

Abstract
Objective
To determine if type 2 diabetes mellitus (DM) is protective against giant cell arteritis (GCA) and to estimate the incidence of GCA diagnosis in the Medicare population.
Methods
Medicare 5% claims files from 1991-2011 were used to identify beneficiaries diagnosed with DM, but not GCA, within a three-year ascertainment period. Propensity score matching was used to define a control group of non-diabetics with comparable demographic covariates. Competing-risk regression was then used to assess the impact of DM diagnosis on GCA diagnosis. To allow for a three-year ascertainment period, the analysis sample was limited to beneficiaries over 68 years old at baseline.
Results
A total of 151,041 beneficiaries diagnosed with DM were matched to an equal number of controls. Mean study follow-up was 67.75 months. GCA was diagnosed among 1,116 beneficiaries with DM (0.73%) versus 465 (0.30%) controls. The risk of receiving a GCA diagnosis among patients with DM was increased by 100% (sub hazard ratio (SHR): 2.00; 95% confidence interval (CI): 1.78 2.25). The annual incidence of GCA diagnosis among U.S. Medicare beneficiaries over 68 was 93 in 100,000.
Conclusion
A DM diagnosis is not protective against a GCA diagnosis in the Medicare population. Our data suggests that a DM diagnosis increases the risk of GCA diagnosis within 5.7 years for Medicare beneficiaries over 68.
Introduction
Giant cell arteritis (GCA) is a T-cell dependent vasculitis of medium and large arteries, almost exclusively affecting patients over age 50(1). Histologically, GCA causes a panarteritis with infiltration of the vessel wall by activated T-cells and macrophages(1). Destruction of the internal elastic lamina and intimal hyperplasia lead to an occlusive vasculitis, which can result in irreversible blindness, myocardial infarction, or stroke if untreated. Diagnosis of GCA requires careful consideration of clinical presentation and serum inflammatory markers. Ultimately, the diagnostic gold standard for GCA remains temporal artery biopsy (TAB).
Clinically, the diagnosis can be quite challenging, especially in the absence of headache, jaw claudication, or other systemic GCA symptoms. Physicians must weigh their clinical index of suspicion in the context of serum erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and platelet levels to decide when to start corticosteroids and proceed with TAB. Unfortunately, ESR and CRP are not 100% sensitive or specific, and equivocal serum inflammatory markers can occur in biopsy proven GCA. Conversely, chronically elevated ESR and CRP can occur in patients without GCA who have diabetes mellitus (DM), cardiovascular disease, and metabolic syndromes (2-7).
Previous reports have identified a potentially protective effect of DM against GCA (5, 8-9). Proposed mechanisms include an altered cytokine profile, impaired T cell response, and impaired dendritic cell function due to excessive antigen glycosylation(5, 8-9). One recent retrospective study found a lower prevalence of DM among patients with a positive TAB than with a negative TAB(5). A meta-analysis of eight GCA studies found a low prevalence of biopsy positive GCA among patients with DM(5). The authors suggested that DM may actually lower the risk of GCA. Additionally, they implied that elevated ESR and CRP may result from DM and may not increase the likelihood of GCA diagnosis. If this is true, our GCA practice patterns for patients with DM may change dramatically. Therefore, we sought to explore this hypothesis in a larger patient population. Given the low incidence of GCA, it is not practical to study the relationship between DM and GCA prospectively (10-19). Therefore, we queried the Medicare 5% claims and enrollment data, and then followed beneficiaries through time to compare the incidence of GCA diagnosis among individuals with and without a DM diagnosis.
Methods
Institutional review board approval was obtained for this study from Duke University Medical Center.
Data
Medicare is a national health insurance program that serves Americans who live within the geographical borders of the U.S. Most Medicare beneficiaries qualify at age 65. Younger individuals with a qualifying disability may also be enrolled. The Medicare 5% claims dataset is a nationally representative, random sample of the overall Medicare beneficiary population containing information on services paid for by Medicare Part A (covering hospital and other facility-based services) and Medicare Part B (covering professional services, mostly services provided by physicians). Claims information is not available for beneficiaries who enroll in a Medicare Advantage (MA) plan, a private alternative to traditional Medicare. The 5% Medicare claims and enrollment data from 1991-2011 was queried for diagnoses of DM and GCA. Demographic characteristics, enrollment information, International Classification of Diseases, 9th Revision (ICD-9) codes, and Current Procedure Terminology (CPT) codes were available for the entire sample. Beneficiaries enrolled in Medicare Advantage (MA) plans were excluded since the 5% sample does not include claims for care provided by MA.
Sample Selection
To assess the incidence of GCA diagnosis, study inclusion required an ascertainment period of three-years during which a diagnosis of GCA was not documented. Since TAB results are not part of the Medicare 5% sample data, GCA was defined as 1) any beneficiary who received a GCA diagnosis (ICD-9 446.5) following a temporal artery biopsy (CPT 37609), or 2) any patient who received two GCA diagnoses (ICD-9 446.5) within 180 days. To ensure the availability of a three-year ascertainment period, beneficiaries were required to be age 68 or older at baseline. Baseline was defined as the date of the first type 2 DM diagnosis (ICD-9 250.xx) for the DM group and the first date, after the beneficiary's 68th birthday, a Medicare claim appeared in the claims data for the non-DM group. All beneficiaries diagnosed with DM received this diagnosis prior to being diagnosed with GCA. The non-DM control group was identified using propensity score matching (PSM) to select a matched sample of beneficiaries who were never diagnosed with type 2 DM during the three-year ascertainment period or during follow up.
Current standards of PSM research accept a standardized difference of roughly 10% between the treatment and control groups for all covariates of interest (20-23). Matching covariates were: gender; black race; other race (white race omitted); baseline year; age at baseline; and binary variables for hypertension (ICD-9 401.xx) and hyperlipidemia (ICD-9 272.0x–272.4x) at baseline. We used logistic regression to estimate the propensity score and nearest neighbor matching (without replacement) within a caliper of 0.001 to identify the match. The size of the caliper was well below the 20% of the standard deviation of the propensity score guideline suggested in the literature (24).
Beneficiaries meeting the inclusion criteria were followed until they were 1) diagnosed with GCA, 2) died, or 3) otherwise censored. Beneficiaries were censored if they 1) joined a MA plan, 2) moved outside the U.S., or 3) had no Medicare claims during follow-up.
Statistical Analysis
The DM and non-DM group were compared using a competing-risk regression model based on the method of Fine and Gray(25). The underlying rationale for competing risk regression has been discussed elsewhere (26-28). Briefly, competing risk regression uses the cumulative incidence function to model probability of failure from a specific cause (the primary risk), in the presence of other events that could prevent the outcome of interest (the competing risks). GCA diagnosis was the primary risk, and death was the competing risk. The sub-hazard ratios (SHR) are interpreted like hazard ratios in Cox regression. Given the advanced age and morbidity of the Medicare population, competing-risk regression was chosen over Cox regression because the risk of death from all other causes could potentially censor the observations of interest prematurely.
The incidence of GCA was calculated by dividing the number of GCA diagnoses for the PSM matched cohort by the total number of person years, which was calculated by multiplying the number of PSM matched beneficiaries by the average study time.
Results
A total of 151,041 beneficiaries with a type 2 DM diagnosis were matched to an equal number of beneficiaries without a type 2 DM diagnosis. In the absence of PSM, the beneficiary mix in the DM and non-DM groups was not comparable with respect to hyperlipidemia, hypertension, and baseline year, as these covariates exceeded the 10% standardized difference criterion(20-23). PSM substantially improved the comparability of both groups, bringing all covariates well within the 10% guideline (Table 1).
Table 1. Sample characteristics before and after propensity score matching.
| Before Matching | After Matching | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| DM Group (treatment) | No DM Group (control) | Standardized difference | DM Group (treatment) | No DM Group (control) | Standardized difference | |
|
|
|
|
|
|
|
|
| Covariate | ||||||
| Male | 0.40 | 0.36 | 7.88 | 0.40 | 0.42 | -4.54 |
| Black | 0.08 | 0.07 | 7.10 | 0.09 | 0.11 | -6.42 |
| Other | 0.07 | 0.06 | 5.19 | 0.07 | 0.08 | -6.11 |
| Age at baseline | 77.93 | 77.41 | 7.44 | 77.89 | 77.56 | 4.42 |
| Hyperlipidemia | 0.71 | 0.05 | 188.28 | 0.28 | 0.26 | 4.80 |
| Hypertension | 0.85 | 0.14 | 204.35 | 0.58 | 0.61 | -5.95 |
| Baseline year | 2002 (+8mo) | 1995 (+6mo) | 120.46 | 2000 (+3mo) | 2000 | 5.35 |
|
| ||||||
| n=536,115 | n=1,069,735 | n=151,041 | n=151,041 | |||
Note: standardized differences, greater than 10% are in bold. Values presented are group means, which correspond to the percentages of the population for the binary variables.
In the pooled sample, the majority of subjects were Caucasian (82.3%) and female (58.8%). A total of 9.9% were black, and 7.8% were identified as other race. The mean beneficiary age at baseline was 77.7 years. The mean age for black and non-black beneficiaries was 77.6 years and 77.7, respectively (p<0.01). Substantial beneficiary fractions were diagnosed with hyperlipidemia (26.8%) and hypertension (59.1%).
Mean study time was similar for the DM and non-DM groups (67.4 months, and 68.1 months, respectively). Median follow up was 56 months for the DM group and 57 months for the non-DM group. GCA was diagnosed among 1,116 (0.73%) beneficiaries in the DM group versus 465 (0.30%) beneficiaries in the non-DM group. Death, the competing-risk event occurred in 94,273(62.42%) of the DM group and 80,512(46.39%) of the non-DM group. The risk of receiving a GCA diagnosis among beneficiaries diagnosed with type 2 DM increased by 100% (SHR: 2.00; 95% confidence interval (CI): [1.78, 2.25]). Non-white race and male gender were primarily protective. Hyperlipidemia and hypertension increased the risk of GCA (Table 2, Figure 1).
Table 2. Risk factors for giant cell arteritis.
| Giant Cell Arteritis, SHR(95% CI) | |
|---|---|
|
|
|
| Diabetic | 2.00** |
| (1.78 2.25) | |
| Male | 0.47** |
| (0.41 0.53) | |
| Black | 0.76* |
| (0.62 0.93) | |
| Hyperlipidemia | 1.27** |
| (1.12 1.42) | |
| Hypertension | 1.22* |
| (1.09 1.36) | |
| Age at baseline | 0.97** |
| (0.96 0.98) | |
| Baseline year | 0.98** |
| (0.97 0.99) | |
| N | 302,082 |
| % with condition | 0.52 |
p<0.05;
p<0.01 Confidence intervals are reported at 95%
Abbreviations: SHR, sub hazard ratio; CI, confidence interval
Note: Other race, including Hispanic ethnicity were also controlled for in the analysis.
Figure 1. Cumulative Incidence of GCA by whether or not a beneficiary was diagnosed with Diabetes Mellitus.
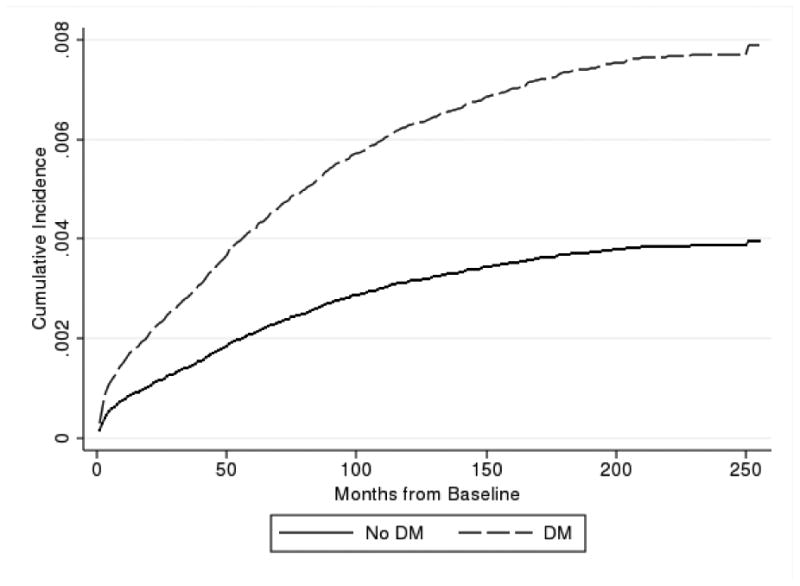
Of the 302,082 PSM-matched beneficiaries included in this study, 1,581 were diagnosed with GCA (GCA group). The majority (85%) of the GCA group had a GCA diagnosis following TAB, while only 15% of the GCA group had two diagnoses of GCA within a 180 day period, but no TAB. We conducted a sensitivity analysis omitting the two diagnosis only group and observed no notable changes in the results. The mean study time for the entire matched cohort was 67.8 months, or 5.7 years. The annual incidence of GCA diagnosis was, therefore, 92 diagnoses per 100,000 beneficiaries. The incidence of GCA diagnosis was significantly higher among white beneficiaries than black beneficiaries (98 per 100,000 vs. 77 per 100,000, respectively [p<0.001]).
Discussion
Contrary to previous reports suggesting a protective effect of DM on GCA, we found that Medicare beneficiaries over age 68 with a type 2 DM diagnosis were more likely to receive a GCA diagnosis than beneficiaries over age 68 without a DM diagnosis. Therefore, DM is not protective against GCA in this population, and DM actually increases the risk of GCA by 100%.
Pathophysiologic links between DM and GCA have been proposed, but not well defined. Previous studies have suggested that an impaired T-cell response in DM is protective against GCA, however, our results suggest the opposite (5,8-9). The study by Matthews et al was a retrospective study of 215 patients at one academic institution (5). One strength of the Matthews study is that all subjects underwent TAB. However, only 44 patients had biopsy-positive GCA, and only 4 of these patients had DM. Similarly, Cid et al studied 200 biopsy-positive GCA patients at 3 hospitals, and Gonzalez-Gay et al studied 215 biopsy-positive patients at one hospital (8-9). The relatively small sample sizes in these studies compared to our sample of over 300,000 Medicare beneficiaries may explain our conflicting results. Moreover, our random sampling of the national database removes the selection bias inherent in the single center studies. Because our inclusion criteria did not mandate TAB results like the aforementioned studies, we acknowledge our GCA incidence may be overestimated. However, the overwhelming majority (85%) of our GCA group received a GCA diagnosis following TAB, implying that the biopsy was positive.
Mounting evidence suggests a common abnormality between cell-mediated immune dysfunction in patients with DM and GCA, which may support our findings. Specifically, regulatory T-cell (Treg) deficiency has been implicated in various autoimmune and metabolic disorders including rheumatoid arthritis, atherosclerosis, diabetes type-1, metabolic syndrome, and multiple sclerosis(30). Patients with GCA and polymyalgia rheumatic (PMR) may have decreased serum levels of these immunosuppressant Treg cells(31). Conceivably, an impaired immune response, possibly due to Treg deficiency, may represent a common thread between DM and GCA that could explain the increased risk of GCA diagnosis we found in type 2 diabetic Medicare beneficiaries over 68.
The annual incidence of GCA has been previously reported in geographically and ethnically homogenous populations over age 50. Retrospective studies of primarily Scandinavian populations from Northern Europe and Olmsted County, Minnesota indicate an annual incidence around 20 per 100,000 individuals over age 50(10-15). A lower GCA incidence of 10 per 100,000 was reported in Mediterranean populations over age 50(14, 16-19). An accurate annual incidence of GCA across the entire U.S. population has not been firmly established. The study herein demonstrates the overall incidence of GCA diagnosis is 92 per 100,000 U.S. Medicare beneficiaries over age 68. This is not necessarily inconsistent with previous published series, but cannot be compared directly due to the different age of the populations studied. Despite the inherent age restrictions encountered in Medicare-based studies like ours, the incidence of GCA diagnosis in this large, heterogeneous, nationally representative sample defines the annual GCA in the patient population most afflicted by this potentially devastating disease.
GCA rarely occurs among black individuals(19,32). In our study, the incidence of GCA diagnosis was significantly higher among white beneficiaries than black beneficiaries. The reasons for this difference are unclear, and further research is needed to determine the effect of race on GCA diagnosis.
We acknowledge the inherent limitations of using billing records to measure disease incidence. Medicare claims data are designed for administrative rather than for clinical purposes. However, use of these data has been shown to provide valid measures of underlying clinical phenomena. We also recognize that not all providers use strict diagnostic criteria when coding for GCA. Moreover, we assumed that 1) a GCA diagnostic billing code following a TAB CPT code implies that the TAB was positive, and 2) two GCA ICD-9 codes within 180 days indicates the presence of GCA. However, the Medicare database yielded a nationwide sample of more than 300,000 beneficiaries, which represents a large, diverse cohort adequate to study the incidence of GCA diagnosis. Using this national sample of real world practicing providers makes our data highly generalizable to the elderly population. Conceivably, we may be overestimating the incidence of GCA by including beneficiaries with two GCA diagnoses within 180 days, and not requiring a GCA diagnosis after TAB for study inclusion. This study's retrospective design is also a limitation, however, the relative rarity of GCA makes a true prospective study comparing the incidence of GCA in diabetics and non-diabetics nearly impossible. Finally, PSM does not completely eliminate selection bias or replace random assignment.
In conclusion, we found that DM is not protective against GCA in the Medicare population. Moreover, our data suggest that a diagnosis of DM increases the risk of GCA diagnosis in Medicare beneficiaries over 68. Contrary to previous studies, our results suggest that elevated ESR and CRP are just as concerning in diabetic patients as they are in non-diabetic patients undergoing GCA evaluation. Ideally, a prospective study would further clarify the relationship between DM and GCA. To our knowledge, this is the largest population based study to report the incidence of GCA in the Medicare population.
Acknowledgments
Financial Support: Partial support for this study was obtained from a grant from the National Institute on Aging (NIA) to Duke University (R01-AG017473) and from a grant from Research to Prevent Blindness (New York, NY), Lions Club of Minnesota (MSL). The funding organizations had no role in the design or conduct of this research.
Footnotes
Conflict of Interest: The author(s) have no proprietary or commercial interest in any materials discussed in this article. No conflicting relationship exists for any author.
A portion of this research was presented at the North American Neuro-Ophthalmology Society Annual Meeting, Rio Grande, Puerto Rico, March 1-6, 2014.
References
- 1.Weyand CM, Goronzy JJ. Medium-and large-vessel vasculitis. N Engl J Med. 2003;349(2):160–9. doi: 10.1056/NEJMra022694. [DOI] [PubMed] [Google Scholar]
- 2.Elias A, Domurat E. Erythrocyte sedimentation rate in diabetic patients: relationship to glycosylated hemoglobin and serum proteins. J Med. 1989;20(3-4):297–302. [PubMed] [Google Scholar]
- 3.Fröhlich M, Imhof A, Berg G, Hutchinson WL, Pepys MB, Boeing H, Muche R, Brenner H, Koenig W. Association between c-reactive protein and features of the metabolic syndrome: a population-based study. Diabetes Care. 2000;23(12):1835–9. doi: 10.2337/diacare.23.12.1835. [DOI] [PubMed] [Google Scholar]
- 4.Fronczyk A, Molęda P, Safranow K, Piechota W, Majkowska L. Increased concentration of C-reactive protein in patients with type 2 diabetes is associated with obesity and presence of diabetes but not with macorvascular and microvascular complications or glycemic control Inflammation. 2013 doi: 10.1007/s10753-013-9746-4. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthews JL, Gilbert DN, Farris BK, Siatkowski RM. Prevalence of diabetes mellitus in biopsy-positive giant cell arteritis. J Neuroophthalmol. 2012;32(3):202–6. doi: 10.1097/WNO.0b013e31825103cb. [DOI] [PubMed] [Google Scholar]
- 6.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286(3):327–34. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 7.Yousuf O, Mohanty BD, Martin SS, Joshi PH, Blaha MJ, Nasir K, Blumenthal RS, Budoff MJ. High-sensitivity C-reactive protein and cardiovascular disease: a resolute belief or an elusive link? J Am Coll Cardiol. 2013;62(5):397–408. doi: 10.1016/j.jacc.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Cid MC, Font C, Oristrell J, de la Sierra A, Coll-Vinent B, López-Soto A, Vilaseca J, Urbano-Marquez A, Grau JM. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. 1998;41(1):26–32. doi: 10.1002/1529-0131(199801)41:1<26::AID-ART4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Gay MA, Piñeiro A, Gomez-Gigirey A, Garcia-Porrua C, Pego-Reigosa R, Dierssen-Sotos T, Llorca J. Influence of traditional risk factors of atherosclerosis in the development of severe ischemic complications in giant cell arteritis. Medicine. 2004;83(6):342–7. doi: 10.1097/01.md.0000145369.25558.b5. [DOI] [PubMed] [Google Scholar]
- 10.Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland: an epidemiologic and histopathalogic analysis. Arthritis Rheum. 1994;37(7):1007–12. doi: 10.1002/art.1780370705. [DOI] [PubMed] [Google Scholar]
- 11.Franzen P, Sutinen S, Von Knorring J. Giant cell arteritis and polymyalgia rheumatica in a region of Finland: an epidemiologic, clinical and pathologic study, 1984-1988. J Rheumatol. 1992;19(2):273–6. [PubMed] [Google Scholar]
- 12.Gran JT, Myklebust G. The incidence of polymyalgia rheumatica and temporal arteritis in the county of Aust Agder, south Norway: a prospective study 1987-94. J Rheumatol. 1997;24(9):1739–43. [PubMed] [Google Scholar]
- 13.Haugeberg G, Paulsen PQ, Bie RB. Temporal arteritis in Vest Agder County in southern Norway: incidence and clinical findings. J Rheumatol. 2000;27(11):2624–7. [PubMed] [Google Scholar]
- 14.Hunder GG. Epidemiology of giant-cell arteritis. Cleve Clin J Med. 2002;69(Suppl 2):SII79–82. doi: 10.3949/ccjm.69.suppl_2.sii79. [DOI] [PubMed] [Google Scholar]
- 15.Salvarani C, Crowson CS, O'Fallon WM, Hunder GG, Gabriel SE. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Care Res. 2004;51(2):264–8. doi: 10.1002/art.20227. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez-Gay M, Garcia-Porrua C, Rivas M, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis. 2001;60(4):367–71. doi: 10.1136/ard.60.4.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salvarani C, Macchioni P, Rossi F, Castri C, Capozzoli N, Baricchi R, Boiardi L, Chiaravallot F. Epidemiologic and immunogenetic aspects of polymyalgia rheumatica and giant cell arteritis in northern Italy. Arthritis Rheum. 1991;34(3):351–6. doi: 10.1002/art.1780340313. [DOI] [PubMed] [Google Scholar]
- 18.Sonnenblick M, Nesher G, Friedlander Y, Rubinow A. Giant cell arteritis in Jerusalem: a 12-year epidemiological study. Rheumatology. 1994;33(10):938–41. doi: 10.1093/rheumatology/33.10.938. [DOI] [PubMed] [Google Scholar]
- 19.Liu NH, LaBree LD, Feldon SE, Rao NA. The epidemiology of giant cell arteritis: A 12-year retrospective study. Ophthalmology. 2001;108(6):1145–9. doi: 10.1016/s0161-6420(01)00574-7. [DOI] [PubMed] [Google Scholar]
- 20.Austin PC, Grootendorst P, Anderson GM. A comparison of the ability of different propensity score models to balance measured variables between treated and untreated subjects: a Monte Carlo study. Stat Med. 2007;26(4):734–53. doi: 10.1002/sim.2580. [DOI] [PubMed] [Google Scholar]
- 21.d'Agostino RB. Tutorial in biostatistics: propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Stat Med. 1998;17(19):2265–81. doi: 10.1002/(sici)1097-0258(19981015)17:19<2265::aid-sim918>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 22.Normand S-LT, Landrum MB, Guadagnoli E, Ayanian JZ, Ryan TJ, Cleary PD, McNeil BJ. Validating recommendations for coronary angiography following acute myocardial infarction in the elderly: a matched analysis using propensity scores. J Clin Epidemiol. 2001;54(4):387–98. doi: 10.1016/s0895-4356(00)00321-8. [DOI] [PubMed] [Google Scholar]
- 23.Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41–55. [Google Scholar]
- 24.Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46:399–424. doi: 10.1080/00273171.2011.568786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94(446):496–509. [Google Scholar]
- 26.Allignol A, Schumacher M, Wanner C, Drechsler C, Beyersmann J. Understanding competing risks: a simulation point of view. BMC Med Res Methodol. 2011;11(1):86–98. doi: 10.1186/1471-2288-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dignam JJ, Zhang Q, Kocherginsky M. The use and interpretation of competing risks regression models. Clin Cancer Res. 2012;18(8):2301–8. doi: 10.1158/1078-0432.CCR-11-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18(6):695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 29.Austin PG. A critical appraisal of propensity-score matching in the medical literature between 1996 and 2003. Statist Med. 27:2037–49. doi: 10.1002/sim.3150. [DOI] [PubMed] [Google Scholar]
- 30.Chen X, Wu Y, Wang L. Fat-resident Tregs: an emerging guard protecting from obesity-associated metabolic disorders. Obes Rev. 2013;14(7):568–78. doi: 10.1111/obr.12033. [DOI] [PubMed] [Google Scholar]
- 31.Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, Ciudad M, Leguy V, Berthier S, Vinit J, Manckoundia P, Maillefert JF, Besancenot JF, Aho-Glele S, Olsson NO, Lorcerie B, Guillevin L, Mouthon L, Saas P, Bateman A, Martin L, Janikashvili N, Larmonier N, Bonnotte B. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. 2012;64(11):3788–98. doi: 10.1002/art.34647. [DOI] [PubMed] [Google Scholar]
- 32.Sanford R, Berney S. Polymyalgia rheumatica and temporal arteritis in blacks-clinical features and HLA typing. J Rheumatol. 1977;4(4):435–42. [PubMed] [Google Scholar]