

Abstract
Objectives
Aromatase deficiency is a rare disorder resulting in estrogen insufficiency in humans. It has been reported in remarkably few men with loss-of-function mutations in the CYP19A1 gene encoding the aromatase, a cytochrome P450 enzyme that plays a crucial role in the biosynthesis of estrogens from androgens. We investigated a non-consanguineous family including an adult man with clinical features of aromatase deficiency, and studied the effects of estrogen replacement in the man.
Methods
We investigated the clinical and biochemical phenotype, performed CYP19A1 mutational analysis in the family and 50 unrelated persons, studied the effects of CYP19A1 mutations on aromatase protein structure, functionally characterized the mutations by cell-based aromatase activity assays, and studied the effects of estrogen replacement on the bone, lipid, liver and glucose metabolism.
Results
The man with clinical features of aromatase deficiency had novel compound heterozygous CYP19A1 mutations (Y81C and L451P) that were not found in 50 unrelated persons. Three-dimensional modeling predicted that Y81C and L451P mutants disrupted protein structure. Functional studies on the basis of in vitro expression showed that Y81C and L45P mutants significantly decreased the aromatase activity and catalytic efficiency. Estrogen replacement in the man increased bone mineral density, accelerated bone maturation, improved lipid profile and liver steatosis, and improved glucose levels but not insulin resistance.
Conclusions
We have identified two novel CYP19A1 missense mutations in an aromatase-deficient man. Estrogen replacement in the man shows great impact on recovering the impairments in the bone, lipid, liver and glucose metabolism, but fails to improve insulin resistance.
Keywords: Aromatase, CYP19A1, Aromatase deficiency, Metabolic syndrome, Insulin resistance, Estrogen replacement
1. Introduction
Aromatase, an enzyme of the cytochrome P450 superfamily, plays a critical role in the biosynthesis of estrogens (C18 steroids) from androgens (C19 steroids) in vertebrate species (Simpson et al., 2002). In humans, aromatase is encoded by the CYP19A1 gene located at chromosome 15q21.2 (Chen et al., 1988). In fertile women the ovary represents the major source of circulating estrogens, whereas in men the testes accounts for up to 15% of circulating estrogens. The remaining 85% comes from peripheral aromatization of circulating androgen precursors in different tissues, including the adipose tissue, brain, skin, endothelium and bone (Gennari et al., 2004).
Aromatase has important biological functions. Aromatase deficiency, which leads to estrogen deficiency, exerts major effects on bone metabolism, adiposity, lipid profiles, liver function, glucose metabolism, insulin sensitivity and sexual behavior (Santen et al., 2009). The aromatase knockout (ArKO) mouse presents with elevated gonadotropins and testosterone levels, loss of bone mass, obesity, dyslipidemia, liver steatosis, insulin resistance and hyperglycemia, defects in folliculogenesis and spermatogenesis, infertility and impaired sexual behavior (Fisher et al., 1998; Honda et al., 1998; Jones et al., 2000; Nemoto et al., 2000; Robertson et al., 1999).
Aromatase deficiency is an extremely rare disorder in humans. To date, only a small number of cases of women (Bouchoucha et al., 2014; Hauri-Hohl et al., 2011; Zirilli et al., 2008) and men (Morishima et al., 1995; Carani et al., 1997; Deladoey et al., 1999; Herrmann et al., 2002; Maffei et al., 2004, 2007; Lanfranco et al., 2008) have been reported, and most of them are Caucasians. Affected women present with ambiguous genitalia at birth, elevated androgens and undetectable estrogens, primary amenorrhea, and failure of breast development at puberty. Men with aromatase deficiency usually present after puberty with continuing linear growth, tall stature, unfused epiphyses, delayed bone age, eunuchoid skeletal proportions, genu valgum, decreased bone mineral density, overweight or obese, dyslipidemia, liver steatosis, insulin resistance, and impaired fertility. Interestingly, one man and one unrelated woman with estrogen resistance caused by mutations in the estrogen receptor α (ESR1) gene have been reported, and their clinical presentations are similar to that of aromatase-deficient men and women (Quaynor et al., 2013; Smith et al., 1994). Due to the observations of women and men with mutations in the CYP19A1 and ESR1 genes, as well as the studies of animal models such as ArKO mice and estrogen receptor-knockout mice (Jones et al., 2006), the importance of estrogens in the male has been appreciated recently.
We present here a case of a 24-year-old man from a non-consanguineous family with clinical features of aromatase deficiency caused by novel compound heterozygous mutations in the CYP19A1 gene, and the effects of estrogen replacement therapy in the man on the bone, lipid, liver and glucose metabolism.
2. Methods
2.1. Ethics
The study was approved by the Institutional Review Board of the Peking Union Medical College Hospital. Written informed consents were obtained from the patient and family, and 50 unrelated persons.
2.2. Hormonal and biochemical measurements
Serum estradiol, testosterone, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were determined by chemiluminescent immunoassays (Bayer Diagnostics, East Walpole, USA). Osteocalcin and 24 h urine hydroxyproline were measured by radioimmunoassay (Beijing North Institute of Biological Technology, Beijing, China). Serum c-telopeptide of type I collagen (β-CTX) was determined by chemiluminescent immunoassays (E170; Roche Diagnostics, Basel, Switzerland). Alkaline phosphatase, calcium, phosphate, hemoglobin A1c, total cholesterol, low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, triglycerides, aminoleucine transferase (ALT), aspartate aminotransferase (AST), γ-glutamy transferase (GGT) were assayed by a multichannel automatic biochemical analyzer (AU5800; Beckman Coulter, Mishima, Japan).
2.3. X-ray, bone densitometry and ultrasound
X-ray of hands, wrists, lumbar spine and knees were performed. Bone mineral density was assessed at the lumbar spine (L2–4) and femoral neck using dual-energy X-ray absorptiometry (DEXA) (Lunar Prodigy; GE Healthcare, Madison, USA). The T-score was defined as the deviation from the mean bone mineral density of healthy young adults of the same sex and ethnicity. The Z-score was defined as the deviation from the mean bone mineral density of age, sex and ethnicity matched population. Ultrasound of the liver was performed.
2.4. Oral glucose-tolerance testing
The patient underwent a 75-g oral glucose-tolerance test (OGTT) after fasting for 12 h. Plasma glucose and serum insulin levels were measured in blood samples obtained at 0, 30, 60, 120 and 180 minutes after oral glucose loading. Insulin resistance was estimated from the OGTT result and the fasting insulin level, the area under the curve (AUC) for insulin, the homeostasis model assessment (HOMA) of insulin resistance (Wallace et al., 2004), the Matsuda index (Matsuda and DeFronzo, 1999) and the quantitative insulin sensitivity check index (QUICKI) (Katz et al., 2000). Insulin secretion was estimated on the basis of the disposition index, which adjusts insulin secretion for the insulin sensitivity as measured by multiplying the insulinogenic index by the Matsuda index, with the insulinogenic index calculated as the ratio of incremental insulin to incremental glucose response during the first 30 minutes of the OGTT.
2.5. DNA sequence analysis
Genomic DNA was extracted from peripheral blood leukocytes of the patient, his parents and his brother, and 50 unrelated persons using QIAGEN DNA extraction kit (QIAGEN, Hilden, Germany). All nine coding exons and proximal splice sites of the CYP19A1 gene (NCBI Reference Sequence: NM_000103.3) were amplified by PCR with CYP19A1-specific primers (Supplementary Table S1) (Mullis et al., 1997). PCR was carried out in 50 μg reactions containing 100 ng genomic DNA, 0.2 mM of each deoxynucleotide triphosphate (dNTP), 20 pmol of each primer and 1.2 U Taq-DNA polymerase for 35 cycles. DNA sequences of both the sense and antisense strands of each PCR product were sequenced in duplicate with an ABI3730 automated sequencer (Applied Biosystems, Foster City, USA), followed by analysis with the sequence of the CYP19A1 gene using Basic Local Alignment Search Tools (BLAST) software. Sequence changes were assessed for the occurrence of polymorphisms in 50 unrelated persons and in the exome sequence data from approximately 6500 unrelated persons that were obtained from the Exome Sequencing Project of the National Heart, Lung, and Blood Institute (NHLBI-ESP) (http://evs.gs.washington.edu/EVS/).
2.6. Prediction of missense variants, protein sequence alignments and three-dimensional structure modeling
The Sorting Intolerant From Tolerant (SIFT) (Kumar et al., 2009) and the Polymorphism Phenotyping, version 2 (PolyPhen2) (Adzhubei et al., 2010) programs were used to predict whether a missense mutation affects protein function. Sequences of aromatase orthologs and paralogs were obtained from the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov) (Supplementary Table S2), and aligned using the ClustalW multiple sequence alignment program (http://www.ebi.ac.uk). Three-dimensional homology modeling of human aromatase mutants was based on the crystal structure of Cytochrome P450 19A1 (Protein Data Bank code 3EQM) (Ghosh et al., 2009) with the use of the PyMOL Molecular Graphics System (http://www.pymol.org).
2.7. Site-directed mutagenesis
A wild-type human aromatase clone, pMG-H2-Aro, was used as a template to generate the mutated clones (pMG-H2-Aro-Y81C and pMG-H2-Aro-L451P) by the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, USA). The mutagenic primers were designed using Stratagene’s web-based QuikChange Primer Design Program available online (Supplementary Table S3). PCR reactions were performed to synthesize the aromatase mutant strands using PfuUltra High-Fidelity DNA polymerase (2.5 U/μl, Stratagene) under the following conditions: step 1: 95 °C, 30 s; step 2: 20 cycles of 95 °C, 30 s; 55 °C, 1 minute; 68 °C, 6 minutes 15 s. Dpn I restriction enzyme (10 U/μl; Stratagene) was used to digest the parental supercoiled dsDNA at 37 °C for 3 h. The entire cDNA sequences of all mutants were verified by DNA sequencing.
2.8. Western blotting
Chinese hamster ovary (CHO) cells were seeded in 60-mm dishes and were transiently transfected with wild-type or mutant cDNA (5 μg/well) using Lipofectamine 2000 (Invitrogen, Carlsbad, USA). After 24 h, transfected cells were washed with PBS and lysed in 300 μl ice cold RIPA buffer (Cell Signaling, Danvers, USA). Sixty micrograms of cell lysate was resolved on a 10% SDS-PAGE gel and then transferred onto a polyvinylidene difluoride membrane under 18 V for 60 minutes using a Semi Dry Transfer Cell (Bio-Rad, Hercules, USA). The membrane was blocked in 5% w/v nonfat dry milk in TBST for 1 h at room temperature, incubated with aromatase antiserum, which was generated against the human recombinant aromatase produced by the Chen laboratory (Hong et al., 2007), at 1:250 dilution at 4 °C overnight. The membrane then incubated with a horseradish peroxidase conjugated goat anti-rabbit secondary antibody at 1:2500 dilution for 1 h, followed by SuperSignal West Pico Chemiluminescent (Thermo Scientific, Rockford, USA) substrate visualization.
2.9. In-cell aromatase activity assay
CHO cells were seeded in 12-well plates and were transiently transfected with wild-type or mutant constructs (1.25 μg/well) using Lipofectamine 2000 (Invitrogen, Carlsbad, USA). In-cell aromatase activity was determined 24 h later by the release of tritiated water from the substrate [1β-3H(N)]-androstenedione (Perkin Elmer, Waltham, USA) using modified methods described previously (Zhou et al., 1991). Briefly, 1 ml serum-free media containing 100 nM [1β-3H(N)]-androstenedione and 500 nM progesterone were added to each well. After incubating cells for 1.5 h at 37 °C, the media were mixed with charcoal-dextran to remove any trace amount of unreacted substrate, and were centrifuged at 14,000 rpm for 5 minutes. The radioactivity of the supernatant was measured with a liquid scintillation counter (LS 6500; Beckman Coulter, Fullerton, USA). Cells remaining in each well were solubilized with 0.5 M NaOH, and the protein concentration was determined by standard Bradford assay. Aromatase activity was calculated by the radioactivity and adjusting for total protein concentration. At least three independent experiments were performed in triplicate, and results were expressed as mean and standard deviation.
2.10. Aromatase kinetic analysis
Transient transfection of wild-type or mutant constructs were performed in CHO cells the same as the in-cell aromatase activity assay. Aromatase activity was measured 24 h later by the release of tritiated water from the substrate [1β-3H(N)]-androstenedione (Perkin Elmer, Waltham, USA). A saturated curve assay was performed by incubating transfected cells with 0, 2.5, 5,10,15, 25,35, and 65 nM final concentration of [1β-3H(N)]-androstenedione in serum free medium for 1.5 h at 37 °C. The maximum reaction rate (Vmax) and the Michaelis constant (Km) values of aromatase were calculated from using nonlinear regression of the Michaelis-Menten equation by the GraphPad Prism 5 Software. At least three independent experiments were performed in triplicate, and results were expressed as mean and standard error of mean.
2.11. Estrogen replacement therapy
The patient received 25 μg transdermal estradiol (Estraderm TTS; Novartis, Basel, Switzerland) twice per week. To investigate the effects of estradiol replacement in the patient, the following measures were performed for 6 months: sex steroids, gonadotropins, bone mineral density, bone age, bone turnover markers, lipid profile, liver function, glucose levels, insulin resistance, and semen analysis.
3. Results
3.1. Case report
The patient was a 24-year-old Chinese man. He was the first of two siblings. His parents were not cognate. His mother did not have clinical signs of virilization during the pregnancy. The patient’s childhood and pubertal development were unremarkable. At the age of 20 years, he was 172 cm tall (50th percentile) and he continued to grow thereafter. At the age of 22 years, he got genu valgum, which deteriorated gradually. The patient stated that he had normal libido and had no erectile dysfunction. At first observation, the 24-year-old patient weighed 88.2 kg with eunuchoid proportions (the ratio of the upper segment to the lower segment was 0.92) (Fig. 1A). He was 182.5 cm tall (97th percentile) and had an arm span of 190 cm. His blood pressure was 130/80 mmHg. His body mass index (BMI) was 26.5 and waist circumference was 96.5 cm. His pubic hair was normal (Tanner’s stage IV). The sizes (both 15 ml) and consistency of the testis were normal. He had bilateral genu valgum (Fig. 1A). He had acanthosis nigricans on the neck and axillae (Fig. 1B). Palpation of the abdomen indicated hepatomegaly.
Fig. 1.
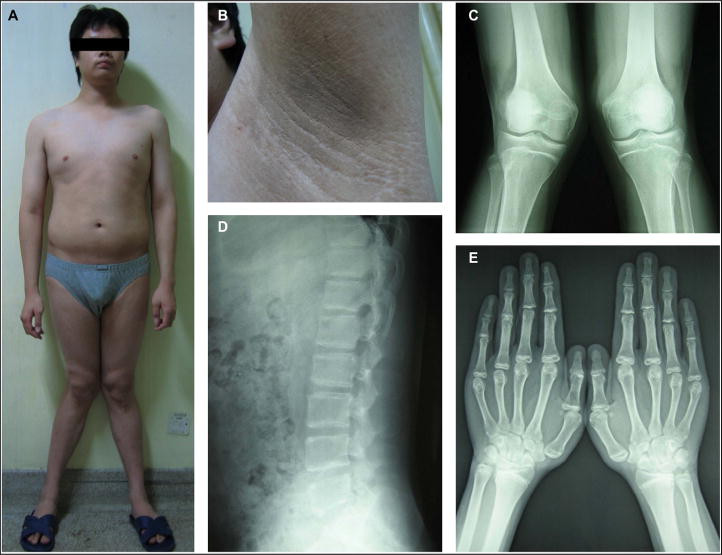
Photographs and X-rays of the patient with aromatase deficiency. (A) Eunuchoid proportions, long arms, visceral adiposity, and bilateral genu valgum are present in the 24-year-old man. (B) Acanthosis nigricans on the axilla in the man. (C) X-ray of knees reveals incompletely fused epiphyses in the tibiae and fibulae, and bilateral genu valgum. (D) X-ray of the lumbar spine reveals osteopenia. (E) X-ray of hands reveals delayed bone age by 6–8 years.
X-ray revealed incompletely fused epiphyses (Fig. 1C), osteopenia (Fig. 1D) and bone age was delayed by 6–8 years (Fig. 1E). Bone densitometry showed a T-score of −1.4 and a Z-score of −2.9 in the lumbar spine. Bone turnover markers (serum alkaline phosphatase, serum osteocalcin, 24 h urine hydroxyproline, and serum β-CTX) were elevated. Gonadotropins (FSH and LH) and the testosterone were in normal ranges but the estradiol was undetectable (Table 1, column Baseline). Lipid profile showed that the triglyceride, total cholesterol and LDL cholesterol levels were increased, whereas the HDL cholesterol level was decreased. Serum ALT, AST and GGT levels were increased, and abdominal ultrasound revealed severe steatohepatitis. OGTT indicated impaired glucose tolerance and hyperinsulinemia. The hemoglobin A1c was 6.9%. The sperm count and vitality were normal (Table 1, column Baseline).
Table 1.
Efficacy of estrogen replacement therapy in the aromatase-deficient man.
| Variable | Baseline | 1 month | 2 months | 3 months | 6 months | Reference range |
|---|---|---|---|---|---|---|
| Estradiol (pmol/l) | Undetectable | 24.81 | 57.59 | 184.61 | 100.37 | 43.9–249.5 |
| Testosterone (nmol/l) | 20.5 | 8 | 2.3 | 2 | 4.7 | 11.1–35.1 |
| Follicle-stimulating hormone (IU/l) | 14.4 | 6.4 | 2.3 | 1 | 3.4 | 0–20.3 |
| Luteinizing hormone (IU/l) | 4.8 | 5.8 | 1.3 | 2 | 2.6 | 0–11.1 |
| Bone age (years) | 16–18 | 16–18 | 16–18 | 18 | 18 | |
| Lumbar spine (L2–4) (DEXA) | ||||||
| Bone mineral density (g/cm2) | 1.021 | 1.041 | 1.087 | 1.105 | 1.137 | |
| T-score | −1.4 | −1.2 | −0.9 | −0.7 | 0.2 | ≥ −1 |
| Z-score | −2.9 | −2.8 | −2.3 | −2.1 | −0.4 | ≥ −2.0 |
| Femoral neck (DEXA) | ||||||
| Bone mineral density (g/cm2) | 0.939 | 0.973 | 0.949 | 0.992 | 1.008 | |
| T-score | −0.1 | 0.2 | 0.3 | 0.3 | 0.2 | ≥ −1 |
| Z-score | −0.9 | −0.7 | −0.8 | −0.4 | −0.2 | ≥ −2.0 |
| Alkaline phosphatase (U/l) | 109 | 118 | 119 | 79 | 94 | 27–107 |
| Osteocalcin (ng/ml) | 12.5 | 10.5 | 11.9 | 7.8 | 7.7 | 1.0–10.2 |
| 24 h urine hydroxyproline (mg) | 52.7 | 50.1 | 20–40 | |||
| C-telopeptide of type I collagen (ng/ml) | 1.2 | 1.0 | 0.016–0.584 | |||
| Calcium (mmol/l) | 2.42 | 2.39 | 2.43 | 2.38 | 2.37 | 2.13–2.70 |
| Phosphorous (mmol/l) | 1.58 | 1.87 | 1.77 | 1.46 | 1.55 | 0.74–1.37 |
| Total cholesterol (mmol/l) | 6.35 | 6.57 | 6.05 | 5.46 | 5.76 | 2.85–5.7 |
| LDL cholesterol (mmol/l) | 4.72 | 5.06 | 4.5 | 3.99 | 4.48 | 2.07–3.63 |
| HDL cholesterol (mmol/l) | 0.78 | 0.82 | 0.9 | 0.93 | 0.98 | 0.93–1.81 |
| Triglycerides (mmol/l) | 2.14 | 1.68 | 1.67 | 1.99 | 1.47 | 0.45–1.7 |
| Aminoleucine transferase (U/l) | 178 | 137 | 127 | 106 | 90 | 5–40 |
| Aspartate aminotransferase (U/l) | 72 | 63 | 47 | 36 | 33 | 5–37 |
| γ-Glutamy transferase (U/l) | 105 | 97 | 84 | 79 | 71 | 10–67 |
| Hemoglobin A1c (%) | 6.9 | 7.5 | 6.8 | 6.3 | 6.1 | 4.5–6.3 |
| Fasting plasma glucose (mmol/l) | 5.8 | 5.9 | 5 | 4.4 | 4.5 | 3.6–6.1 |
| 2 h postload glucose (mmol/l) | 10.6 | 13.8 | 9.9 | 8.3 | 12.1 | <7.8 |
| AUC for glucose | 31.2 | 33.9 | 27.7 | 27.1 | 30.0 | |
| Fasting insulin (μIU/ml) | 17.71 | 36.97 | 49.88 | 50.69 | 47.12 | 5.2–17.2 |
| 2 h insulin (μIU/ml) | 147.45 | 268.87 | 148.64 | >300 | >300 | |
| AUC for insulin | 347.82 | 537.56 | 402.78 | 829.35 | 836.78 | |
| HOMA for insulin resistance | 4.51 | 9.69 | 11.08 | 9.91 | 9.42 | <2.5 |
| QUICKI score | 0.31 | 0.28 | 0.27 | 0.28 | 0.28 | >0.31 |
| Matsuda index score | 1.52 | 0.87 | 0.92 | 0.75 | 0.73 | ≥ 2.5 |
| Insulinogenic index | 0.66 | 0.57 | 1.12 | 3.42 | 3.26 | >0.4 |
| Disposition index | 1.01 | 0.5 | 1.03 | 2.56 | 2.4 | >1 |
| Sperm count (million/ml) | 129 | 84.19 | >20 | |||
| Sperm vitality (%) | 97 | 70.26 | >70 |
DEXA denotes dual-energy X-ray absorptiometry, AUC area under the curve, HOMA homeostasis model assessment, QUICKI quantitative insulin sensitivity check index, LDL low-density lipoprotein, and HDL high-density lipoprotein.
3.2. Genetic analysis of CYP19A1 mutations
DNA sequence analysis of the CYP19A1 1511-bp coding regions and 16 exon-intron boundaries in the proband revealed compound heterozygous sequence abnormalities comprising an A→G transition at c.384 in exon 3 and a T→C transition at c.1494 in exon 10, which predicted the missense mutations Tyr81Cys (Y81C) and Leu451Pro (L451P), respectively. CYP19A1 mutational analysis in the patient’s family identified a heterozygous Y81C missense substitution in his mother, and a heterozygous L451P missense substitution in his father and brother (Fig. 2A and B).
Fig. 2.
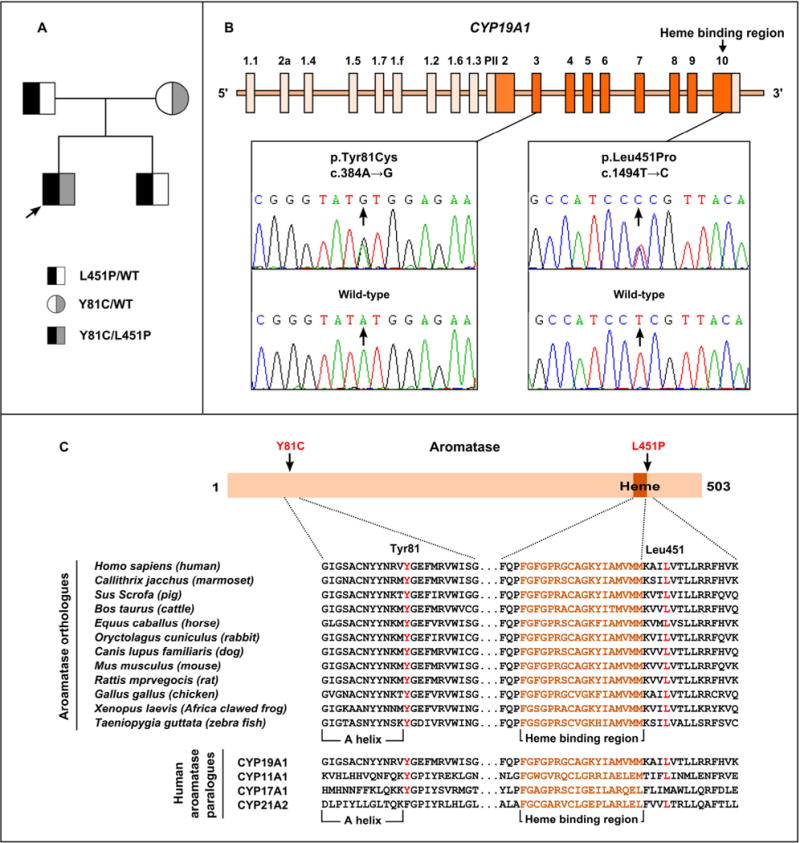
Genetic analysis of the CYP19A1 gene. (A) Pedigree of the patient family with two CYP19A1 missense mutations. (B) The DNA sequence chromatogram of the proband compare with that of a normal unrelated individual (wild-type) reveals the proband to have compound heterozygous CYP19A1 mutations comprising an A→G transition at c.384 (arrowed) in exon 3 and a T→C transition at c.1494 (arrowed) in exon 10, which predicts the missense mutations Tyr81Cys (Y81C) and Leu451Pro (L451P), respectively. (C) Evolutionary conservation of aromatase residues with identified aromatase mutants. Multiple protein sequence alignments are done in aromatase orthologs and paralogs. The Tyr81 residue (red), which is adjacent to the A helix, is highly conserved in aromatase orthologs and is conserved in paralogs. The Leu451 residue (red), which is close to the heme binding region, is highly conserved in aromatase orthologs and is conserved in paralogs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Exon 3 and exon 10 of the CYP19A1 gene of 50 unrelated normal controls were amplified, and both the Y81C and L451P missense variants were not found. In addition, these DNA sequence abnormalities were absent in approximately 6500 exome (in data obtained from the NHLBI-ESP). Multiple protein sequence alignments revealed that both the Tyr81 and Leu451 residues were highly conserved in vertebrate aromatase orhologs, and were also conserved in human aromatase paralogs (Fig. 2C). Taken together, the Y81C and L451P abnormalities were probably CYP19A1 mutations rather than polymorphic variants.
3.3. Predicted effects of aromatase mutant proteins
Both the Tyr81 and Leu451 residues were highly conserved, and Y81C and L451P missense variants were predicted to be “probably damaging” (likely to disrupt protein function) on the basis of SIFT scores of 0.00 (the range for the score is 0–1.0, with scores ≤0.05 predicting damage to protein function) and PolyPhen2 scores of 1.0 (the range for the score is 0–1.0, with 0 indicating that a change is predicted to be neutral and 1.0 indicating that it is most likely to be deleterious).
To further predict the effects of aromatase mutant proteins, we generated the three-dimensional modeling of aromatase mutants based on the reported crystal structure of cytochrome P450 19A1 (PDB code 3EQM) (Ghosh et al., 2009). The Tyr81 residue is located in the N terminus of the molecule. It is adjacent to helixes A (residues 69–80) and A′ (residues 57–68) that are considered to be responsible for the membrane integration of aromatase, which position the entrance to the active site access channel on the membrane surface and allow steroids to enter the aromatase active site (Ghosh et al., 2009, 2010). The Y81C mutation replaced a hydrophobic residue with a neutral polar amino acid. An analysis of the predicted effect of the Y81C on the structure of aromatase indicated that it would lead to a loss of five hydrogen bonds and cause steric clashes in helixes A and A′, thereby disrupting helixes A and A′ and affecting the entrance to the active site access channel, which would limit the entry of steroids to the active site (Fig. 3A).
Fig. 3.
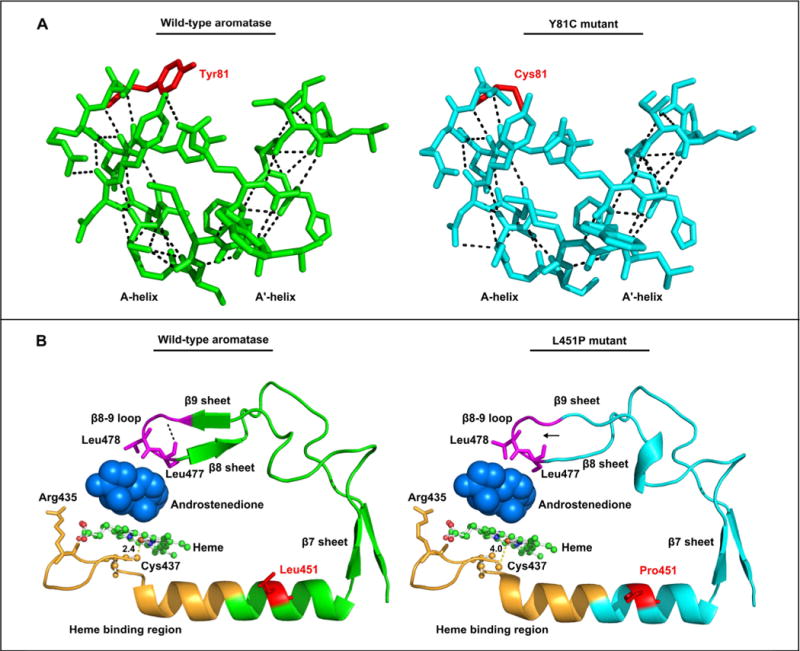
Three-dimensional modeling of aromatase mutant proteins. (A) A three-dimensional modeling of aromatase Y81C mutant compared with the wild-type aromatase. The model is based on the reported crystal structure of human aromatase in complex with androstenedion (PDB code 3EQM). The affected residue Tyr81 (red, left panel), replaced with a cysteine (Cys81, red, right panel) in the mutant, is ajacent to helixes A and A′. An analysis of the predicted structural effect of the Y81C mutant on hydrogen bonds (broken lines) in the helixes A and A′ region shows that Y81C leads to a loss of five hydrogen bonds and causes steric clashes in helixes A and A′ of the Y81C mutant (blue, right panel) as compared with the wild-type aromatase (green, left panel). (B) A three-dimensional modeling of aromatase L451P mutant compared with the wild-type aromatase. The affected residue Leu451 (red, left panel), replaced with a proline (Pro451, red, right panel) in the mutant, is located in the L-helix and is close to the heme binding region (orange) and β7–β9 sheets that contains the β8–β9 loop (purple). The β8–β9 loop contributes important residues (Leu477and Ser478) to the active site/catalytic center and the substrate/product passage channel. An analysis of the predicted effect of the L451P on the structure of aromatase shows that it leads to a loss of a hydrogen bond (arrowed, right panel) in the β8–β9 loop and shift the position of the residue Cys437, the ligand to the heme ion, in the heme binding region. The distance between the Cys437 and the heme ion is 4.0 Å in the L451P mutant (right panel) as compare with 2.4 Å in the wild-type aromatase (left panel). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The Leu451 residue is located in the L-helix (residues 440–455), close to the heme binding region (residues 430–447) and β7–β9 sheets. In the heme binding region, the Arg435 residue is involved in heme coordination, and the Cys437 is the ligand to the heme ion. The β8–β9 loop (residues 475–478) contributes important residues to the active site/catalytic center (Leu477and Ser478), and the channel that permits the passage of the substrate and product as well as exits to the exterior of the protein surface (Ser478) (Ghosh et al., 2009, 2010). The L451P mutation changed a hydrophobic residue to a proline, which not only lacks an amide hydrogen for hydrogen bonding but also has strong sterochemical constraints (Richardson, 1981). A similar analysis of the predicted effect of the L451P on the structure of aromatase indicated that it would lead to a loss of a hydrogen bond in the β8–β9 loop, thereby disrupting the β8–β9 loop and affecting the residues to the active site and the substrate/product passage channel (Fig. 3B). Moreover, it is predicted to shift the position of Cys437, the ligand to the heme ion, in the heme binding region. The distance between the Cys437 and the heme ion is 4.0 Å in the L451P mutant as compare with 2.4 Å in the wild-type aromatase (Fig. 3B). Therefore, it would likely lead to a loss of aromatase function.
3.4. Functional characterization of aromatase mutants
Three-dimensional modeling predicted that the Y81C and L451P mutations would probably result in loss of aromatase function, and we hypothesized that these mutations would result in decreased enzyme activity of aromatase. To investigate this hypothesis, we used transiently transfected CHO cells with wild-type or mutated pMG-H2-Aro expression constructs or with empty vector. We then accessed the enzyme activity by the in-cell aromatase activity assay. Expression of aromatase protein was detected by western blotting analysis. Expression levels for the wild-type aromatase, Y81C and L451P mutants were similar (Fig. 4A). In-cell aromatase activity assay revealed greatly reduced activity in both aromatase mutants. The Y81C mutant was found to have 14.3% wild-type activity, whereas the L451P mutant was found to have 3.1% wild-type activity (Fig. 4B).
Fig. 4.
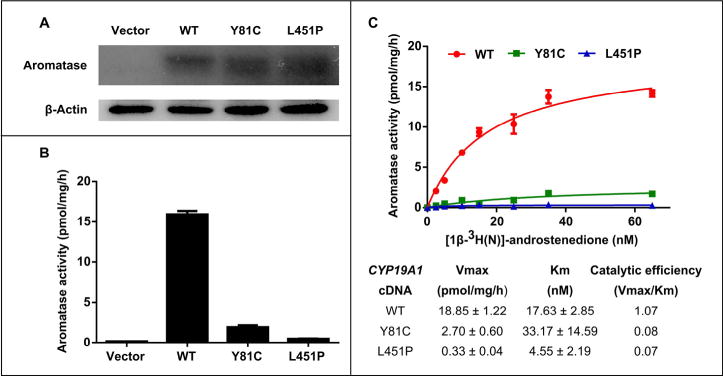
Functional characterization of aromatase mutants. (A) Expression levels for the wild-type aromatase, Y81C and L451P mutants are similar in transfected CHO whole-cell lysates on Western blotting (B) .B. In-cell aromatase activity assay shows greatly reduced activity in the aromatase mutants. The Y81C mutant has 14.3% wild-type activity, whereas the L451P mutant has 3.1% wild-type activity. (C). Aromatase kinetic analysis of the wild-type aromatase, Y81C and L451P mutants. The upper panel shows the Michaelis–Menton model of the kinetic analysis; the lower panel shows the calculated maximum reaction rate (Vmax), Michaelis constant (Km) and catalytic efficiency (Vmax/Km). The catalytic efficiencies are reduced by 92.5% and 93.5%, respectively, in the Y81C and L451P mutants as compare with the wild-type aromatase.
To further access the kinetic properties of the Y81C and L451P mutants compared with the wild-type aromatase, we performed the aromatase kinetic analysis. Based on the Michaelis–Menton model, the Y81C mutant showed lower Vmax, higher Km, and lower catalytic efficiency (Vmax/Km) as compared with wild-type aromatase, indicating decreased affinity to the substrate and reduced reaction rate (Fig. 4C). Interestingly, the L451P mutant had lower Vmax, Km and catalytic efficiency as compared with the wild-type aromatase, indicating that it probably binds to the substrate tighter but the reaction rate is slower (Fig. 4C). The catalytic efficiencies were reduced by 92.5% and 93.5%, respectively, in the Y81C and L451P mutants as compare with the wild-type aromatase.
3.5. Effects of estrogen replacement therapy
The patient received 25 μg transdermal estradiol (Estraderm TTS, Novartis) twice per week for 6 months. The serum estradiol level was increased to normal range, whereas the serum testosterone level decreased after 2–3 months of therapy. The serum levels of gonadotropins (FSH and LH) decreased (Table 1).
The bone mineral density was increased and the bone maturation was accelerated. At 6 months, the bone mineral density in the lumbar spine and the femoral neck increased by 11.4% and 7.3%, respectively, as compare with baseline. The Z-scores of the lumbar spine and the femoral neck at 6 months were 2.5 and 0.7 higher, respectively, than that at baseline; and the T-scores, 1.6 and 0.3 higher, respectively. His bone age reached to 18 years old at 3 months. Elevated bone turnover markers (alkaline phosphatase and osteocalcin) returned to normal range after 3 months. Serum calcium and phosphorous levels did not change significantly during treatment (Table 1).
There were improvements in the measures of both the lipid profile and liver function. At 3 months, the total cholesterol level was 14% lower than that at baseline; and the LDL cholesterol level, 16.4% lower. At 6 months, there were an increase of 20% of the HDL cholesterol level and a decrease of 31.3% of the triglyceride level, as compare with baseline. All measures of liver function were significantly decreased. The ALT level was 49.4% lower at 6 months than that at baseline; the AST level, 54.2% lower; and the GGT level, 32.4% lower (Table 1). Abdominal ultrasound confirmed that the patient had improved steatohepatitis at 6 months.
There were improvements in the measures of glucose levels but not insulin resistance. At 6 months, his hemoglobin A1c level was 6.1% (normal range, 4.5–6.3%) and his fasting glucose level was 4.5 mmol/l, with a reduction of 11.6% and 24.1%, respectively, as compared with baseline. The AUC for glucose was 13.1% lower at 3 months than that at baseline. However, all measures of insulin resistance were significantly increased, and measures of insulin sensitivity were decreased after the estradiol therapy. At 6 months, the fasting insulin level was 2.66 times greater than that at baseline; the AUC for insulin, 2.41 times greater; and the HOMA of insulin resistance, 2.09 times greater. With regard to insulin sensitivity, the QUICKI score at 6 months was 9% lower than that at baseline; and the Matsuda index score, 52.0% lower. Intriguingly, insulin secretion, as measured by the disposition index, was 2.38 times greater at 6 months than that at baseline (Table 1).
We evaluated the patient’s sex function and semen during treatment. He did not have sexual dysfunction. Testicular volumes did not change significantly. Although the sperm count and the sperm vitality at 6 months were 34.5% and 27.6% lower, respectively, than that at baseline, they remained in normal ranges (Table 1).
The patient complained of swollen breasts at 6 months. On physical examination, he had gynecomastia (Tanner stage II), but no breast discharge or tenderness was found. In addition, the levels of testosterone and LH fell significantly despite normal estradiol level, suggesting that the estrogen replacement may have been excessive. He started to receive conjugated estrogens (Premarin) 0.3 mg daily instead of transdermal estradiol after 6 months. The gynecomastia went away. Measures of sex steroids and gonadotropins were normal after 3 months of conjugated estrogens therapy (data not shown).
4. Discussion
In this report, we described a 24-year-old aromatase-deficient man treated with estrogen replacement. To our knowledge, this is the first report of a Chinese patient with aromatase deficiency. Our patient had clinical features of defects in the bone, lipid, liver, body fat and glucose metabolism. Hormonal measurements revealed normal testosterone level but undetectable estradiol level, indicating defects in the aromatase enzyme. We subsequently identified novel compound heterozygous CYP19A1 mutations (Y81C/L451P) in the patient. Both CYP19A1 missense mutations predicted disrupted protein structures, and decreased the activity of aromatase enzyme on the basis of in vitro expression. We also investigated the effects of estrogen replacement therapy in the patient on the bone, lipid, liver and glucose metabolism.
The heterozygous CYP19A1 mutations (Y81C/L451P) in our patient are novel, and moreover result in only partial loss of the enzymatic activity of aromatase. The Y81C mutation retains 14.3% wild-type activity, whereas the L451P mutation retains 3.1% wild-type activity. This is likely the explanation for the normal levels of testosterone and gonadotropins, the normal testicular weights, and the normal testis function including sperm count and vitality. On the other hand, despite residual aromatase activity our patient had severe hepatic steatosis, which is characteristic of the phenotype of the male ArKO mouse that is totally aromatase deficient (Jones et al., 2000; Nemoto et al., 2000). This suggests that there is differential responsiveness to aromatase insufficiency in different organs or cells. In men, estrogen is produced through the aromatization of androgen and mainly acts locally at the sites of synthesis as a paracrine or intracrine factor to maintain important tissue-specific function (Simpson et al., 2002). These sites include the mesenchymal cells of adipose tissue, osteoblasts and chondrocytes of bone, numerous sites in the brain, and the Leydig cells and germ cells of testes. Aromatase is highly expressed in the fetal liver but is undetectable in the adult liver in humans, and hence it is unlikely that localized estrogen synthesis occurs in adult liver (Simpson et al., 2002).
Maternal virilization during pregnancy is present in the majority of aromatase-deficient patients (Grumbach and Auchus, 1999), but not in our patient. Massive placenta estrogens are produced in pregnant women, especially during the third trimester of gestation. Androgens from fetal adrenal glands are a major source of placenta estrogens. Fetal aromatase deficiency prevents the placenta from converting androgens to estrogens, and results in excessive androgen levels, leading to ambiguous genitalia in female fetuses and virilization in pregnant mothers (Grumbach and Auchus, 1999). In samples of umbilical cord blood from aromatase-deficient fetuses, levels of androgens are elevated and levels of estrogens are decreased (Mullis et al., 1997; Shozu et al., 1991). The failure of the mother to virilize during pregnancy suggests either that the fetal adrenals have lower than normal androgen secretion or else enough estrogens are being produced by the placenta to counter the increase in androgens resulting from insufficient placental aromatase, or both of these. It seems that as little as 1% wild-type aromatase activity is enough to protect the mother from virilization (Grumbach and Auchus, 1999). No virilization of mother during pregnancy in our patient is probably due to some partial aromatase activity, which is consistent with quite a few reported cases (Bouchoucha et al., 2014; Ito et al., 1993).
Our analysis of structure and function highlighted critical aromatase residues. Aromatase, a heme-thiolate-containing enzyme, catalyzes the conversion of androgens to estrogens through a sequential three-step reaction (Simpson et al., 2002). A recently solved three-dimensional crystal structure of aromatase in complex with androstenedione reveals a relatively small active site (Ghosh et al., 2009). Our results of the three-dimensional structure modeling based on this reported structure indicate that the Y81C mutation probably disrupts helixes A and A′ and affects the entrance to the active site access channel, whereas the L451P mutation probably disrupts the active site and the substrate/product passage channel as well as impairs the heme binding region. Assessment on the basis of in vitro expression showed that both mutations significantly reduced the activity and catalytic efficiency of the aromatase enzyme. The Y81C mutant also had higher Km as compared with the wild-type aromatase, indicating decreased affinity to the substrate. Thus, our studies of disease-causing aromatase mutants with loss of function provide support for the roles of important regions and residues in the aromatase enzyme.
Our patient had multiple defects in the bone, such as continuing linear growth, genu valgum, delayed-fusion of epiphyses, osteopenia, and eunuchoid proportion, suggesting that estrogen is directly involved in the bone maturation, accrual of bone mineral density, and maintenance of the skeleton. The bone phenotype is similar to other aromatase-deficient men and ArKO male mice (Santen et al., 2009). Aromatase is widely expressed in bone, primarily in osteoblasts and chondrocytes (Oz et al., 2001), and thus aromatase deficiency affects bone metabolism. Estrogen replacement therapy in our patient accelerated the bone maturation and increased the bone mineral density, which confirmed the roles of aromatase and estrogen in the bone in men. The patient had genu valgum, similar to that of other aromatase-deficient men (Carani et al., 1997; Herrmann et al., 2002; Maffei et al., 2004, 2007; Morishima et al., 1995). The mechanisms of genu valgum in these patients are unknown, probably due to the unfused epiphyses in the lower extremities, decreased bone mineral density, and degenerative changes in the knee joints, all of which lead to alterations of mechanics. The abdominal obesity, which is common in such patients, might also increase the weight load on the knees and contribute to the skeletal malformation.
Our patient met all the International Diabetes Federation criteria for the diagnosis of metabolic syndrome (visceral obesity, hypertension, hyperglycemia, and dyslipidemia) (Alberti et al, 2005). Metabolic syndrome-like phenotype has been described in adult male and female patients with aromatase deficiency. As shown in Table 2, we summarized a total of eight reported adult patients, including seven men and one woman (Baykan et al., 2013; Carani et al., 1997; Gagliardi et al., 2014; Herrmann et al., 2002; Lanfranco et al., 2008; Maffei et al., 2004, 2007; Morishima et al., 1995). The mean age is 27.6 years (range, 24–32). At least seven patients (87.5%) are overweight or obese on the basis of the BMI; and at least five patients (62.5%) have central obesity as measured by the waist circumference. At least two patients (25%) have hypertension. Three patients (37.5%) have raised fasting glucose; and at least three patients (37.5%) have elevated hemoglobin A1c. Seven patients (87.5%) have insulin resistance as measured by the HOMA of insulin resistance; of these patients, six (85.7%) have increased fasting insulin. Six patients (75%) have raised total cholesterol and LDH cholesterol; six patients (75%) have reduced HDL cholesterol; and seven patients (87.5%) have increased triglycerides. At least five patients (62.5%) have elevated ALT and/or AST; and at least five patients (62.5%) have been confirmed to have liver steatosis. Therefore, metabolic syndrome probably develops in aromatase-deficient patients at the relatively young age of 20s or 30s.
Table 2.
Metabolic syndrome-like phenotype in adult male and female patients with aromatase deficiency
| Variable | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | IDF criteria for the metabolic syndrome | Normal range |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Male | Male | Male | Male | Male | Female | ||
| Age (years) | 24 | 31 | 27 | 29 | 25 | 26 | 27 | 32 | ||
| CYP19A1 mutation(s) | P.R375C | P.R365Q | c.IVS5-3C>A | P.E210K | P.M127R; P.R375H | c.312–334del; C.IVS9 + 1G>T | P.R375H | p.A306_S314dup | ||
| Body mass index (BMI) | 32.5 | 27.6 | 30.9 | 27.6 | 35.8 | 29.3 | 25.7 | <25 | ||
| Waist circumference (cm) | 124 | 122 | 121 | 102 | 131 | ≥ 94 (men); ≥ 80 (women)* | ||||
| Blood pressure (mmHg) | 158/72 | Normal | 125/80 | 125/75 | 130–145/80–100 | ≥ 130/ ≥ 85 | ||||
| Fasting glucose (mmol/l) | 3.9 | 4.9 | Normal | 10 | 5.6 | 4.1 | 5.4 | 6.1 | ≥ 5.6 | |
| Fasting insulin (μIU/ml) | 52 | 22 | 94 | 42 | 62.8 | 9.89 | 57 | 5–25 | ||
| HOMA for insulin resistance | 9 | 4.8 | 3.6 | 41.7 | 10.6 | 11.3 | 2.4 | 15.5 | <2.5 | |
| Hemoglobin A1c (%) | 7.4 | 8.3 | 5.5 | 5.6 | 6.7 | 4.5–6 | ||||
| Total cholesterol (mmol/l) | 6.2 | 7.9 | 4.6 | 4.6 | 5.6 | 5.6 | 6.4 | 5.9 | <5.2 | |
| LDL cholesterol (mmol/l) | 3.6 | 5.4 | 2.9 | 2.8 | 3.4 | 3.7 | 4.2 | 3.6 | <3.3 | |
| HDL cholesterol (mmol/l) | 0.93 | 1.11 | 0.54 | 0.8 | 0.82 | 0.98 | 1.04 | 1.4 | <1.03 (men); <1.29 | |
| Triglycerides (mmol/l) | 3.6 | 3.4 | 3 | 2.2 | 2.9 | 1 | 2.2 | 2 | (women) ≥ 1.7 | |
| Aminoleucine transferase (U/l) | Normal | 195 | 104 | 72 | 41 | <37 | ||||
| Aspartate aminotransferase (U/l) | 74 | 108 | 60 | 24 | 38 | <40 | ||||
| Liver steatosis | Yes | Yes | Yes | Yes | Yes | |||||
| Reference | Morishima et al., 1995 | Carani et al., 1997 | Herrmann et al., 2002 | Maffei et al., 2004 | Maffei et al., 2007 | Lanfranco et al., 2008 | Baykan et al., 2013 | Gagliardi et al., 2014 | Alberti et al., 2005 |
The waist circumference cutoff is for white people of European origin; IDF denotes International Diabetes Federation, HOMA homeostasis model assessment, LDL low-density lipoprotein, and HDL high-density lipoprotein.
Our observations of the effects of estrogen replacement in the patient demonstrate that estrogen plays important roles in lipid regulations, fatty acid homeostasis and glucose metabolism. The abnormal lipid profile, characterized by increased levels of total cholesterol, triglycerides and LDL cholesterol and decreased level of HDL cholesterol, was reversed by estrogen replacement. The liver steatosis in our patient was significantly improved after estrogen replacement. These observations are consistent with the reported aromatase-deficient adult men and the ArKO male mice (Jones et al., 2006). Liver steatosis due to aromatase deficiency is associated with impaired gene expression and enzyme activities of fatty acid β-oxidation as well as increased expression of fatty acid biosynthesis enzymes (Hewitt et al., 2004; Nemoto et al., 2000). Alterations of these enzymes are recovered by estradiol treatment in ArKO male mice (Hewitt et al., 2004; Toda et al., 2001).
Estrogen replacement therapy in our patient improved glucose levels, but failed to improve insulin resistance. The fasting insulin level, the AUC for insulin, and the HOMA of insulin resistance were significantly higher at 6 months than that at baseline, whereas measures of insulin sensitivity, the QUICKI and the Matsuda index were lower. It is consistent with two cases of aromatase-deficient men (Herrmann et al., 2002; Maffei et al., 2004). In contrast, estrogen replacement reduced basal serum insulin levels in two other aromatase-deficient men (Bilezikian et al., 1998; Carani et al., 1997). Paradoxically, despite decreased insulin sensitivity, his hemoglobin A1c and fasting glucose levels were returned to normal ranges at 6 months. We thus measured his insulin secretion, which was significantly increased in the patient as indicated by the disposition index. The relationship between sex steroids and insulin resistance in men is complicated. In our patient, he had normal testosterone level but undetectable estradiol level before he was treated. After estrogen replacement therapy, his estradiol level increased to normal range, but his testosterone level decreased significantly. These observations are consistent with a report that shows acute testosterone withdrawal reduces insulin sensitivity in men (Rubinow et al., 2012). Interestingly, the report also shows decreased insulin levels in the estradiol withdrawal group. Taken together, testosterone appears to play important roles in maintaining insulin sensitivity in men, whereas estrogen may enhance insulin secretion. However, the optimal dose and route of estrogen replacement therapy in such cases are still unclear due to the scarcity of the disease. Although it has been suggested a serum estradiol threshold around 18 pg/ml for bone health in men (Khosla, 2010), the optimal estrogen level for maintaining glucose homeostasis is unknown.
In conclusion, we have identified two novel CYP19A1 missense mutations in an adult man with aromatase deficiency. Estrogen replacement in the patient shows great impact on recovering the impairments in the bone, lipid, liver and glucose metabolism, but fails to improve insulin resistance. In addition to remarkably few other men with aromatase deficiency, our studies on this aromatase-deficient man furthers our understanding of the roles of aromatase and estrogen in men.
Supplementary Material
Acknowledgments
The study was supported by the Scientific Research Foundation for Young Investigators of Peking Union Medical College Hospital (Grant No. 2006118) and the National Institutes of Health (ES08258 to SC).
Abbreviations
- ALT
aminoleucine transferase
- ArKO
aromatase knockout
- AST
aspartate aminotransferase
- AUC
area under the curve
- β-CTX
c-telopeptide of type I collagen
- BLAST
Basic Local Alignment Search Tools
- BMI
body mass index
- CHO
Chinese hamster ovary
- DEXA
dual-energy X-ray absorptiometry
- dNTP
deoxynucleotide triphosphate
- FSH
follicle-stimulating hormone
- GGT
γ-glutamy transferase
- HDL
high density lipoprotein
- HOMA
homeostasis model assessment
- Km
Michaelis constant
- LDL
low density lipoprotein
- LH
luteinizing hormone
- NCBI
National Center for Biotechnology Information
- NHLBI-ESP
Exome Sequencing Project of the National Heart Lung and Blood Institute
- OGTT
oral glucose-tolerance test
- PolyPhen2
Polymorphism Phenotyping version 2
- QUICKI
quantitative insulin sensitivity check index
- SIFT
Sorting Intolerant From Tolerant
- Vmax
maximum reaction rate
Appendix: Supplementary material
Supplementary data to this article can be found online at doi:10.1016/j.mce.2014.09.016.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti KG, Zimmet P, Shaw J, IDF Epidemiology Task Force Consensus Group The metabolic syndrome – a new worldwide definition. Lancet. 2005;366:1059–1062. doi: 10.1016/S0140-6736(05)67402-8. [DOI] [PubMed] [Google Scholar]
- Baykan EK, Erdogan M, Ozen S, Darcan S, Saygili LF. Aromatase deficiency, a rare syndrome: case report. J Clin Res Pediatr Endocrinol. 2013;5:129–132. doi: 10.4274/Jcrpe.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilezikian JP, Morishima A, Bell J, Grumbach MM. Increased bone mass as a result of estrogen therapy in a man with aromatase deficiency. N Engl J Med. 1998;339:599–603. doi: 10.1056/NEJM199808273390905. [DOI] [PubMed] [Google Scholar]
- Bouchoucha N, Samara-Boustani D, Pandey AV, Bony-Trifunovic H, Hofer G, Aigrain Y, et al. Characterization of a novel CYP19A1 (aromatase) R192H mutation causing virilization of a 46,XX newborn, undervirilization of the 46,XY brother, but no virilization of the mother during pregnancies. Mol Cell Endocrinol. 2014;390:8–17. doi: 10.1016/j.mce.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, et al. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med. 1997;337:91–95. doi: 10.1056/NEJM199707103370204. [DOI] [PubMed] [Google Scholar]
- Chen SA, Besman MJ, Sparkes RS, Zollman S, Klisak I, Mohandas T, et al. Human aromatase: cDNA cloning, southern blot analysis, and assignment of the gene to chromosome 15. DNA. 1988;7:27–38. doi: 10.1089/dna.1988.7.27. [DOI] [PubMed] [Google Scholar]
- Deladoey J, Fluck C, Bex M, Yoshimura N, Harada N, Mullis PE. Aromatase deficiency caused by a novel P450arom gene mutation: impact of absent estrogen production on serum gonadotropin concentration in a boy. J Clin Endocrinol Metab. 1999;84:4050–4054. doi: 10.1210/jcem.84.11.6135. [DOI] [PubMed] [Google Scholar]
- Fisher CR, Graves KH, Parlow AF, Simpson ER. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci USA. 1998;95:6965–6970. doi: 10.1073/pnas.95.12.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi L, Scott HS, Feng J, Torpy DJ. A case of aromatase deficiency due to a novel CYP19A1 mutation. BMC Endocr Disord. 2014;14:16. doi: 10.1186/1472-6823-14-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennari L, Nuti R, Bilezikian JP. Aromatase activity and bone homeostasis in men. J Clin Endocrinol Metab. 2004;89:5898–5907. doi: 10.1210/jc.2004-1717. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Griswold J, Erman M, Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature. 2009;457:219–223. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Griswold J, Erman M, Pangborn W. X-ray structure of human aromatase reveals an androgen-specific active site. J Steroid Biochem Mol Biol. 2010;118:197–202. doi: 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumbach MM, Auchus RJ. Estrogen: consequences and implications of human mutations in synthesis and action. J Clin Endocrinol Metab. 1999;84:4677–4694. doi: 10.1210/jcem.84.12.6290. [DOI] [PubMed] [Google Scholar]
- Hauri-Hohl A, Meyer-Boni M, Lang-Muritano M, Hauri-Hohl M, Schoenle EJ, Biason-Lauber A. Aromatase deficiency due to a functional variant in the placenta promoter and a novel missense mutation in the CYP19A1 gene. Clin Endocrinol (Oxf) 2011 doi: 10.1111/j.1365-2265.2011.04012.x. [DOI] [PubMed] [Google Scholar]
- Herrmann BL, Saller B, Janssen OE, Gocke P, Bockisch A, Sperling H, et al. Impact of estrogen replacement therapy in a male with congenital aromatase deficiency caused by a novel mutation in the CYP19 gene. J Clin Endocrinol Metab. 2002;87:5476–5484. doi: 10.1210/jc.2002-020498. [DOI] [PubMed] [Google Scholar]
- Hewitt KN, Pratis K, Jones ME, Simpson ER. Estrogen replacement reverses the hepatic steatosis phenotype in the male aromatase knockout mouse. Endocrinology. 2004;145:1842–1848. doi: 10.1210/en.2003-1369. [DOI] [PubMed] [Google Scholar]
- Honda S, Harada N, Ito S, Takagi Y, Maeda S. Disruption of sexual behavior in male aromatase-deficient mice lacking exons 1 and 2 of the cyp19 gene. Biochem Biophys Res Commun. 1998;252:445–449. doi: 10.1006/bbrc.1998.9672. [DOI] [PubMed] [Google Scholar]
- Hong Y, Yu B, Sherman M, Yuan YC, Zhou D, Chen S. Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase. Mol Endocrinol. 2007;21:401–414. doi: 10.1210/me.2006-0281. [DOI] [PubMed] [Google Scholar]
- Ito Y, Fisher CR, Conte FA, Grumbach MM, Simpson ER. Molecular basis of aromatase deficiency in an adult female with sexual infantilism and polycystic ovaries. Proc. Natl Acad Sci USA. 1993;90:11673–11677. doi: 10.1073/pnas.90.24.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci USA. 2000;97:12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME, Boon WC, Proietto J, Simpson ER. Of mice and men: the evolving phenotype of aromatase deficiency. Trends Endocrinol Metab. 2006;17:55–64. doi: 10.1016/j.tem.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402–2410. doi: 10.1210/jcem.85.7.6661. [DOI] [PubMed] [Google Scholar]
- Khosla S. Update in male osteoporosis. J Clin Endocrinol Metab. 2010;95:3–10. doi: 10.1210/jc.2009-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lanfranco F, Zirilli L, Baldi M, Pignatti E, Corneli G, Ghigo E, et al. A novel mutation in the human aromatase gene: insights on the relationship among serum estradiol, longitudinal growth and bone mineral density in an adult man under estrogen replacement treatment. Bone. 2008;43:628–635. doi: 10.1016/j.bone.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, et al. Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J Clin Endocrinol Metab. 2004;89:61–70. doi: 10.1210/jc.2003-030313. [DOI] [PubMed] [Google Scholar]
- Maffei L, Rochira V, Zirilli L, Antunez P, Aranda C, Fabre B, et al. A novel compound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin Endocrinol (Oxf) 2007;67:218–224. doi: 10.1111/j.1365-2265.2007.02864.x. [DOI] [PubMed] [Google Scholar]
- Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80:3689–3698. doi: 10.1210/jcem.80.12.8530621. [DOI] [PubMed] [Google Scholar]
- Mullis PE, Yoshimura N, Kuhlmann B, Lippuner K, Jaeger P, Harada H. Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab. 1997;82:1739–1745. doi: 10.1210/jcem.82.6.3994. [DOI] [PubMed] [Google Scholar]
- Nemoto Y, Toda K, Ono M, Fujikawa-Adachi K, Saibara T, Onishi S, et al. Altered expression of fatty acid-metabolizing enzymes in aromatase-deficient mice. J Clin Invest. 2000;105:1819–1825. doi: 10.1172/JCI9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz OK, Millsaps R, Welch R, Birch J, Zerwekh JE. Expression of aromatase in the human growth plate. J Mol Endocrinol. 2001;27:249–253. doi: 10.1677/jme.0.0270249. [DOI] [PubMed] [Google Scholar]
- Quaynor SD, Stradtman EW, Jr, Kim HG, Shen Y, Chorich LP, Schreihofer DA, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor alpha variant. N Engl J Med. 2013;369:164–171. doi: 10.1056/NEJMoa1303611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- Robertson KM, O’Donnell L, Jones ME, Meachem SJ, Boon WC, Fisher CR, et al. Impairment of spermatogenesis in mice lacking a functional aromatase (cyp 19) gene. Proc Natl Acad Sci USA. 1999;96:7986–7991. doi: 10.1073/pnas.96.14.7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinow KB, Snyder CN, Amory JK, Hoofnagle AN, Page ST. Acute testosterone deprivation reduces insulin sensitivity in men. Clin Endocrinol (Oxf) 2012;76:281–288. doi: 10.1111/j.1365-2265.2011.04189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A. History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev. 2009;30:343–375. doi: 10.1210/er.2008-0016. [DOI] [PubMed] [Google Scholar]
- Shozu M, Akasofu K, Harada T, Kubota Y. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab. 1991;72:560–566. doi: 10.1210/jcem-72-3-560. [DOI] [PubMed] [Google Scholar]
- Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, et al. Aromatase – a brief overview. Annu Rev Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- Toda K, Takeda K, Akira S, Saibara T, Okada T, Onishi S, et al. Alternations in hepatic expression of fatty-acid metabolizing enzymes in ArKO mice and their reversal by the treatment with 17beta-estradiol or a peroxisome proliferator. J Steroid Biochem Mol Biol. 2001;79:11–17. doi: 10.1016/s0960-0760(01)00135-2. [DOI] [PubMed] [Google Scholar]
- Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- Zhou DJ, Pompon D, Chen SA. Structure-function studies of human aromatase by site-directed mutagenesis: kinetic properties of mutants Pro-308—Phe, Tyr-361—Phe, Tyr-361—Leu, and Phe-406—Arg. Proc Natl Acad Sci USA. 1991;88:410–414. doi: 10.1073/pnas.88.2.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirilli L, Rochira V, Diazzi C, Caffagni G, Carani C. Human models of aromatase deficiency. J Steroid Biochem Mol Biol. 2008;109:212–218. doi: 10.1016/j.jsbmb.2008.03.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.