

Abstract
The dentate gyrus is a region subject to intense study in epilepsy because of its posited role as a ‘gate’, acting to inhibit overexcitation in the hippocampal circuitry through its unique synaptic, cellular and network properties that result in relatively low excitability. Numerous changes predicted to produce dentate hyperexcitability are seen in epileptic patients and animal models. However, recent findings question whether changes are causative or reactive, as well as the pathophysiological relevance of the dentate in epilepsy. Critically, direct in vivo modulation of dentate ‘gate’ function during spontaneous seizure activity has not been explored. Therefore, using a mouse model of temporal lobe epilepsy with hippocampal sclerosis, a closed-loop system and selective optogenetic manipulation of granule cells during seizures, we directly tested the dentate ‘gate’ hypothesis in vivo. Consistent with the dentate gate theory, optogenetic gate restoration through granule cell hyperpolarization efficiently stopped spontaneous seizures. By contrast, optogenetic activation of granule cells exacerbated spontaneous seizures. Furthermore, activating granule cells in non-epileptic animals evoked acute seizures of increasing severity. These data indicate that the dentate gyrus is a critical node in the temporal lobe seizure network, and provide the first in vivo support for the dentate ‘gate’ hypothesis.
Key points
A key mechanistic concept in epilepsy is the dentate gate hypothesis, which argues that the dentate gyrus protects hippocampal circuits from overexcitation and that a breakdown of this gate leads to epilepsy.
Direct in vivo evidence for the dentate gate hypothesis is lacking and it is therefore unclear whether interventions selectively targeting the dentate gyrus would inhibit seizures.
We demonstrate that on-demand optogenetic restoration of the dentate gate through selective inhibition of granule cells is sufficient to inhibit spontaneous seizures in a mouse model of temporal lobe epilepsy.
By contrast, activation of granule cells worsens spontaneous seizures and can even induce acute seizures in non-epileptic animals.
These data provide direct evidence for the dentate gate hypothesis, indicate that the dentate gyrus is indeed a critical node in temporal lobe seizure circuitry, and illustrate that the dentate gyrus can be an effective target for seizure inhibition.
Introduction
Epilepsy manifests with recurrent bursts of hypersynchronous neuronal activity that can arise from a number of brain regions. The most commonly affected regions are areas where recurrent excitatory networks pre-exist and can serve as a substrate for this type of activity, such as the temporal lobe, where recurrent excitatory connections of the hippocampus are required for normal spatial navigation, learning and memory (Eichenbaum & Cohen, 2014; Hartley et al. 2014). Thus, the dentate gyrus (DG), classically considered as the first stop in the trisynaptic loop of entorhinal-hippocampal circuitry within the temporal lobe (van Strien et al. 2009), has been extensively studied in both healthy and pathological circumstances. Both the microcircuitry of the DG and unique features of granule cells (GCs; the main principal cell type of the DG) make them less easily excitable than other types of principal cells: in healthy tissue, GCs not only have extensive feedforward and feedback inhibition, but also intrinsic properties, such as a particularly hyperpolarized resting membrane potential, low input resistance and a relatively high threshold for firing (Heinemann et al. 1992; Lothman et al. 1992; Mody et al. 1992; Heinemann et al. 1993; Acsady et al. 1998; Coulter, 1999; Henze et al. 2002; Lysetskiy et al. 2005; Coulter & Carlson, 2007; Leutgeb et al. 2007; Morgan et al. 2007; de Almeida et al. 2009; Armstrong et al. 2011; Armstrong et al. 2012; Yu et al. 2013). Therefore, although substantial barrages of excitatory input may arrive at the DG, there is normally only sparse activation of GCs, limiting the input to downstream regions of the hippocampus, where pyramidal cells can generate action potentials more easily and where there are abundant intrinsic recurrent excitatory connections. The limited activation of GCs is important for pattern separation functions during normal brain activity (Leutgeb et al. 2007; de Almeida et al. 2009) but, in addition, this may help protect the vulnerable downstream hippocampal formation and prevent seizure activity. The latter idea is the basis of the dentate ‘gate’ theory of temporal lobe epilepsy (TLE), which posits that seizures occur when the gate function of the DG is disrupted such that excess excitation emerges from or passes through the DG to downstream regions (Heinemann et al. 1992; Lothman et al. 1992).
The dentate gate hypothesis has been a major and frequently cited mechanistic concept in epilepsy for over 20 years (Heinemann et al. 1992; Lothman et al. 1992) and, further illustrating the importance of this concept, the National Institutes of Health has invested millions of dollars in projects related to the DG and epilepsy in this fiscal year alone (http://projectreporter.nih.gov). A wealth of evidence in support of the theory has been found in the numerous changes in this region in TLE that are predicted to cause hyperexcitability. These changes include, amongst many others, mossy fibre sprouting (such that GCs form aberrant recurrent synapses onto other GCs, a feature not observed in healthy tissue; Nadler et al. 1980; Zhang et al. 2012), altered intrinsic properties and receptor expression (Coulter, 1999; Stegen et al. 2012) and reduced GABAergic inhibition (Acsady et al. 1998; Bouilleret et al. 2000; Kobayashi & Buckmaster, 2003; Coulter & Carlson, 2007). A recent study found that dysregulation in GCs of molecular pathways known to be important in epileptogenesis can result in epilepsy (Pun et al. 2012) and correlations have been reported between seizure frequency and changes seen in the DG, including mossy fibre sprouting (Marucci et al. 2010; Hester & Danzer, 2013).
However, other work has demonstrated that, unexpectedly, it is possible to suppress certain changes considered to lead to hyperexcitability, such as mossy fibre sprouting, without also suppressing seizures, questioning the causality of the morphological changes observed in the dentate in TLE (Buckmaster & Lew, 2011). Additionally, calcium imaging of hippocampal slices suggests only an initial, transient breakdown in the dentate gate prior to the emergence of spontaneous seizures in a rat model of TLE, with the temporoammonic pathway to the CA1 region instead showing the greatest dysregulation at a time when the first spontaneous seizures appear (Wozny et al. 2005; Ang et al. 2006; Pathak et al. 2007; Coulter et al. 2011). These data raise the possibility that there is a primary alternate pathway by which seizures spread that bypasses the DG altogether. If this were the case, therapeutic interventions targeting the DG in an attempt to restore the dentate gate in TLE would be ineffective at controlling seizures. Importantly, direct in vivo evidence supporting the dentate gate hypothesis in TLE has been conspicuously lacking.
For the dentate gate hypothesis to be demonstrated in vivo, inhibition of GCs during spontaneous temporal lobe seizures to restore gate function should prevent overexcitation of the rest of the hippocampal formation and effectively stop seizure activity. If, however, an alternate pathway bypassing the DG is the critical pathway involved in TLE, targeting GCs would be ineffective at inhibiting ongoing seizure activity. Similarly, overexcitation of GCs to mimic a breakdown of the gate might be expected to cause seizures. Optogenetics is a powerful tool that can be harnessed to study epilepsy (Krook-Magnuson et al. 2014a) and has been successfully applied in animal models of focal cortical epilepsy (Wykes et al. 2012), cortical stroke-induced thalamocortical epilepsy (Paz et al. 2011) and TLE (Krook-Magnuson et al. 2013), as well as to inhibit acute seizures in vivo (Sukhotinsky et al. 2013; Berglind et al. 2014; Chiang et al. 2014) and epileptiform activity in vitro (Tonnesen et al. 2009; Berglind et al. 2014; Ledri et al. 2014). Importantly, by allowing direct and selective manipulation of GCs in vivo, on-demand optogenetic techniques (Armstrong et al. 2013) provide a straightforward way of directly testing the dentate gate hypothesis.
Methods
Ethical approval
All procedures were approved by the UC Irvine Animal Care and Use Committee and were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council.
Animals
The molecular Cre-lox system permitted the selective introduction of the inhibitory halorhodopsin (HR) or the excitatory channelrhodopsin (ChR2) into specific cell populations. Mice were generated by crossing Cre lines expressing Cre either in DG GCs selectively [B6.FVB-Tg(Pomc-cre)1Stl/J; stock 010714; Jackson Laboratories, Bar Harbor, ME, USA] (McHugh et al. 2007) or in principal cells broadly (including CA1 pyramidal cells: CamK-Cre; B6.Cg-Tg(Camk2a-cre)T29-1Stl/J; stock 005359; Jackson Laboratories) (Tsien et al. 1996) with either floxed-STOP HR mice [Ai39; B6;129S-Gt(ROSA)26Sortm39(CAGHOP/EYFP)Hze/J; generated by Hongkui Zeng; stock 014539; Jackson Laboratories] (Madisen et al. 2012) or floxed-STOP ChR mice (Ai32; Rosa-CAG-LSLChR2H134R-EYFP-deltaNeo generated by Hongkui Zeng; stock 012569; obtained from the Allen Institute, Seattle, WA, USA; now available from Jackson Laboratories) (Madisen et al. 2012). Ai39 and Ai32 lines were maintained by crossing with C57BL/6 J mice (stock 000664; Jackson Laboratories). For visualization, the GC-Cre line was also crossed with a line expressing the red fluorescent protein tdTomato in a Cre-dependent fashion [B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J; stock 007905; Jackson Laboratories] (Madisen et al. 2012). Although Cre is transiently expressed in developing GCs in the B6.FVB-Tg(Pomc-cre)1Stl/J line, recombination leads to permanent opsin or tdTomato expression in GCs with this approach. Images of expression were acquired using either an Axioskop 2 plus (Carl Zeiss, Oberkochen, Germany) and post-acquisition colouring, or the confocal facility of the Optical Biology Shared Resource centre at the University of California, Irvine. GC-ChR2, GC-HR and GC-Tomato mice demonstrated occasional scattered cortical cells with expression. The location of these varied between animals and was not within the path of light. However, in a subset of GC-ChR2, GC-HR and GC-Tom animals, we found widespread non-specific expression (potentially a result of transient Cre expression during early development of the animal). Therefore, the expression in all GC mice was examined post hoc, and animals with such non-selective expression were not included in the analysis. This resulted in the exclusion of 10 animals. Negative littermates were used for opsin-negative controls. Animals were housed under a 12:12 h light/dark cycle with access to food and water available ad libitum. Male and female mice were used. At the end of experiments, animals were deeply anaesthetized with isoflurane, followed by rapid decapitation.
Epilepsy induction and monitoring
The procedures employed are similar to those described in Krook-Magnuson et al. (2013) and a detailed protocol is provided in Armstrong et al. (2013). We used the unilateral intrahippocampal kainate model of TLE (Cavalheiro et al. 1982), which best mimics unilateral hippocampal sclerosis. Kainic acid (KA; 50–100 nL, 20 mm in saline; Tocris Bioscience, St Louis, MO, USA) was stereotaxically injected directly into the left dorsal hippocampal formation (posterior 2.0 mm, lateral 1.25 mm, ventral 1.6 mm with respect to bregma) of mice under isoflurane anaesthesia on or after postnatal day 46. After at least 2 weeks, allowing for the emergence of spontaneous recurrent seizures, optical fibres and twisted wire bipolar electrodes were implanted into the dorsal hippocampal formation (posterior 2.6 mm, lateral 1.75 mm, ventral 1.4 mm with respect to bregma). Light was delivered ipsilaterally or contralaterally to the site of previous kainate injection, as noted in the Results. Based on previous estimates of the volume of light delivery (Krook-Magnuson et al. 2013) and volume of the GC layer (Peirce et al. 2003), in healthy tissue, this fibre location would result in less than 5% of GCs being illuminated. Seizures were detected and recorded from electrodes located in the hippocampal formation ipsilateral to previous kainate injection; direct information about the source or spatial profile of seizures was not obtained in our experiments. The selection of our electrode placement was based on previous studies indicating that, in this model of epilepsy, spontaneous seizures typically arise ipsilateral, and slightly posterior, to the site of previous kainate injection (Cavalheiro et al. 1982; Bragin et al. 1999; Haussler et al. 2012), such that the light stimulation was directed at seizure activity in an area of the DG where gate function was expected to be most compromised. The extent of hippocampal sclerosis is variable between animals; an example of the extent of sclerosis is provided by Krook-Magnuson et al. (2013), who illustrate GC dispersion and CA1 cell loss; note that the optical fibre was not placed in the area of maximal hippocampal sclerosis but rather where sclerotic and non-sclerotic tissue tended to interface. In separate experiments, animals not previously injected with kainate (non-epileptic, kainate-naïve animals), were similarly implanted with optical fibres and bipolar depth electrodes. Data for some of the Cam-HR mice used for comparison with the effects seen in GC-HR mice were reported previously (in six animals). These animals had optrodes targeted to above the CA1 region (ventral 1.25 mm with respect to bregma), with light reaching an estimated depth of at least 0.55 mm from the tip of the fibre, and thus also reaching the DG (Krook-Magnuson et al. 2013). In two additional Cam-HR animals, the placement of the optical fibre was lowered to match that of GC-HR animals. The results obtained at this location were not substantially different, and the results from all Cam-HR animals were combined. After recovery, 24 h video and EEG monitoring for seizures and subsequent closed-loop seizure detection and/or light delivery were initiated. On average, KA-injected animals were implanted 15.9 ± 2.3 weeks after KA injection and the effect of light on seizures was examined 19.9 ± 2.6 weeks after KA injection. There was no correlation between seizure duration reduction and time subsequent to kainate injection (P = 0.34, Spearman test).
Closed-loop seizure detection and light delivery
Closed-loop seizure detection and light delivery was performed in a manner similar to that described by Krook-Magnuson et al. (2013) and in more detail by Armstrong et al. (2013). For experiments using on-demand light delivery, the hippocampal EEG signal was analysed in real-time by a PC running a custom MATLAB (http://www.mathworks.com) seizure detection algorithm. A version of this software is available for download from Armstrong et al. (2013). Once the presence of spontaneous recurrent seizures in individual animals was established, an experimenter identified features of the early ictal electrographic signal to be used in triggering the real-time closed-loop seizure detection software. The recording software has been modified slightly and interfaces with a custom-built trigger control box for a more accurate timing of light pulses. A fibre-coupled diode laser (Shanghai Laser & Optics Century Co., Ltd, Shanghai, China) of an appropriate wavelength to activate the opsin expressed (473 nm for ChR2, 589 nm for HR) was used. Steady state power measured post hoc from the tip of the optical fibres was 9.3 ± 0.6 mW. When a seizure was detected, it was flagged for later review and, for a preset percentage of events (in a random sequence), light delivery (30 s of 50 ms on, 100 ms off for 473 nm; 30 s of 2000 ms on, 50 ms off for 589 nm) was immediately triggered. This allowed for each animal to serve as its own control, in addition to opsin-negative controls. In non-epileptic animals, triggering was instead performed in a scheduled manner, with triggers occurring every 15 min and light being delivered for 50% of triggers.
Slice electrophysiology
Whole-cell patch-clamp recordings were made at 36°C from coronal slices using artificial cerebrospinal fluid (aCSF) containing (in mm) 2.5 KCl, 10 glucose, 126 NaCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl, 26 NaHCO3. The intracellular solution contained (in mm): 90 potassium gluconate, 27.4 KCl, 1.8 NaCl, 1.7 MgCl2, 0.05 EGTA, 10 Hepes, 2 Mg-ATP, 0.4 Na2-GTP, 10 phosphocreatine, 8 biocytin; pH 7.2; 270–290 mosmol l–1; pipette resistance: 3–4.5 MΩ. Recordings were made a using a Multiclamp 700B, Digidata 1322A (Axon Instruments, Foster City, CA, USA), a 4–10 kHz low pass filter and a sampling rate of 10–50 kHz. Light was delivered through the epifluorescence port of a Eclipse FN-1 (Nikon, Tokyo, Japan), using a Lambda DG-4 with smart shutter and Lambda SC controller (Sutter Instruments, Novato, CA, USA) and TTL input from a Digidata 1322A (Axon Instruments). Where noted in the text, 1 μm of TTX was added to the aCSF.
Scoring of behavioural seizures
The extended Racine scale of Pinel and Rovner (1978), which also captures more severe behavioural seizures, was modified to include additional phenotypes observed (Racine, 1971; Pinel & Rovner, 1978; Luttjohann et al. 2009): stage 1: a change in behavioural state (sudden behavioural arrest or sudden motion); stage 2: head nodding; stage 3: forelimb clonus; stage 4: rearing, or clonus when on belly, or strong hindlimb clonus (bucking); stage 5: falling, or clonus when on side; stage 6: multiple sequences of rearing and falling, or brief jumps; stage 7: violent jumping; and stage 8: class seven, followed by a period of tonus lasting longer than 5 s.
Statistical analysis
Electrographic seizure durations after the time of the trigger and the time to next seizure were analysed offline by reviewers who were blinded to the light condition and genotype of the animal, and behavioural seizures were confirmed by video and EEG analysis. Post-detection seizure durations for light and no light conditions in GC-HR animals were compared in each animal using a two-sample Kolmogorov–Smirnov test and a two-tailed Mann–Whitney test (93 ± 5 seizure events per animal were analysed to assess the effect of ipsilateral light delivery; 102 ± 2 seizure events per animal were analysed to assess the effect of contralateral light delivery). Group level statistics for duration reduction and time to next seizure used a Wilcoxon signed ranks test (light vs. no light) and a Mann–Whitney test (opsin-expressing vs. opsin-negatives; GC-HR vs. Cam-HR). Comparisons for the frequency of events progressing to behavioural seizures in epileptic GC-ChR2 animals with and without light delivery was made using chi-squared tests (137 ± 44 events per animal were examined to assess the effect of ipsilateral light delivery; 75 ± 22 events per animal for contralateral light delivery). In kainate-naïve animals, a correlation between the number of light deliveries (for light deliveries one to eleven) and seizure duration or behavioural seizure score was tested using Spearman's correlation coefficient. Note that this is a non-parametric test and does not assume a linear relationship. Values presented are the mean ± SEM. P < 0.05 was considered statistically significant. Statistical analysis was conducted using Excel 2007 and 2013 (Microsoft, Redmond, WA, USA), OriginPro 8 and 9 (OriginLab Corp., Northampton, MA, USA) and Google documents (https://docs.google.com).
Results
On-demand inhibition of dentate GCs
To gain selective optogenetic inhibition of GCs, we generated chronically epileptic mice expressing the inhibitory opsin HR in DG GCs (GC-HR) (Fig.1) utilizing a model of TLE with unilateral hippocampal sclerosis as described in the Methods. GC-HR mice were injected with KA in the left dorsal hippocampus, and the effect of inhibition of GCs on spontaneous seizures was examined during the chronic phase of the disorder. Expression of HR selectively in GCs remained after chronic, spontaneous seizures emerged (specificity of expression was confirmed post hoc for all animals included in the present study). Light delivery produced robust inhibitory currents, hyperpolarizing GCs (Fig.1D and E).
Figure 1.
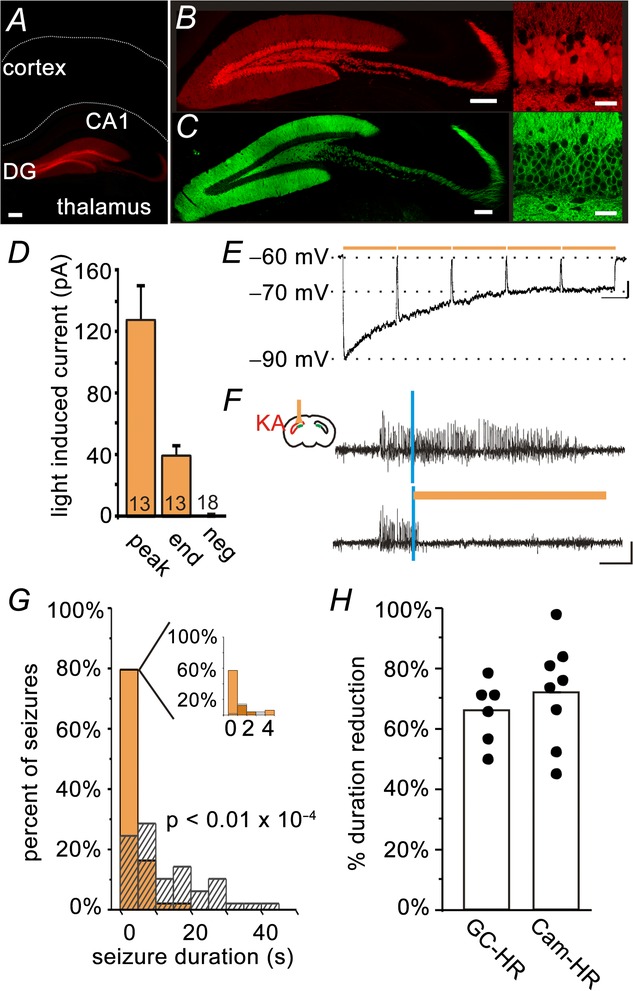
On-demand restoration of the dentate gate inhibits spontaneous temporal lobe seizures
Crossing a mouse line expressing Cre in DG GCs (visualized in A and B, by crossing with a tdTomato reporter line; red in the online version) with a mouse line expressing the inhibitory opsin HR in a Cre-dependent fashion, produced mice with HR expressed selectively in DG GCs (GC-HR). Selectivity of opsin expression was maintained in epileptic animals (C, yellow fluorescent protein tagged HR; green in the online version). The edge of the slice and the border between CA1 and the alveus are drawn in A for reference. D and E, whole-cell patch-clamp recordings from epileptic brain slices revealed robust light-induced inhibition of GCs of opsin-expressing but not opsin-negative animals. (Peak, peak-induced currents; End, current measured at the end of 10 s of pulsed light delivery; Neg, opsin-negative controls. The number of cells recorded is indicated in each bar. D, summary voltage clamp data. E, example current clamp recording). F–G, in vivo online detection of spontaneous seizures allowed on-demand light delivery, which rapidly truncated seizures when delivered to the hippocampus ipsilateral to prior KA injection (example animal: vertical blue lines indicate seizure detection; amber bars indicate light delivery; hashed bars indicate events not receiving light; G, inset: expansion of the first 5 s after light delivery; 100 seizures; colour is shown in the online version). H, the inhibition of seizure duration achieved with selective inhibition of GCs is comparable to that achieved with broader inhibition of excitatory cells including CA1 pyramidal cells (Cam-HR; shown for reference in H; each dot represents one animal). Scale bars: A, 200 μm; B and C, left 200 μm, right 30 μm; E, 5 mV, 1 s; F, 0.2 mV, 5 s.
During closed-loop spontaneous seizure detection and intervention in vivo, amber light was immediately delivered upon seizure detection above the DG (reaching a depth of ∼0.55 mm from the tip of the optical fibre; Krook-Magnuson et al. 2013; thus reaching both blades of the DG) for 50% of detected events in a random fashion, allowing each animal to serve as its own internal control. As noted above, seizures in this model typically arise ipsilateral to the site of prior kainate injection (Bragin et al. 1999), and thus the region of the hippocampal formation with the greatest probablility of having a compromised dentate gate was the region both recorded from and targeted with light. Supporting the dentate gate hypothesis, we found that ipsilateral on-demand light delivery in GC-HR mice dramatically truncated seizures (75 ± 7% stopping within 5 s of light delivery; post-detection seizure duration, light vs. no light, P < 0.05 Wilcoxon test; six animals) (Fig.1F–H). On average, there was a 66 ± 4% duration reduction (P < 0.01, opsin-positive vs. opsin-negative controls, Mann–Whitney). There was no effect on time to next seizure, indicating a lack of any rebound effect (P = 0.2). These data illustrate that selective inhibition of dentate GCs is able to inhibit temporal lobe seizure activity arising from regions of the hippocampus where there is probably most deficient DG function.
We previously demonstrated significant seizure control with on-demand light delivery to the hippocampal formation in mice broadly expressing HR in excitatory cells, including CA1 pyramidal cells (Cam-HR mice; Krook-Magnuson et al. 2013). We therefore also directly compared the seizure control obtained with this broader inhibition with the seizure control obtained with selective inhibition of GCs. Selectively inhibiting GCs produced comparable seizure control (Cam-HR 72 ± 6% duration reduction; eight animals; GC-HR 66 ± 4% duration reduction, as discussed above; Cam-HR vs. GC-HR: P = 0.33, Mann–Whitney) (Fig.1H), indicating that, by identifying a key component of the network, improved intervention specificity can be achieved without sacrificing efficacy, and that the DG is a necessary participant in ongoing seizure activity.
We additionally tested the efficacy of light delivered contralateral to the site of previous kainate injection and recording electrode in GC-HR mice and found that it did not affect seizure duration (light vs. no light, GC-HR: P = 1.0, Wilcoxon). Therefore, inhibition of GCs ipsilaterally (and not contralaterally) inhibits seizures. Given that seizures in this model typically arise ipsilateral and in relatively close proximity to the site of prior kainate injection (Bragin et al. 1999; Haussler et al. 2012), and that GCs do not have contralateral projections, in the context of the dentate gate hypothesis, these findings suggest that inhibiting GCs can prevent seizure activity only where changes indicative of dentate gate breakdown occur.
Direction of modulation is a critical factor
Recent work examining cerebellar directed intervention for TLE unexpectedly found that the direction of modulation of cerebellar neurons was not a critical factor in achieving a reduction in seizure duration, with optogenetic excitation or inhibition of cerebellar neurons inhibiting seizures (Krook-Magnuson et al. 2014b). We thus investigated whether the direction of modulation is a critical factor in achieving seizure inhibition when targeting DG GCs in the hippocampal formation, or whether a disruption of on-going activity through on-demand optogenetic activation rather than inhibition of GCs could also inhibit seizures. To be able to selectively excite GCs, we generated mice expressing the excitatory opsin ChR2 selectively in GCs (GC-ChR2) and tested the effect of on-demand optogenetic intervention in these animals. Light delivery produced strong direct excitation of GCs (Fig.2A and B; light-induced action potentials in 6 of 6 GCs recorded in current clamp; on average, light induced 820 ± 150 pA in 7 GCs recorded in voltage clamp), which remained in the presence of TTX (Fig.2B, inset; 90 ± 5% of induced current remained in TTX; 3 GCs), as well as indirect excitation of downstream CA3 pyramidal cells, which was blocked in the presence of TTX (Fig.2C; average peak current in regular aCSF: 270 ± 110 pA; abolished by TTX; 3 CA3 pyramidal cells).
Figure 2.
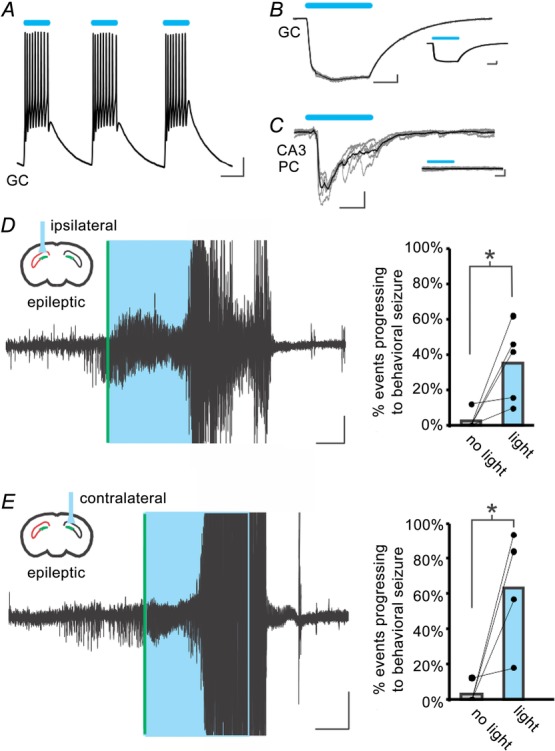
Excitation of GCs worsens spontaneous seizures
A, light-induced excitation of a GC in a slice from an epileptic GC-ChR2 animal. B, robust light-induced currents in a GC, which remain in the presence of TTX (inset). Grey traces: individual sweeps; black traces: average. C, optogenetic activation of GCs produces postsynaptic currents in a downstream CA3 pyramidal cell (PC), which are eliminated by the application of TTX (inset). D and E, on-demand light delivery to the dentate gyrus in mice expressing the excitatory opsin ChR2 in GCs (GC-ChR2 mice) ipsilateral (D) or contralateral (E) to previous kainate injection causes electrographic seizures to progress to large behavioural seizures. Right: each dot pair represents one animal. *P < 0.001 (chi-squared). Scale bars: A, 10 mV, 50 ms; B and C, 100 pA, 20 ms; D and E, 0.1 mV, 10 s. Boxes (coloured blue in the online version) denote light delivery. Vertical lines (coloured green in the online version) indicate online seizure detection. Large amplitude signals include a movement artefact and have been truncated.
Rather than truncating spontaneous seizures, in vivo on-demand light delivery to chronically epileptic GC-ChR2 mice increased the probability of electrographic seizures becoming large behavioural seizures (seizures were 90 times more probable to become a large behavioural seizure with light delivery; light vs. no light, P < 0.001, χ2; behaviour included rearing with forelimb clonus, falling and violent jumping; five animals; Fig.2D). Light delivery to the contralateral hippocampus also induced behavioural seizures (Fig. 2E; light vs. no light, P < 0.001, χ2; four animals). These data indicate that increased excitatory drive to GCs selectively is sufficient to push the network to behavioural seizures in epileptic animals, and that this occurs not only when excitatory drive is increased in areas of expected dentate gate compromise, but also in the contralateral hippocampus where dentate gate restoration had no effect on ongoing seizure activity.
Seizure induction in kainate-naïve animals
Given the finding that activation of GCs worsened, rather than inhibited, seizures in epileptic animals, and that this occurred even with contralateral activation (distant from the site of dentate gate injury, in a location where inhibition had no effect on seizure activity), we reasoned that overexcitation of GCs, effectively selectively collapsing or bypassing the dentate gate in the targeted region, may be sufficient to induce seizures in kainate-naïve non-epileptic animals. To achieve this, we implanted optrodes in non-epileptic (kainate-naïve) GC-ChR2 animals and repetitively stimulated DG GCs unilaterally for 30 s with at least 15 min between stimulation periods. This protocol of 30 s of pulsed light delivery was chosen to mimic the previous on-demand experiments performed in epileptic animals and determine whether the DG is a sufficiently powerful region to generate seizures in kainate-naïve animals. As such, the parameters do not represent an attempt to mimic the overexcitation of GCs probably occurring spontaneously in epileptic animals during endogenously occurring seizure activity.
Light delivery to the DG in kainate-naïve GC-ChR2 animals was capable of inducing seizures (Fig.3) (in total, 30 of 47 light deliveries produced behavioural seizures in three animals). The duration and severity of seizure activity in response to light increased with repeated light delivery, resembling a kindling effect seen with electrical stimulation (Racine et al. 1973; Pinel & Rovner, 1978), and seizure duration outlasted light delivery (Spearman's correlation coefficient for seizure duration: 0.50; P < 0.01; Spearman's correlation coefficient for behavioural seizure score: 0.69; P < 0.001; Fig.3). Light never produced a seizure in opsin-negative kainate-naïve controls (367 light deliveries in four animals). These findings indicate that the selective excitation of GCs, mimicking a breakdown of the dentate gate, is sufficient to induce seizures even in non-epileptic animals.
Figure 3.
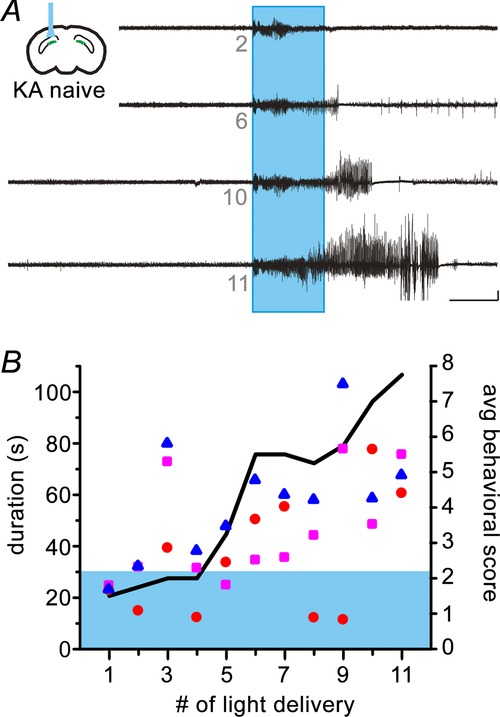
Excitation of GCs is sufficient to induce seizures in kainate-naïve animals
A, example responses to repeated light delivery in a kainate-naïve GC-ChR2 animal. Small grey numbers under traces indicate the number of light delivery. Boxes (coloured blue in the online version) denote 30 s of pulsed light delivery. Large amplitude signals include a movement artefact and have been truncated. Scale bar: 0.1 mV, 20 s. B, seizure duration and severity increases with repeated light delivery. Each symbol represents the seizure duration from a given animal; shading (coloured blue in the online version) indicates the duration of light delivery (left axis). The black line indicates the average behavioural seizure score (right axis). Seizure scoring was based on the extended Racine scale of Pinel & Rover (1978), as detailed in the Methods.
Discussion
Using optogenetics to selectively manipulate GCs in the DG, we demonstrate that (i) on-demand selective inhibition of GCs ipsilateral to kainate injection effectively stops spontaneous temporal lobe seizures; (ii) the direction of modulation is a critical factor, such that excitation of GCs worsens, rather than inhibits, seizures; and (iii) selective optogenetic activation of GCs can induce acute seizures in kainate-naïve, non-epileptic animals. These findings provide direct support for the dentate gate hypothesis because inhibition of the DG in areas of hippocampal sclerosis, and thus of predicted dentate gate breakdown, inhibits spontaneous temporal lobe seizures being propagated through the region. In addition, the DG is a sufficiently powerful node in the circuit such that driving GCs can generate seizures even in healthy, non-sclerotic tissue.
Our findings suggest that, although blocking some of the changes in the dentate associated with TLE may not be sufficient in isolation to stop the generation of seizures (Buckmaster & Lew, 2011), and although alternative pathways may exist (Wozny et al. 2005; Coulter et al. 2011), the DG is a critical component of the seizure circuitry. The numerous changes seen in hippocampal sclerosis that lead to DG hyperexcitability are indeed probably pathogenic, and the DG can be an effective target for the inhibition of seizures. The fact that selective inhibition of GCs produced a robust inhibition of seizures comparable to the broad inhibition of excitatory neurons in the hippocampal formation (including CA1 pyramidal cells) illustrates that, by identifying a key component in the network, increased specificity of intervention can be achieved without greatly sacrificing efficacy.
In considering these findings, it is important to emphasize that, although dysfunction of the dentate clearly allows the propagation of seizure activity through the hippocampal formation in temporal lobe seizures, our findings have limited spatial implications about the propagation patterns, and certainly do not imply that upstream regions such as the entorhinal cortex are not involved in seizure initiation, nor do they indicate a unique or privileged role of the dentate in stopping seizure activity. Modulating any essential node within the seizure circuitry, or even modulating other brain regions that have strong influence on any of the essential nodes, may effectively inhibit seizures as well. Indeed, modulating the activity of certain extrahippocampal regions is known to modulate temporal lobe seizures (Fisher, 2013; Bertram, 2014; Krook-Magnuson et al. 2014b), and changes seen in other brain regions, including in the CA1 and the temporoammonic pathway, may also contribute to seizure propagation. Alternative potential sites of intervention do not detract from the significance of the dentate gate hypothesis, nor from the finding that the DG is a critical node in the network and an effective target for seizure inhibition. With the recent approval of medical devices in human patients that are capable of delivering closed-loop electrical interventions to specific brain regions in response to seizure activity (Heck et al. 2014), understanding seizure circuitry becomes even more beneficial because the findings may be more readily translated for clinical use. Future work examining other nodes or modulatory regions will provide increasing insight into the mechanisms and underlying architecture of seizures and epilepsy, and will also identify new intervention strategies.
Traditional medications, which have broad effects in numerous brain areas, can have major negative side effects, and restricted intervention strategies may reduce any negative side effects. Although our understanding of the exact changes that critically contribute to the breakdown of the dentate gate is still incomplete, the findings obtained from numerous studies of this region in epilepsy hint at a number of unique changes that may make excellent targets for interventions. The results of the present study clearly demonstrate that the DG is an essential node in TLE with hippocampal sclerosis, and indicate that an intervention approach selectively targeting GCs or otherwise improving dentate gate function may be an effective strategy for inhibiting seizures and also reducing side-effects.
Acknowledgments
We thank Cecilia Lozoya, Rose Zhu, Judit Vargane, Chris Krook-Magnuson and Dhrumil Vyas for providing technical support. This work was made possible, in part, through access to the confocal facility of the Optical Biology Shared Resource of the Cancer Centre Support Grant (CA-62203) at the University of California, Irvine.
Glossary
- aCSF
artificial cerebrospinal fluid
- ChR2
channelrhodopsin
- DG
dentate gyrus
- GC
granule cell
- HR
halorhodopsin
- KA
kainic acid
- TLE
temporal lobe epilepsy
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
EK-M, CA and IS designed the experiments. EK-M, CA, AB, SL and MO contributed to data collection and analysis. EK-M, CA, AB, MO and IS contributed to scientific discussions. EK-M, CA and IS drafted and revised the article. Experiments were conducted at the University of California, Irvine.
Funding
This work was supported by Citizens United for Research in Epilepsy (CURE) Taking Flight Award (to EK-M), a US National Institutes of Health grant F31NS086429 (to AB), the Epilepsy Foundation (to CA) and a US National Institutes of Health grant NS074432 (to IS).
References
- Acsady L, Kamondi A, Sik A, Freund T. Buzsaki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang CW, Carlson GC. Coulter DA. Massive and specific dysregulation of direct cortical input to the hippocampus in temporal lobe epilepsy. J Neurosci. 2006;26:11850–11856. doi: 10.1523/JNEUROSCI.2354-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C, Krook-Magnuson E, Oijala M. Soltesz I. Closed-loop optogenetic intervention in mice. Nat Protoc. 2013;8:1475–1493. doi: 10.1038/nprot.2013.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C, Krook-Magnuson E. Soltesz I. Neurogliaform and Ivy cells: a major family of nNOS expressing GABAergic neurons. Front Neural Circuits. 2012;6:23. doi: 10.3389/fncir.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C, Szabadics J, Tamas G. Soltesz I. Neurogliaform cells in the molecular layer of the dentate gyrus as feed-forward gamma-aminobutyric acidergic modulators of entorhinal-hippocampal interplay. J Comp Neurol. 2011;519:1476–1491. doi: 10.1002/cne.22577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglind F, Ledri M, Sorensen AT, Nikitidou L, Melis M, Bielefeld P, Kirik D, Deisseroth K, Andersson M. Kokaia M. Optogenetic inhibition of chemically induced hypersynchronized bursting in mice. Neurobiol Dis. 2014;65:133–141. doi: 10.1016/j.nbd.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Bertram EH. Extratemporal lobe circuits in temporal lobe epilepsy. Epilepsy Behav. 2014 doi: 10.1016/j.yebeh.2014.07.012. [DOI] [PubMed] [Google Scholar]
- Bouilleret V, Loup F, Kiener T, Marescaux C. Fritschy JM. Early loss of interneurons and delayed subunit-specific changes in GABA(A)-receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus. 2000;10:305–324. doi: 10.1002/1098-1063(2000)10:3<305::AID-HIPO11>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Jr, Wilson CL, Vizentin E. Mathern GW. Electrophysiologic analysis of a chronic seizure model after unilateral hippocampal KA injection. Epilepsia. 1999;40:1210–1221. doi: 10.1111/j.1528-1157.1999.tb00849.x. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS. Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–2347. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalheiro EA, Riche DA. Le Gal La Salle G. Long-term effects of intrahippocampal kainic acid injection in rats: a method for inducing spontaneous recurrent seizures. Electroencephalogr Clin Neurophysiol. 1982;53:581–589. doi: 10.1016/0013-4694(82)90134-1. [DOI] [PubMed] [Google Scholar]
- Chiang CC, Ladas TP, Gonzalez-Reyes LE. Durand DM. Seizure suppression by high frequency optogenetic stimulation using in vitro and in vivo animal models of epilepsy. Brain Stimul. 2014;7:890–899. doi: 10.1016/j.brs.2014.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA. Chronic epileptogenic cellular alterations in the limbic system after status epilepticus. Epilepsia. 1999;40(Suppl 1):S23–33. doi: 10.1111/j.1528-1157.1999.tb00875.x. ; discussion S40-21. [DOI] [PubMed] [Google Scholar]
- Coulter DA. Carlson GC. Functional regulation of the dentate gyrus by GABA-mediated inhibition. Prog Brain Res. 2007;163:235–812. doi: 10.1016/S0079-6123(07)63014-3. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Yue C, Ang CW, Weissinger F, Goldberg E, Hsu FC, Carlson GC. Takano H. Hippocampal microcircuit dynamics probed using optical imaging approaches. J Physiol. 2011;589:1893–1903. doi: 10.1113/jphysiol.2010.202184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida L, Idiart M. Lisman JE. The input–output transformation of the hippocampal granule cells: from grid cells to place fields. J Neurosci. 2009;29:7504–7512. doi: 10.1523/JNEUROSCI.6048-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H. Cohen NJ. Can we reconcile the declarative memory and spatial navigation views on hippocampal function? Neuron. 2014;83:764–770. doi: 10.1016/j.neuron.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS. Deep brain stimulation for epilepsy. Handb Clin Neurol. 2013;116:217–234. doi: 10.1016/B978-0-444-53497-2.00017-6. [DOI] [PubMed] [Google Scholar]
- Hartley T, Lever C, Burgess N. O'Keefe J. Space in the brain: how the hippocampal formation supports spatial cognition. Philos Trans R Soc Lond B Biol Sci. 2014;369:20120510. doi: 10.1098/rstb.2012.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler U, Bielefeld L, Froriep UP, Wolfart J. Haas CA. Septotemporal position in the hippocampal formation determines epileptic and neurogenic activity in temporal lobe epilepsy. Cereb Cortex. 2012;22:26–36. doi: 10.1093/cercor/bhr054. [DOI] [PubMed] [Google Scholar]
- Heck CN, King-Stephens D, Massey AD, Nair DR, Jobst BC, Barkley GL, Salanova V, Cole AJ, Smith MC, Gwinn RP, Skidmore C, Van Ness PC, Bergey GK, Park YD, Miller I, Geller E, Rutecki PA, Zimmerman R, Spencer DC, Goldman A, Edwards JC, Leiphart JW, Wharen RE, Fessler J, Fountain NB, Worrell GA, Gross RE, Eisenschenk S, Duckrow RB, Hirsch LJ, Bazil C, O'Donovan CA, Sun FT, Courtney TA, Seale CG. Morrell MJ. Two-year seizure reduction in adults with medically intractable partial onset epilepsy treated with responsive neurostimulation: final results of the RNS System Pivotal trial. Epilepsia. 2014;55:432–441. doi: 10.1111/epi.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J. Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273–280. [PubMed] [Google Scholar]
- Heinemann U, Zhang CL. Eder C. Entorhinal cortex-hippocampal interactions in normal and epileptic temporal lobe. Hippocampus. 1993;3(Spec No):89–97. [PubMed] [Google Scholar]
- Henze DA, Wittner L. Buzsaki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Hester MS. Danzer SC. Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. J Neurosci. 2013;33:8926–8936. doi: 10.1523/JNEUROSCI.5161-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M. Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Oijala M. Soltesz I. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun. 2013;4:1376. doi: 10.1038/ncomms2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Ledri M, Soltesz I. Kokaia M. a How might novel technologies such as optogenetics lead to better treatments in epilepsy? Adv Exp Med Biol. 2014;813:319–336. doi: 10.1007/978-94-017-8914-1_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Szabo GG, Armstrong C, Oijala M. Soltesz I. b Cerebellar directed optogenetic intervention inhibits spontaneous hippocampal seizures in a mouse model of temporal lobe epilepsy. eNeruo. 2014 doi: 10.1523/ENEURO.0005-14.2014. 2014 Dec;1(1). pii: e.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, Kirik D. Kokaia M. Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci. 2014;34:3364–3377. doi: 10.1523/JNEUROSCI.2734-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB. Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL. Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–313. [PubMed] [Google Scholar]
- Luttjohann A, Fabene PF. van Luijtelaar G. A revised Racine's scale for PTZ-induced seizures in rats. Physiol Behav. 2009;98:579–586. doi: 10.1016/j.physbeh.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Lysetskiy M, Foldy C. Soltesz I. Long- and short-term plasticity at mossy fiber synapses on mossy cells in the rat dentate gyrus. Hippocampus. 2005;15:691–696. doi: 10.1002/hipo.20096. [DOI] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ, 3rd, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsaki G, Ramirez JM, Jones AR, Svoboda K, Han X, Turner EE. Zeng H. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci. 2012;15:793–802. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marucci G, Rubboli G. Giulioni M. Role of dentate gyrus alterations in mesial temporal sclerosis. Clin Neuropathol. 2010;29:32–35. doi: 10.5414/npp29032. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA. Tonegawa S. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–99. doi: 10.1126/science.1140263. [DOI] [PubMed] [Google Scholar]
- Mody I, Kohr G, Otis TS. Staley KJ. The electrophysiology of dentate gyrus granule cells in whole-cell recordings. Epilepsy Res Suppl. 1992;7:159–168. [PubMed] [Google Scholar]
- Morgan RJ, Santhakumar V. Soltesz I. Modeling the dentate gyrus progress in brain research. In: Scharfman HE, editor; The Dentate Gyrus: A Comprehensive Guide to Structure, Function, and Clinical Implications. Vol. 163. Elsevier; 2007. pp. 639–658. , Volume, Amsterdam. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Gentry C. Cotman CW. Loss and reacquisition of hippocampal synapses after selective destruction of CA3-CA4 afferents with kainic acid. Brain Res. 1980;191:387–403. doi: 10.1016/0006-8993(80)91289-5. [DOI] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ. Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K. Huguenard JR. A new mode of corticothalamic transmission revealed in the Gria4(−/−) model of absence epilepsy. Nat Neurosci. 2011;14:1167–1173. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce JL, Chesler EJ, Williams RW. Lu L. Genetic architecture of the mouse hippocampus: identification of gene loci with selective regional effects. Genes Brain Behav. 2003;2:238–252. doi: 10.1034/j.1601-183x.2003.00030.x. [DOI] [PubMed] [Google Scholar]
- Pinel JP. Rovner LI. Experimental epileptogenesis: kindling-induced epilepsy in rats. Exp Neurol. 1978;58:190–202. doi: 10.1016/0014-4886(78)90133-4. [DOI] [PubMed] [Google Scholar]
- Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, Schmeltzer SN, Faulkner C, Bronson SL, Murphy BL, Richards DA, Holland KD. Danzer SC. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. 2012;75:1022–1034. doi: 10.1016/j.neuron.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R( Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1971;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Racine RJ, Burnham WM, Gartner JG. Levitan D. Rates of motor seizure development in rats subjected to electrical brain stimulation: strain and inter-stimulation interval effects. Electroencephalogr Clin Neurophysiol. 1973;35:553–556. doi: 10.1016/0013-4694(73)90033-3. [DOI] [PubMed] [Google Scholar]
- Stegen M, Kirchheim F, Hanuschkin A, Staszewski O, Veh RW. Wolfart J. Adaptive intrinsic plasticity in human dentate gyrus granule cells during temporal lobe epilepsy. Cereb Cortex. 2012;22:2087–2101. doi: 10.1093/cercor/bhr294. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Chan AM, Ahmed OJ, Rao VR, Gradinaru V, Ramakrishnan C, Deisseroth K, Majewska AK. Cash SS. Optogenetic delay of status epilepticus onset in an in vivo rodent epilepsy model. PloS ONE. 2013;8:e62013. doi: 10.1371/journal.pone.0062013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnesen J, Sorensen AT, Deisseroth K, Lundberg C. Kokaia M. Optogenetic control of epileptiform activity. Proc Natl Acad Sci U S A. 2009;106:12162–12167. doi: 10.1073/pnas.0901915106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER. Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- van Strien NM, Cappaert NL. Witter MP. The anatomy of memory: an interactive overview of the parahippocampal-hippocampal network. Nat Rev Neurosci. 2009;10:272–282. doi: 10.1038/nrn2614. [DOI] [PubMed] [Google Scholar]
- Wozny C, Gabriel S, Jandova K, Schulze K, Heinemann U. Behr J. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobiol Dis. 2005;19:451–460. doi: 10.1016/j.nbd.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S. Kullmann DM. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med. 2012;4:161ra152. doi: 10.1126/scitranslmed.3004190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EP, Dengler CG, Frausto SF, Putt ME, Yue C, Takano H. Coulter DA. Protracted postnatal development of sparse, specific dentate granule cell activation in the mouse hippocampus. J Neurosci. 2013;33:2947–2960. doi: 10.1523/JNEUROSCI.1868-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huguenard JR. Buckmaster PS. Increased excitatory synaptic input to granule cells from hilar and CA3 regions in a rat model of temporal lobe epilepsy. J Neurosci. 2012;32:1183–1196. doi: 10.1523/JNEUROSCI.5342-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]