

Abstract
For more than 20 years, reverse pharmacology has been the preeminent strategy to discover the activating ligands of orphan G protein-coupled receptors (GPCRs). The onset of a reverse pharmacology assay is the cloning and subsequent transfection of a GPCR of interest in a cellular expression system. The heterologous expressed receptor is then challenged with a compound library of candidate ligands to identify the receptor-activating ligand(s). Receptor activation can be assessed by measuring changes in concentration of second messenger reporter molecules, like calcium or cAMP. The fluorescence-based calcium mobilization assay described here is a frequently used medium-throughput reverse pharmacology assay. The orphan GPCR is transiently expressed in human embryonic kidney 293T (HEK293T) cells and a promiscuous Gα16 construct is co-transfected. Following ligand binding, activation of the Gα16 subunit induces the release of calcium from the endoplasmic reticulum. Prior to ligand screening, the receptor-expressing cells are loaded with a fluorescent calcium indicator, Fluo-4 acetoxymethyl. The fluorescent signal of Fluo-4 is negligible in cells under resting conditions, but can be amplified more than a 100-fold upon the interaction with calcium ions that are released after receptor activation. The described technique does not require the time-consuming establishment of stably transfected cell lines in which the transfected genetic material is integrated into the host cell genome. Instead, a transient transfection, generating temporary expression of the target gene, is sufficient to perform the screening assay. The setup allows medium-throughput screening of hundreds of compounds. Co-transfection of the promiscuous Gα16, which couples to most GPCRs, allows the intracellular signaling pathway to be redirected towards the release of calcium, regardless of the native signaling pathway in endogenous settings. The HEK293T cells are easy to handle and have proven their efficacy throughout the years in receptor deorphanization assays. However, optimization of the assay for specific receptors may remain necessary.
Keywords: Cellular Biology, Issue 89, G protein-coupled receptor (GPCR), calcium mobilization assay, reverse pharmacology, deorphanization, cellular expression system, HEK293T, Fluo-4, FlexStation
Introduction
G protein-coupled receptors (GPCRs) constitute one of the largest and most diverse families among all cell surface proteins. Their presence in vertebrates, invertebrates, plants, yeast, and slime mold, as well as in Protozoa and the earliest diploblastic Metazoa indicates that GPCRs are among the oldest molecules linked with signal transduction1. Their natural activating ligands comprise a wide diversity of external stimuli including peptides, biogenic amines, odorants, glycoproteins, and photons2. As such, these receptor-ligand signaling systems are involved in a great variety of physiological processes. The broad functional spectrum makes them ideally suited for the development of therapeutic drugs that cover a wide range of human diseases. About 50-60% of the current drug targets are represented by GPCRs3,4. Besides their great importance in the pharmaceutical industry, GPCRs are also in the spotlight for the development of a new generation of species-specific insecticides5,6 and pesticides in general. Because the natural ligands of many GPCRs are still unidentified, they are classified as orphan GPCRs. The deorphanization of these receptors will improve the understanding of their physiological roles in organisms and may uncover putative targets for new drug applications7.
Since the genomic era, the reverse pharmacology strategy is widely applied for the deorphanization of GPCRs8. The approach implies that an orphan receptor is used as a ‘hook’ to ‘fish out’ its activating ligand from a biological extract or from a library of synthetic compounds. The GPCR of interest is therefore cloned and subsequently transfected in a cellular expression system. In the most commonly used methods, receptor activation is determined by measuring changes in the concentration of second messenger molecules9. The main receptor screening assays rely on calcium-sensitive bioluminescent proteins (e.g., aequorin)10 or fluorescent calcium indicators (e.g., Fluo-4)11. The fluorescence-based assays, in which receptor-expressing cells are loaded with a fluorescent calcium indicator prior to ligand screening, have the advantage that they allow high-throughput screening due to their ease of use, short reading time, and the flexibility of screening multiple orphan receptors on a single plate12.
Here, the fluorescence-based calcium mobilization assay is thoroughly described and illustrated by the deorphanization process of the Drosophila melanogaster short neuropeptide F (sNPF) receptor. This neuropeptidergic signaling system was originally characterized by Mertens et al. in 200213 with a calcium bioluminescence assay performed in Chinese hamster ovary (CHO) cells14 and by Feng et al. in 2003 with an electrophysiological assay using Xenopus oocytes15. The presence of the sNPF signaling system seems to be limited to the phylum of Arthropoda, where it is implicated in a wide range of processes including the regulation of feeding, growth, stress reactions, locomotion, and circadian rhythms16.
Research on neuropeptidergic signaling systems in insects may not only lead to new targets for the development of insecticides, but the knowledge of their functioning may also be extrapolated towards other organisms as many signaling systems have been generally well conserved throughout evolution17. In the last decade, great progress has been made in the deorphanization process of insect neuropeptide GPCRs. Despite these efforts, only a small number of receptors have been matched to their cognate ligand, and loads of sequence information for new orphan GPCRs has become available due to the booming of genomics18. The availability of medium/high-throughput screening approaches, like the fluorescence-based calcium mobilization assay that has proven to be a widely applied technique9,18, is therefore invaluable.
The fluorescence-based calcium mobilization assay as described here is performed in the human embryonic kidney 293T (HEK293T) cell line and uses a fluorescent probe to determine changes in intracellular calcium concentrations upon receptor activation. To guarantee high expression and translation levels of the receptor, a Kozak consensus sequence19 is added to the 5’ end of the receptor-coding sequence, which is subsequently cloned in an expression vector (e.g., pcDNA vector series for mammalian cell lines). As it is difficult to predict the endogenous G protein-coupling of an orphan GPCR based on sequence information alone, the second messenger molecules (e.g., calcium or cAMP) that are modulated after receptor activation often remain unknown prior to ligand identification. To circumvent this problem, promiscuous G proteins of the Gq family (e.g., murine Gα15 or human Gα16 [used here]) or chimeric G proteins (e.g., Gαqi5) that interact with most GPCRs and induce the release of calcium can be co-expressed20,21,22. Upon binding of the ligand to its receptor, the GPCR undergoes a conformational change that leads to the activation of specific intracellular pathways. The guanosine diphosphate (GDP) molecule, bound under resting conditions to the Gα16 subunit, will be replaced by a guanosine triphosphate (GTP) molecule. This provokes the dissociation of the heterotrimeric G protein in a Gα16 and Gβγ subunit. The Gα16 subunit activates phospholipase Cβ (PLCβ), which in turn hydrolyzes the membrane-bound phosphatidylinositol bisphosphate (PIP2) resulting in diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 will spread throughout the cytoplasm and activates IP3-dependent calcium channels present in the membrane of the endoplasmic reticulum, which induces the release of calcium into the cytoplasm.
The calcium release upon receptor activation occurs within seconds and can be detected by loading cells before the screening assay with a calcium-sensitive dye, like Fluo-4 acetoxymethyl (AM)11. The AM ester group enables the fluorophore to cross the cell membrane and is cleaved off by cytoplasmic esterases once inside the cell. Consequently, the negative charges of the fluorescent dye are unmasked, preventing it from diffusing out of the cell and allowing it to interact with calcium ions. The fluorescent signal of Fluo-4 is negligible in cells under resting conditions only containing calcium concentrations in the nanomolar range. However, when calcium is released upon receptor activation, the signal can increase concentration-dependently to more than a 100-fold, hereby ensuring a large signal-to-noise ratio. Fluo-4 also exhibits a large dynamic range for reporting [calcium] around a Kd(calcium) of 345 nM, making it suitable to measure physiologically relevant calcium changes in a wide range of cells. The excitation of Fluo-4 occurs at 488 nm and the emission fluorescence is measured at 525 nm11. Fluorimeters like the fluorescence imaging plate reader (FLIPR)23, the NOVOstar, or the FlexStation (station device)12 are medium/high-throughput systems that allow simultaneous compound addition and the detection of the Fluo-4 signal upon receptor activation for every well in an assay plate. The calcium mobilization assay described here relies on the station device 96-well microplate system.
The SoftMax Pro software (software) is used to operate the station device as well as for data analysis. The program immediately displays the results as graphs in 96-well format. Multiple wells can be selected simultaneously to compare the outcome of these wells on the same graph. The relative fluorescent unit (RFU) values of wells in each column are simultaneously measured for a period of two minutes, starting before the addition of compounds to the wells and continuing after measurement of the fluorescent signal following receptor activation. Typically, the trend of an agonist curve aligns with the baseline until an activating compound is added to the cells, resulting in a rapid increase of the fluorescent signal. The peak height is correlated with the final agonist concentration in the well. After the peak, the fluorescent signal slowly drops towards the baseline level. The RFU measurements can be converted into concentration-response curves to determine the EC50 value (half maximal effective concentration) of a ligand. In general, at least three independent screens, each including three replicas of a concentration series, should be performed to compose a reliable concentration-response curve.
It is recommended to include several positive and negative controls in the experimental design. First of all, a transfection control, i.e. the implementation of a receptor with a known ligand, should be tested. This allows verifying whether the transfection agent was operational. Incorporation of a control experiment with an agonist for an endogenous receptor of the cell line and a negative control (e.g., wash buffer) are also recommended to monitor the health and viability of the cells and to exclude the possibility that the wash buffer was contaminated with a factor that could elicit an auto-fluorescent response. Frequently used agonists are a peptide derived from the protease-activated receptor-1 (PAR1), which acts as a PAR1 selective agonist, or carbachol, which activates the acetylcholine receptor. Cells transfected with an empty expression vector should also be tested to exclude that active compounds interact with the cell’s endogenous receptors. Optimization of several parameters described in the protocol below may be required for different signaling systems. A schematic figure of the complete fluorescence-based calcium mobilization assay is depicted in Figure 1.
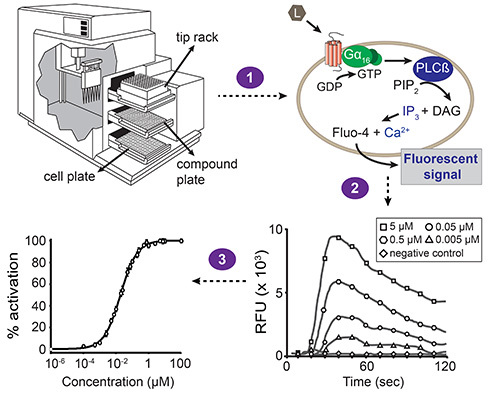 Figure 1. Overall scheme of the fluorescence-based calcium mobilization assay. Automated liquid handling and simultaneous fluorescence measurements are performed with the station device microplate reader, driven by the software. The station device contains three drawers: one for the cell plate, compound plate and tip rack. The build-in pipettor transfers compounds from one column of the compound plate to the corresponding column of the cell plate (step 1). Each well of the cell plate contains a monolayer of HEK293T cells that have been co-transfected with the GPCR of interest and the promiscuous Gα16 subunit. When a compound activates the receptor, the Gα16-bound GDP is replaced by GTP. The Gα16 subunit subsequently dissociates from the Gβγ complex and activates phospholipase Cβ (PLCβ), which in turn hydrolyzes phosphatidylinositol bisphosphate (PIP2) resulting in diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 activates IP3-dependent calcium channels present in the membrane of the endoplasmic reticulum, inducing the release of calcium into the cytoplasm. The interaction of calcium with Fluo-4 (with which the cells are loaded prior to compound addition) results in a fluorescent signal (step 2). The software presents the results as relative fluorescent unit (RFU) values in function of time, and peak heights correlate with the ligand concentration in a concentration-dependent manner. These data can then be converted into a concentration-response curve to determine the EC50 value of a ligand-receptor pair (step 3).
Figure 1. Overall scheme of the fluorescence-based calcium mobilization assay. Automated liquid handling and simultaneous fluorescence measurements are performed with the station device microplate reader, driven by the software. The station device contains three drawers: one for the cell plate, compound plate and tip rack. The build-in pipettor transfers compounds from one column of the compound plate to the corresponding column of the cell plate (step 1). Each well of the cell plate contains a monolayer of HEK293T cells that have been co-transfected with the GPCR of interest and the promiscuous Gα16 subunit. When a compound activates the receptor, the Gα16-bound GDP is replaced by GTP. The Gα16 subunit subsequently dissociates from the Gβγ complex and activates phospholipase Cβ (PLCβ), which in turn hydrolyzes phosphatidylinositol bisphosphate (PIP2) resulting in diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 activates IP3-dependent calcium channels present in the membrane of the endoplasmic reticulum, inducing the release of calcium into the cytoplasm. The interaction of calcium with Fluo-4 (with which the cells are loaded prior to compound addition) results in a fluorescent signal (step 2). The software presents the results as relative fluorescent unit (RFU) values in function of time, and peak heights correlate with the ligand concentration in a concentration-dependent manner. These data can then be converted into a concentration-response curve to determine the EC50 value of a ligand-receptor pair (step 3).
Protocol
Note: All actions in which cells are involved should be carried out in a sterile environment by working in a laminar flow.
1. Maintenance of the HEK293T Cell Line
Grow the HEK293T cells in a T-75 flask at 37 °C in a 5% CO2 humidified incubator.
- Passage the cells when a confluence of 80% is reached. Note: This normally takes 3 to 4 days. Cells in continuous culture allow 20-25 usable passages for screening.
- Place the Dulbecco’s phosphate buffered saline (PBS) without calcium chloride and magnesium chloride, PBS-trypsin-ethylenediaminetetraacetic acid (EDTA) (500 ml PBS supplemented with 10 ml trypsin-EDTA solution and 4.5 ml 4% EDTA) and growth medium (500 ml Dulbecco’s modified eagle’s medium – high glucose [DMEM] supplemented with 10% fetal bovine serum [FBS] and 1 mM penicillin-streptomycin [P-S]) at room temperature half an hour in advance.
- Remove the old growth medium from the cells. Rinse off dead cells with 3 ml PBS and remove the PBS again.
- Add 3 ml PBS-trypsin-EDTA, incubate for 1 min at room temperature and remove the solution Note: The morphology of the cells will change from star-shaped to sphere-shaped.
- Loosen the cells from the bottom of the flask by gentle tapping and collect the cells by rinsing the bottom several times with 10 ml of fresh growth medium.
- Transfer 1 ml of the cell culture into a new T-75 flask containing 14 ml fresh growth medium, and incubate at 37 °C in a 5% CO2 humidified incubator.
2. Transient Transfection of HEK293T Cells
Grow the HEK293T cells in a T-75 flask 3 days before the actual calcium mobilization assay takes place. Make sure to use three T-75 flasks, one for transfection with the receptor construct, one for transfection with an empty expression vector, as negative control, and one for the transfection control. Note: The passaging of the cells is performed as described in step 1.2. When multiple 96-well plates have to be screened, T-150 flasks can be used to get a higher yield of cells (to do this, double all listed quantities in steps 1.2.5, 2.3, 3.1.3 and 3.1.4).
Incubate the flasks at 37 °C in a 5% CO2 humidified incubator for 20-24 hr until the cell cultures approach 50-70% confluence.
- Co-transfect one population of cells with the expression vector encoding the GPCR of interest and the Gα16 construct, the second flask with the empty vector and the Gα16 construct, and the third with the transfection control and the Gα16 construct. Note: In this protocol, the Drosophila sNPF receptor was cloned in a pcDNA3.1 mammalian expression vector13. JetPRIME was used to perform the transfection, but other transfection reagents can be used as well.
- Add a total of 7.8 µg DNA (3.9 µg receptor construct or empty vector, and 3.9 µg Gα16 expression construct) to a 1.5 ml microcentrifuge tube and add 500 µl of the JetPRIME buffer. Mix well by vortexing the tube, and spin down shortly.
- Add 37.5 µl of the JetPRIME reagent, vortex and spin down for 1 min at 14,000 x g. Incubate the transfection mix 10 min at room temperature.
- Add the transfection mix dropwise to the cell culture medium and make sure to pipette directly into the medium avoiding contact with the walls of the culture flask. Incubate the flasks at 37 °C in a 5% CO2 humidified incubator for 20-24 hr.
3. Calcium Mobilization Assay
- Collect the transfected cells and seed them in 96-well black-walled, clear-bottom plates.
- Place the PBS, PBS-Trypsin-EDTA and the DMEM transfer medium (500 ml DMEM supplemented with 10% dialyzed FBS and 1% P-S) at room temperature half an hour in advance.
- Coat the 96-well black-walled, clear bottom plates with 60 µl of PBS with fibronectin (0.0025%) per well (5.85 ml PBS and 150 µl fibronectin [0.1%] per plate). Incubate the plates at room temperature for 1 hr with the lid on. Remove the solution from the wells and incubate the plates again for 1 hr without the lid on at room temperature. Alternatively, it is possible to use pre-coated plates for enhanced cell attachment.
- Take off the old growth medium from the cells and rinse dead cells off with 3 ml PBS and remove the PBS.
- Add 3 ml PBS-Trypsin-EDTA, incubate for 1 min, then remove the solution. Note: The morphology of the cells changes from star-shaped to sphere-shaped.
- Loosen the cells from the bottom of the flask by gentle tapping and collect them by rinsing several times with 10 ml of DMEM transfer medium. Transfer the cells into a 50 ml Falcon tube.
- Count the number of cells per ml (performed with a NucleoCounter here, but a Burker’s chamber is acceptable). Add 100 µl of the cell culture in a 1.5 ml microcentrifuge tube. Add 100 µl lysis buffer and mix by tapping the tube. Add 100 µl stabilizing buffer and mix by tapping.
- Fill a NucleoCassette with the cell suspension and count the cells with the counter. Multiply the number of cells by three, as the cells were diluted by a factor three when adding the lysis and stabilizing buffer in step 3.1.6.
- Dilute the cells to a final concentration of 600,000 cells/ml and seed 150 µl of the cells per well in the coated plates to obtain a cell density of about 90,000 cells per/well. Avoid air bubbles and tap the plate to spread the cells evenly in order to get a contiguous cell layer. Incubate the plates at 37 °C in a 5% CO2 humidified incubator for 16-24 hr.
- Load the cells with the fluorescent dye and prepare the compound plate.
- Prepare Hank’s balanced salt solution (HBSS)/HEPES/Ca2+/bovine serum albumin (BSA) buffer: add 165 µl CaCl2 stock solution (1 M CaCl2 in distilled water [dH2O] – store at room temperature), 500 µl HEPES stock solution (1 M HEPES in dH2O, pH 7.4 – store at room temperature), and 0.05 g BSA to 50 ml HBSS. Note that fatty acid free BSA is used for calcium assays, as fatty acids can activate specific receptors, impairing the calcium signal of the receptor of interest.
- Prepare probenecid solution (100x, 250 mM): dissolve 0.71 g probenecid in 5 ml NaOH (1 M) and add 50 µl HEPES stock solution and 5 ml of the HBSS/HEPES/Ca2+/BSA buffer – always prepare fresh solution. Note: Probenecid inhibits inorganic anion transporters that can dislodge Fluo-4 from the cytoplasm, thereby reducing the fluorescent signal. It is recommended to perform the dye loading in the presence and absence of probenecid to determine whether inorganic anion transporters present a potential problem in the particular cell lines under investigation, as probenecid might even decrease the agonist-mediated signal.
- Prepare wash buffer: add 500 µl probenecid solution to 50 ml HBSS/HEPES/Ca2+/BSA buffer, adjust the pH to 7.4 and filtrate the buffer solution. Always prepare fresh buffer, and 50 ml buffer per plate is required.
- Prepare 10% pluronic acid solution: mix 50 µl pluronic acid (20% w/v in dimethyl-sulfoxide [DMSO]) with 50 µl DMSO – if crystallization occurs, heat to 37 °C and always prepare fresh solution.
- Prepare Fluo-4 AM solution (1mM): add 44 µl 10% pluronic acid solution to a vial that contains 50 µg Fluo-4 AM and vortex until it is completely dissolved. Avoid exposure to light to prevent bleaching of the fluorophore.
- Prepare loading buffer: add 8.8 ml DMEM supplemented with 10 mM HEPES and 2.5 mM probenecid (pH 7.4) to the Fluo-4 AM solution (44 µl) and vortex. Always use fresh buffer.
- Discard medium from the cells, wash the cells with 200 µl PBS per well, and remove PBS.
- Add 55 µl loading buffer per well and incubate for 1 hr at room temperature. Wrap the plate in aluminum foil to prevent exposure of the plate to light.
- Make sure to dry the compounds before the screen if they are dissolved in solvents that are detrimental (e.g., acetonitrile) for the cellular expression system.
- Solubilize the peptides in wash buffer in siliconized 1.5 ml microcentrifuge tubes. Prepare a dilution series to perform a concentration-response analysis and to determine the EC50 value of a ligand. Add 70 µl of the ligand in the corresponding well of the compound plate (V-shaped 96-well plates) and include positive (endogenous agonist) and negative controls (wash buffer) on each plate. Note: If a compound is not soluble in the wash buffer, one can try other solvents that are not harmful for the cells under investigation, such as water or solutions that emulsify and solubilize oils and other water-insoluble substances (e.g., Kolliphor EL).
- Measure all samples in triplicate. Keep in mind that 50 µl of a ligand from the compound plate will be transferred into a well with 100 µl wash buffer of the cell plate during the assay, so prepare the compounds at 3x their desired final concentration.
- Discard the loading buffer from the cell plate and add 100 µl wash buffer to each well. Incubate the plate 15 min at room temperature and prevent exposure to light.
- Discard the wash buffer from the cell plate and add 100 µl new wash buffer to each well.
- Incubate the cell plate, the compound plate, and a tip rack (96-well, station device pipette tips) for 15 min at 37 °C.
4. Assay on the Station Device
Create and load the desired protocol file with the software and activate the temperature control unit for the reading chamber. Here, measure calcium responses at 37 °C for 2 min one row at a time with 1.52 sec interval between successive readings (measuring a complete 96-well plate takes approximately 25 min) at 525 nm. Set the excitation of Fluo-4 to 488 nm and transfer a total volume of 50 µl of compound to the cell plate, 18 sec after the reading start, with the dispense speed set at 26 µl/sec and a pipette height of 135 µl.
Position the compound and cell plate, and the tip rack in the appropriate drawers of the station device.
Perform a single read of the cell plate at the same excitation and emission wavelength in the ENDPOINT mode, before starting the actual screen in FLEX mode. Note: This measurement gives relative fluorescence unit (RFU) values and allows detecting variability between wells of the plate. Values between 20,000 and 40,000 are considered as acceptable.
Start the assay and analyze the data with the software.
Representative Results
Concentration series ranging from 50 µM to 0.001 nM were tested for all four Drosophila sNPF peptides (Drome-sNPF-1: AQRSPSLRLRFamide, Drome-sNPF-2: SPSLRLRFamide, Drome-sNPF-3: PQRLRWamide, Drome-sNPF-4: PMRLRWamide) on HEK293T cells that transiently express the Drosophila sNPF receptor and the promiscuous Gα16 subunit. The GPCR was activated by all four peptides in final concentrations up to 0.1 nM, and receptor activation was concentration-dependent. Figure 2 depicts the graphs corresponding to one of three replicas of the Drome-sNPF-1 concentration series. The negative control (wash buffer) did not induce a fluorescent signal, while the positive control (PAR1 – 1 µM) elicited strong activation of the endogenous protease-activated receptor-1, leading to a high fluorescent signal (± 30,000 RFU). It should be noted that a deviation of the typical curve may indicate abnormalities. For example, an ongoing rise of the curve without returning to the baseline might be related to non-receptor-mediated signals, such as the presence of calcium ionophores, or a disrupted lipid bilayer causing calcium leaks.
The software allows entering formulas to determine the percentage of activation or the standard errors of the different concentrations among others. The resulting data are used to compose preliminary concentration-response curves to estimate the EC50 values, as shown for the four Drome-sNPF peptides in Figure 3. The curves of Figure 3 also include the data for the negative control where the concentration series of the Drome-sNPF peptides were tested on HEK293T cells transfected with an empty pcDNA3.1 vector. The results indicate that the addition of the peptides to these cells have no effect on endogenous receptors, but indeed activate the receptor of interest. The fluorescence-based calcium mobilization assay was then repeated three times independently. Based on the preliminary concentration-response curves of the initial screen, the tested concentrations were adapted to cover the dynamic range of the curve (Figure 4). The EC50 values of Drome-sNPF-1 (2.04 ± 1.48 nM [95% confidence interval]), Drome-sNPF-2 (5.89 ± 3.74 nM), Drome-sNPF-3 (5.55 ± 3.95 nM) and Drome-sNPF-4 (0.50 ± 1.01 nM) are similar, indicating that they are equally potent to activate the receptor.
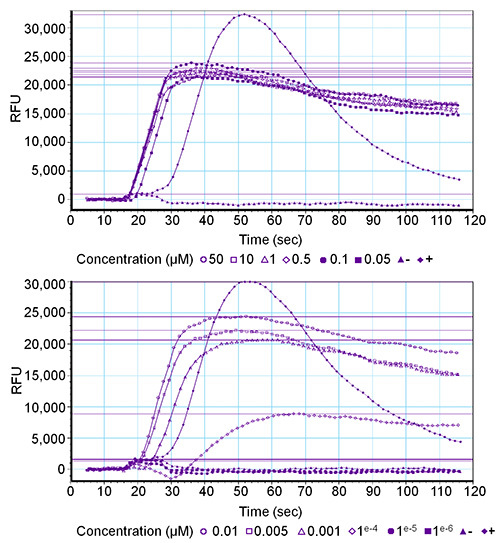 Figure 2. Graphical output of the software for a receptor-mediated fluorescence response of a concentration series. Twelve final concentrations (ranging from 50 µM to 0.001 nM) of the Drome-sNPF-1 peptide were tested on the Drome-sNPF receptor. Activation is expressed in relative fluorescent unit (RFU) values. The upper graph gives the results of the six highest concentrations and the lower graph of the six lowest concentrations. The positive control (+) is PAR1 (1 µM) and the negative control (-) is wash buffer.
Figure 2. Graphical output of the software for a receptor-mediated fluorescence response of a concentration series. Twelve final concentrations (ranging from 50 µM to 0.001 nM) of the Drome-sNPF-1 peptide were tested on the Drome-sNPF receptor. Activation is expressed in relative fluorescent unit (RFU) values. The upper graph gives the results of the six highest concentrations and the lower graph of the six lowest concentrations. The positive control (+) is PAR1 (1 µM) and the negative control (-) is wash buffer.
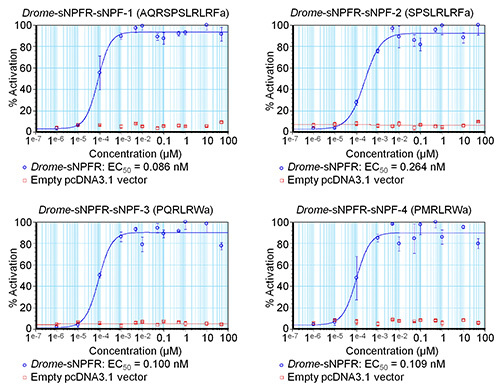 Figure 3. Preliminary concentration-response curves of Drosophila sNPF peptides determined by a single fluorescence-based calcium mobilization assay. The concentration-response curves of the Drosophila sNPF receptor transiently expressed in HEK293T cells for the four Drosophila sNPF peptides are shown in blue. For the negative controls (shown in red), peptides were tested on HEK293T cells transfected with an empty pcDNA3.1 vector. The fluorescent responses are shown as relative (%) to the highest value (100% activation). The curves are the result of one experiment in which each concentration series was measured in triplicate. The vertical bars represent standard errors of the mean (SEM), which sometimes are smaller than the used symbols (in that case, only the symbols are depicted).
Figure 3. Preliminary concentration-response curves of Drosophila sNPF peptides determined by a single fluorescence-based calcium mobilization assay. The concentration-response curves of the Drosophila sNPF receptor transiently expressed in HEK293T cells for the four Drosophila sNPF peptides are shown in blue. For the negative controls (shown in red), peptides were tested on HEK293T cells transfected with an empty pcDNA3.1 vector. The fluorescent responses are shown as relative (%) to the highest value (100% activation). The curves are the result of one experiment in which each concentration series was measured in triplicate. The vertical bars represent standard errors of the mean (SEM), which sometimes are smaller than the used symbols (in that case, only the symbols are depicted).
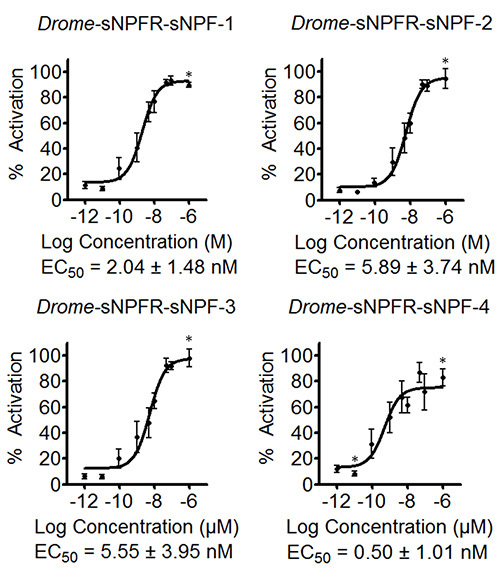 Figure 4. Concentration-response curves and corresponding EC50 values of Drosophila sNPF peptides determined by three independent fluorescence-based calcium mobilization assays. The concentration response curves of the Drosopihla sNPF receptor transiently expressed in HEK293T cells for the four Drosophila sNPF peptides are the result of three independent measurements each performed in triplicate (n ≥ 9). The fluorescent responses are shown as relative (%) to the highest value (100% activation). Asterisks indicate concentrations for which n ≤ 9. Error bars indicate SEM, which sometimes are smaller than the used symbols (in that case, only the symbols are depicted). EC50 values are shown with their 95% confidence intervals.
Figure 4. Concentration-response curves and corresponding EC50 values of Drosophila sNPF peptides determined by three independent fluorescence-based calcium mobilization assays. The concentration response curves of the Drosopihla sNPF receptor transiently expressed in HEK293T cells for the four Drosophila sNPF peptides are the result of three independent measurements each performed in triplicate (n ≥ 9). The fluorescent responses are shown as relative (%) to the highest value (100% activation). Asterisks indicate concentrations for which n ≤ 9. Error bars indicate SEM, which sometimes are smaller than the used symbols (in that case, only the symbols are depicted). EC50 values are shown with their 95% confidence intervals.
Discussion
The fluorescence-based calcium mobilization assay was successfully applied to confirm the functional characterization of the Drosophila sNPF peptidergic signaling system, which was already performed by Mertens et al. with a bioluminescence assay and by Feng et al. with an electrophysiological assay13,15. The EC50 values obtained with the fluorescence assay in HEK293T cells are about 10-fold less than those obtained with the bioluminescence assay performed in CHO cells (Drome-sNPF-1: fluo = 2.04 nM, lumi = 51 nM; Drome-sNPF-2: fluo = 5.89 nM, lumi = 42 nM; Drome-sNPF-3: fluo = 5.55 nM, lumi = 31 nM; Drome-sNPF-4: fluo = 0.50 nM, lumi = 75 nM). These variations can be explained by several factors, including the fact that one of the used expression systems may be better suited for functional expression of a given receptor, or the folding of some receptors can be less efficient in certain cell types. The EC50 values of all four Drome-sNPF peptides are in the nanomolar range when tested on their receptor with both the fluorescence and the bioluminescence assay, generally supporting the physiological relevance of their peptide-receptor interaction in vivo.
Note that no transfection control with a receptor for which the activating ligand is known was included in the screen presented here, because the Drosophila sNPF signaling system is normally the transfection control in experimental setups. The positive control with an endogenous ligand of the HEK293T cells (PAR1) and the negative control (wash buffer) were included in the screen. The results of PAR1 showed that the cells were in a good condition. The negative control (wash buffer) did not elicit a fluorescent signal, which indicates that the medium in which the peptides are dissolved was free of any contaminants that could influence the results.
The previously characterized Drosophila sNPF signaling system was used here to explain the fluorescence-based calcium mobilization assay. For this purpose, concentration series of the activating ligands were tested immediately. However, when an orphan receptor is brought to overexpression in a screening assay to test a library containing hundreds of compounds, it is recommended to first screen with relatively high final concentrations of the ligands (e.g., 10 or 1 µM). Following the detection of an activating compound, a dilution series of that compound can be screened in order to compose a concentration-response curve and to determine the EC50 value.
Once the activating ligand of a receptor is determined, the intracellular signaling pathway can be further investigated by adapting the protocol. The assay can be performed as described above, but without co-transfecting the Gα16 subunit. When a calcium response is measured, it means that the receptor couples with an endogenous Gαq subunit of the cellular expression system. When no fluorescent signal is observed, protocols to measure changes in concentrations of other secondary messengers (e.g., cAMP) can be applied.
Structure-activity relationship (SAR) studies can also be carried out to define the peptide’s core sequence required for receptor activation. First, truncated sequences are assessed to define the minimal amino acid sequence of the peptide that is still able to activate the receptor. Next, peptides can be tested in which systematically every amino acid has been substituted for an alanine residue. Testing synthetic alanine-substitution series on the receptor allows to determine the importance of each of the amino acids for receptor activation24,25.
Despite its frequent use and proven efficacy, it has to be emphasized that the assay described here may need some adaptations to gain optimal results for specific receptors of interest. The Gα16 subunit has the advantage that it binds to most GPCRs, but may also have a dominant-negative effect on receptors that endogenously couple via Gαq22. In this case, it may be useful to test different combinations of G proteins during the optimization of a novel calcium assay and to compare the results for Gαq-coupled receptors in the absence or presence of Gα16. Alternative assays that are independent of the interacting G protein to detect receptor activation can also be performed, such as the translocation of GFP-labeled arrestin, or the detection of changes in membrane potential (e.g., by the FLIPR Membrane Potential Assay Kit). Besides the Fluo-4 AM used here, a wide array of other calcium-sensitive fluorophores, each with its own spectral and chemical properties, is available. The most appropriate fluorophore can be selected based on the GPCR, cell type and the available plate reader, but experimental verification is necessary. The amounts of transfected DNA and the DNA/transfection reagent ratio need to be determined for each receptor-transfection reagent-cell line combination. Finally, it should be kept in mind that cells in continuous culture only allow 20-25 usable passages to perform the screening assays.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors acknowledge the Research Foundation Flanders (FWO-Vlaanderen, Belgium, G.0601.11) and the KU Leuven Research Foundation GOA/11/002. IB, TJ and LT benefit from a fellowship from the FWO-Vlaanderen.
References
- Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors an evolutionary success. EMBO J. 1999;18(7):1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21(1):90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- Drews J. Drug discovery a historical perspective. Science. 2000;287(5460):1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks emerging paradigms. Trends Pharmacol Sci. 2001;22(7):368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- Bendena WG. Neuropeptide physiology in insects. Adv Exp Med Biol. 2010;692:166–191. doi: 10.1007/978-1-4419-6902-6_9. [DOI] [PubMed] [Google Scholar]
- Van Hiel M B, et al. Neuropeptide receptors as possible targets for development of insect pest control agents. Adv Exp Med Biol. 2010;692:211–226. doi: 10.1007/978-1-4419-6902-6_11. [DOI] [PubMed] [Google Scholar]
- Tang X L, Wang Y, Li D L, Luo J, Liu M Y. Orphan G protein-coupled receptors (GPCRs): biological functions and potential drug targets. Acta Pharmacol Sin. 2012;33(3):363–371. doi: 10.1038/aps.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelli O, Reinscheid RK, Zhang Y, Wang Z, Fredriksson R, Schiöth HB. G protein-coupled receptor deorphanizations. Annu Rev Pharmacol Toxicol. 2013;53:127–146. doi: 10.1146/annurev-pharmtox-010611-134548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens I, Vandingenen A, Meeusen T, De Loof A, Schoofs L. Postgenomic characterization of G-protein-coupled receptors. Pharmacogenomics. 2004;5(6):657–672. doi: 10.1517/14622416.5.6.657. [DOI] [PubMed] [Google Scholar]
- Brough SJ, Shah P. Use of aequorin for G protein-coupled receptor hit identification and compound profiling. Methods Mol Biol. 2009;552:181–198. doi: 10.1007/978-1-60327-317-6_13. [DOI] [PubMed] [Google Scholar]
- Gee KR, Brown KA, Chen WNU, Bishop-Stewart J, Gray D, Johnson I. Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium. 2000;27(2):97–106. doi: 10.1054/ceca.1999.0095. [DOI] [PubMed] [Google Scholar]
- Beets I, Lindemans M, Janssen T, Verleyen P. Deorphanizing G protein-coupled receptors by a calcium mobilization assay. Methods Mol Biol. 2011;789:377–391. doi: 10.1007/978-1-61779-310-3_25. [DOI] [PubMed] [Google Scholar]
- Mertens I, Meeusen T, Huybrechts R, De Loof A, Schoofs L. Characterization of the short neuropeptide F receptor from Drosophila melanogaster. Biochem Biophys Res Commun. 2002;297(5):1140–1148. doi: 10.1016/s0006-291x(02)02351-3. [DOI] [PubMed] [Google Scholar]
- Lu H-L, Kersch CN, Taneja-Bageshwar S, Pietrantonio PV. A calcium bioluminescence assay for functional analysis of mosquito (Aedes aegypti) and tick (Rhipicephalus microplus) G protein-coupled receptors. J. Vis. Exp. 2011. p. e2732. [DOI] [PMC free article] [PubMed]
- Feng G, et al. Functional characterization of a neuropeptide F-like receptor from Drosophila melanogaster. Eur. J. Neurosci. 2003;18(2):227–238. doi: 10.1046/j.1460-9568.2003.02719.x. [DOI] [PubMed] [Google Scholar]
- Nässel DR, Wegener C. A comparative review of short and long neuropeptide F signaling in invertebrates any similarities to vertebrate neuropeptide Y signaling. Peptides. 2011;32(6):1335–1355. doi: 10.1016/j.peptides.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Grimmelikhuijzen CJP, Hauser F. Mini-review The evolution of neuropeptide signaling. Regul Pept. 2012;177:S6–S9. doi: 10.1016/j.regpep.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Caers J, Verlinden H, Zels S, Vandersmissen HP, Vuerinckx K, Schoofs L. More than two decades of research on insect neuropeptide GPCRs an overview. Front Endocrinol (Lausanne. 2012;3(151):1–30. doi: 10.3389/fendo.2012.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5’-noncoding sequences from 699 vertebrate messenger RNAs) Nucleic Acids Res. 1987;15(20):8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Simon MI. Gα15 and Gα16 couple a wide variety of receptors to phospholipase. CJ Biol Chem. 1995;270(25):15175–15180. doi: 10.1074/jbc.270.25.15175. [DOI] [PubMed] [Google Scholar]
- Ral Conklin B, et al. Carboxyl-terminal mutations of Gqα and Gsα that alter the fidelity of receptor activation. Mol Pharmacol. 1996;50(4):885–890. [PubMed] [Google Scholar]
- Kostenis E. Is Gα16 the optimal tool for fishing ligands of orphan G-protein-coupled receptors. Trends Pharmacol Sci. 2001;22(11):560–564. doi: 10.1016/s0165-6147(00)01810-1. [DOI] [PubMed] [Google Scholar]
- Robas NM, Fidock MD. Identification of orphan G protein-coupled receptor ligands using FLIPR assays. Methods Mol Biol. 2005;306:17–26. doi: 10.1385/1-59259-927-3:017. [DOI] [PubMed] [Google Scholar]
- Caers J, Peeters L, Janssen T, De Haes W, Gäde G, Schoofs L. Structure-activity studies of Drosophila adipokinetic hormone (AKH) by a cellular expression system of dipteran AKH receptors. Gen Comp Endocrinol. 2012;177(3):332–337. doi: 10.1016/j.ygcen.2012.04.025. [DOI] [PubMed] [Google Scholar]
- Peeters L, et al. A pharmacological study of NLP-12 neuropeptide signaling in free-living and parasitic nematodes. Peptides. 2012;34(1):82–87. doi: 10.1016/j.peptides.2011.10.014. [DOI] [PubMed] [Google Scholar]