jbc.M410242200. [DOI] [PubMed] [Google Scholar]
- Klann E. Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: Implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Verkaart S, van Emst-de Vries SE, Grefte S, Smeitink JA. Willems PH. Simultaneous quantification of oxidative stress and cell spreading using 5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein. Cytometry A. 2006;69:1184–1192. doi: 10.1002/cyto.a.20348. [DOI] [PubMed] [Google Scholar]
- Leroy P, Nicolas A, Gavriloff C, Matt M, Netter P, Bannwarth B, Hercelin B. Mazza M. Determination of 2-mercaptoproprionyl glycine and its metabolite, 2-mercaptopropionic acid, in plasma by ion-pair reversed-phase high performance liquid-chromatography with postcolumn derivatization. J Chromatogr. 1991;564:258–265. doi: 10.1016/0378-4347(91)80088-t. [DOI] [PubMed] [Google Scholar]
- Müller W. Bittner K. Differential oxidative modulation of voltage-dependent K+ currents in rat hippocampal neurons. J Neurophysiol. 2002;87:2990–2995. doi: 10.1152/jn.2002.87.6.2990. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch-clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Nillsson GE. Lutz PL. Short communication: adenosine release in the anoxic turtle brain: a possible mechanism for anoxic survival. J Exp Biol. 1992;162:345–351. [Google Scholar]
- Nishida M, Maruyama Y, Tanaka R, Kontani K, Nagao T. Kurose H. Gαi and Gαo are target proteins of reactive oxygen species. Nature. 2000;408:492–495. doi: 10.1038/35044120. [DOI] [PubMed] [Google Scholar]
- Nusser Z. Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Pamenter ME. Buck LT. Neuronal membrane potential is mildly depolarized in the anoxic turtle cortex. Comp Biochem Physiol A Mol Integr Physiol. 2008;150:410–414. doi: 10.1016/j.cbpa.2008.04.605. [DOI] [PubMed] [Google Scholar]
- Pamenter ME, Hogg DW, Gu XQ, Buck LT. Haddad GG. Painted turtle cortex is resistant to an in vitro mimic of the ischemic mammalian penumbra. J Cereb Blood Flow Metab. 2012;32:2033–2043. doi: 10.1038/jcbfm.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamenter ME, Hogg DW, Ormond J, Shin DS, Woodin MA. Buck LT. Endogenous GABAA and GABAB receptor-mediated electrical suppression is critical to neuronal anoxia tolerance. Proc Natl Acad Sci U S A. 2011;108:11274–11279. doi: 10.1073/pnas.1102429108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamenter ME, Richards MD. Buck LT. Anoxia-induced changes in reactive oxygen species and cyclic nucleotides in the painted turtle. J Comp Physiol B. 2007;177:473–481. doi: 10.1007/s00360-007-0145-8. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Chan CY, Rosenthal M. Sick TJ. Membrane and synaptic activity during anoxia in the isolated turtle cerebellum. Am J Physiol Regul Integr Comp Physiol. 1992;263:R1057–R1063. doi: 10.1152/ajpregu.1992.263.5.R1057. [DOI] [PubMed] [Google Scholar]
- Pitlik TN, Bulai PM, Denisov AA, Afanasenkov DS. Cherenkevich SN. Redox regulation of ionic homeostasis in neurons. Neurochem J. 2009;3:87–92. [Google Scholar]
- Rice ME. H2O2: a dynamic neuromodulator. Neuroscientist. 2011;17:389–406. doi: 10.1177/1073858411404531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter L, de Graaf C, Sieghart W, Varagic Z, Mörzinger M, de Esch IJ, Ecker GF. Ernst M. Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nat Chem Biol. 2012;8:455–464. doi: 10.1038/nchembio.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS. Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3:941–947. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- Rodgers-Garlick CI, Hogg DW. Buck LT. Oxygen-sensitive reduction in Ca2+-activated K+ channel open probability in turtle cerebrocortex. Neuroscience. 2013;237:243–254. doi: 10.1016/j.neuroscience.2013.01.046. [DOI] [PubMed] [Google Scholar]
- Saliba RS, Kretschmannova K. Moss SJ. Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 2012;31:2937–2951. doi: 10.1038/emboj.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JM. Kriegstein AR. Turtle hippocampal cortex contains distinct cell types, burst-firing neurons, and an epileptogenic subfield. J Neurophysiol. 1986;56:1626–1649. doi: 10.1152/jn.1986.56.6.1626. [DOI] [PubMed] [Google Scholar]
- Shepherd GM. The microcircuit concept applied to cortical evolution: from three-layer to six-layer cortex. Front Neuroanat. 2011;5:30. doi: 10.3389/fnana.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DSH. Buck LT. Effect of anoxia and pharmacological anoxia on whole-cell NMDA receptor currents in cortical neurons from the western painted turtle. Physiol Biochem Zool. 2003;76:41–51. doi: 10.1086/374274. [DOI] [PubMed] [Google Scholar]
- Shin DSH, Wilkie MP, Pamenter ME. Buck LT. Calcium and protein phosphatase receptor activity in 1/2A attenuate N-methyl-d-aspartate the anoxic turtle cortex. Comp Biochem Physiol A Mol Integr Physiol. 2005;142:50–57. doi: 10.1016/j.cbpa.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Shindou T, Nonaka H, Richardson PJ, Mori A, Kase H. Ichimura M. Presynaptic adenosine A2A receptors enhance GABAergic synaptic transmission via a cyclic AMP dependent mechanism in the rat globus pallidus. Br J Pharmacol. 2002;136:296–302. doi: 10.1038/sj.bjp.0704702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigo FF, Bouhours B, Rostaing P, Papageorgiou G, Corrie JE, Triller A, Ogden D. Marty A. Presynaptic miniature GABAergic currents in developing interneurons. Neuron. 2010;66:235–247. doi: 10.1016/j.neuron.2010.03.030. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulinski P. Visual cortex of turtles. In: Kaas JH, editor. Evolution of Nervous Systems. Elsevier; 2007. pp. 195–201. [Google Scholar]
- Ultsch GR. The viability of nearctic freshwater turtles submerged in anoxia and normoxia at 3 and 10°C. Comp Biochem Physiol A Comp Physiol. 1985;81:607–611. doi: 10.1016/0300-9629(85)91035-7. [DOI] [PubMed] [Google Scholar]
- Veal EA, Day AM. Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Wake H, Moorhouse AJ. Nabekura J. Clustering of neuronal K+-Cl− cotransporters in lipid rafts by tyrosine phosphorylation. J Biol Chem. 2009;284:27980–27988. doi: 10.1074/jbc.M109.043620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yowtak J, Lee KY, Kim HY, Wang JG, Kim HK, Chung K. Chung JM. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain. 2011;152:844–852. doi: 10.1016/j.pain.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccarato F, Valente M. Alexandre A. Hydrogen peroxide induces a long lasting inhibition of the Ca2+ dependent glutamate release in cerebralcortical synaptosomes without interfering with cytosolic Ca2+ J Neurochem. 1995;64:2552–2558. doi: 10.1046/j.1471-4159.1995.64062552.x. [DOI] [PubMed] [Google Scholar]