

Summary
Unbiased discovery approaches have the potential to uncover neurobiological insights into CNS disease and lead to the development of therapies. Here we review lessons learned from imaging-based screening approaches and recent advances in these areas, including powerful new computational tools to synthesize complex data into more useful knowledge that can reliably guide future research and development.
Keywords: discovery, screening approaches, novel insights, computation, systems, big data, complexity, therapeutics
Introduction
As integrative biology (Blow, 2009) reshapes paradigms in cell biology, it is increasingly clear that many of the phenotypes we seek to measure in isolation are highly connected to each other (Collinet et al., 2010). In fact, cells and their phenotypes exhibit many features that qualify them as complex systems: phenotypes are often emergent properties of dynamic signaling pathways with nested feedback loops, non-linear signaling relationships, and the capacity to undergo adaptive changes.
Complex systems are challenging to understand using standard hypothesis-driven experimental approaches that aim to manipulate one variable at a time. Holding every other variable constant in complex biological systems may be impossible or require artificial measures that confound results. Certain biases are unavoidable, and the investigator's knowledge and conceptual framework limit the pace of discovery. Indeed, the instincts scientists have developed by studying well-defined simple biological systems may mislead them as much as guide them when applied to complex systems. Breakthroughs and paradigm shifts are infrequent and often result from serendipity rather than intention because the hypotheses that drive experimental design evolve slowly from past results.
Discovery or “hypothesis-free” approaches are an important alternative. Whereas hypothesis-driven research is linear, discovery approaches are massively parallel. Since the “system”—the cell in this case—tells the investigator which perturbation is relevant, discoveries can be unexpected and less biased than findings from hypothesis-driven approaches. One of the most common applications is imaging-based phenotypic screens in cells. Cell-based screens have provided novel biological insights into the genes that control cell morphology (Jones et al., 2009), chromosome segregation and structure (Neumann et al., 2006; Walter et al., 2010), cell division, migration and survival (Neumann et al., 2010), susceptibility to infection (Cronin et al., 2009), and regulators of the protein clearance pathway autophagy (Orvedahl et al., 2011). In neuroscience, cell-based screens (Al-Ali et al., 2013) have been used effectively to investigate regenerative approaches to multiple sclerosis (Deshmukh et al., 2013) and synaptogenesis (Sharma et al., 2013; Shi et al., 2011).
Commensurate analysis tools must be applied that treat cells as defined but complex systems (Freddolino and Tavazoie, 2012; Karr et al., 2012). Fortunately, large-scale computational facilities are changing the nature of data analysis. They have increased the ability to access and search data, improved visualization techniques and technologies, enabled the application of powerful statistical techniques to large complex data sets, and made it possible to apply previously computationally untenable machine learning (ML) techniques to build predictive models of complex biological systems.
In the sections that follow, we will review some of the lessons learned from past efforts with cell-based screens, some important considerations for those pursuing these approaches now and for the future, including the challenges and opportunities created by the massive amounts of data that these screens can generate. Our focus is imaging and cell-based screens applied to neurobiology, though the concepts and approaches described here are widely relevant. We will look at the methods for acquiring images, how images are analyzed, the value of cloud computing and machine learning, and the implications of all of this for the future of biology and medicine.
Model Systems
There are several key components of any screen, and the first is the cells to be examined (Figure 1). This choice is critical and should be driven by the biological question rather than expedience. The basic choice is between immortalized cells and primary cells, and more recently, cells derived from induced pluripotent stem cells (iPSCs).
Figure 1. Generic flow scheme for cell-based high throughput screening.

The basic stages of target discovery and selection are outlined
Ultimately, a compromise between feasibility and biological relevance may be needed to conduct a screen. The trade-offs—what can and can't be learned from the in vitro system and the endpoints examined—need to be understood clearly before starting. For the purposes of this discussion, biological relevance is the extent to which lessons from simple systems that are feasible to use for screening hold true for the more complex systems that they are meant to model. In this regard, validating models for use as screening platforms can be complex. If the screen is focused on an aspect of biology observed in vivo, the emphasis for validation and ultimately model selection must be based on the ability of the in vitro model to replicate the critical in vivo biology. For screens focused on discovering treatments of a disease for which no effective therapies currently exist, the options for true validation are limited. Investigators must generally select a model based on a degree of face validity until an effective therapy is found, which can then be used to help validate and invalidate models. To be clear, no in vitro system will display the complexity of an intact organism and not all biological insights will translate from in vitro to in vivo model systems or ultimately to human patients.
New Options with Induced Pluripotent Stem Cells
The Nobel prize–winning discovery of cellular reprogramming by Yamanaka and colleagues (Takahashi and Yamanaka, 2006), which led to the production of human iPSCs, offers new possibilities for disease models (Takahashi et al., 2007; Yu et al., 2007). Primary cells can be collected from people and reprogrammed into a stem or precursor cell that can be expanded and passaged (Churko et al., 2013; Hayes and Zavazava, 2013; Warren et al., 2010). In turn, iPSCs can be differentiated into cell types relevant to the disease, including subtypes of neurons and glia.
Protocols to make different brain cell types are being rapidly developed and improved. Many protocols involve the delivery of critical instructive factors to cells in culture at specific times and in a particular order to recapitulate key steps in development (Kim et al., 2014). For example, efficient protocols have been developed to make neural crest by dual-SMAD inhibition/WNT activation (Chambers et al., 2013). Protocols have been reported for making many brain cell types from stem cells, including dopaminergic neurons (Studer, 2012; Sun et al., 2013; Sundberg et al., 2013), motor neurons (Bilican et al., 2012; Boulting et al., 2011; Di Giorgio et al., 2007), forebrain-like neurons (HD iPSC Consortium et al., 2012), striatal neurons (Aubry et al., 2008), cortical interneurons (Maroof et al., 2013), retinal cells (Jin and Takahashi, 2012); oligodendrocytes (Czepiel et al., 2011; Ogawa et al., 2011; Wang et al., 2013; Yang et al., 2013) and astrocytes (Emdad et al., 2012; Serio et al., 2013). Neurons and neural progenitors can be produced directly from other types of somatic cells without having to first make those cells pluripotent (Ambasudhan et al., 2011; Kim et al., 2011; Vierbuchen et al., 2010).
The application of iPSCs to studying disease has generated the most excitement (Eglen and Reisine, 2011). For the first time, a skin or blood cell from a patient with a neurological or psychiatric disease can be reprogrammed to become a cell type of the nervous system, thereby creating a genetically faithful human model of disease (Churko et al., 2013; Hayes and Zavazava, 2013; Wray et al., 2012). Already, several models have been developed that exhibit disease-relevant phenotypes (Table 1) for Huntington's disease (HD) (HD iPSC Consortium et al., 2012; Zhang et al., 2010), amyotrophic lateral sclerosis (ALS) (Barmada et al.; Bilican et al., 2012; Burkhardt et al., 2013; Donnelly et al., 2013; Egawa et al., 2012; Sareen et al., 2013; Serio et al., 2013), spinal muscular atrophy (Ebert et al., 2009), Parkinson's disease (Cooper et al., 2012; Skibinski et al., 2014), schizophrenia (Brennand et al., 2011), and Alzheimer's disease (AD)(Israel et al., 2012). In principle, genetic and small-molecule screens can be conducted in what might be the most physiologically relevant cell-based model of neurological disease ever developed.
Table 1. Examples of Phenotypes Detected in Patient-Derived iPSCs.
| Disease | Phenotype (s) | Reference |
|---|---|---|
|
| ||
| Huntington's disease |
|
(HD iPSC Consortium et al., 2012; Zhang et al., 2010) |
| Amyotrophic lateral sclerosis |
|
(Barmada et al., 2014; Bilican et al., 2012; Burkhardt et al., 2013; Donnelly et al., 2013; Egawa et al., 2012; Sareen et al., 2013; Serio et al., 2013; Wen et al., 2014b) |
| Spinal muscular atrophy |
|
(Ebert et al., 2009) |
| Parkinson's disease |
|
(Cooper et al., 2012; Sanders et al., 2014; Skibinski et al., 2014) |
| Schizophrenia |
|
(Brennand et al., 2011; Wen et al., 2014a) |
| Alzheimer's disease |
|
(Choi et al., 2014; Israel et al., 2012) |
iPSCs might also help to solve one of the most vexing problems in drug development. Non-human models of neurological disease have a poor track record for predicting results of putative therapies in clinical trials (McGonigle, 2014; McGonigle and Ruggeri, 2014), including HD (Crook and Housman, 2011), ALS (Perrin, 2014), and AD (Mullane and Williams, 2013). Nearly all the compounds that were tested in human clinical trials and failed to show efficacy were supported by data showing that the drugs were effective in mice. There were two exceptions: tetrabenazine, a symptomatic therapy for HD, showed efficacy in mice, and Riluzole had modest effects in a mouse model of ALS and extended the lives of ALS patients by a few months on average. Some discrepancy can be blamed on the design and execution of preclinical efficacy trials in mice (Perrin, 2014). But worrisome data suggest that more fundamental biological differences between mice and humans may be important. Humans and mice diverged in evolution over 65 million years ago, and many publications show that results from mice fail to reliably predict results from humans in drug absorption, distribution, metabolism, elimination, toxicity, bioavailability, carcinogenicity, teratogenicity, and efficacy as well as disease pathophysiology. Known differences in pharmacodynamics (Richert et al., 2008; Xie et al., 2000) and toxicology (Carlson et al., 2009; Singh and Gupta, 1985) between humans and non-human models could affect drug safety. Differences exist in physiological responses and drug effects in human cells (e.g., neurons), compared to murine or other non-human counterparts (Berger et al., 2006; Castan et al., 1994; Curtis et al., 1997; Derian et al., 1995; Guo et al., 1989; Keshavaprasad et al., 2005; Kopin et al., 1997; Liang et al., 2010; Mattson et al., 1991; Okazaki et al., 1995; Penhoat et al., 1996; Rasakham and Liu-Chen, 2011). Murine and human cells respond differently to neuropeptides, hormones, and drugs, including anesthetics, antiarrhythmics, and ligands for G-protein-coupled and nuclear receptors (Castan et al., 1994; Derian et al., 1995; Guo et al., 1989; Keshavaprasad et al., 2005; Kopin et al., 1997; Liang et al., 2010; Okazaki et al., 1995; Penhoat et al., 1996; Rasakham and Liu-Chen, 2011). For the neurokinin NK1 (Pradier et al., 1995) and the brain cholecystokinin CCK-B (Kopin et al., 1997) receptors, two key drug targets, differences in just one or two amino acids in the orthologous receptors resulted in major differences in the efficacy of ligands for human and murine cells. Thus, an advantage of iPSCs is that both on-target and off-target pharmacology of compounds can be assessed in the context of a fully human system.
That a number of disease-relevant phenotypes have been observed after only weeks or months in culture may be surprising: the symptoms of many of these diseases are not normally evident for decades. However, the symptoms might be misleading. Redundancy and coping mechanisms in the nervous system are extensive and may obscure deficits until substantial cell loss has occurred and coping mechanisms have been exhausted. Indeed, abnormalities at the cellular level may begin much earlier, particularly in the case of genetic causes of these diseases, and may be robustly detected with new and more sensitive instrumentation described below (Clarke et al., 2000; Finkbeiner, 2011). Also the in vitro environment may be less supportive than that in vivo, which may help to uncover differences in the vulnerability of cells differentiated from patient-derived iPSCs and controls.
Yet iPSCs are no universal panacea. More needs to be understood about environmental causes of sporadic disease. If environmental influences are mediated by epigenetic mechanisms and those marks are erased during reprogramming, the utility of the cells for modeling may be limited until it becomes possible to reintroduce those epigenetic marks in the laboratory. Even for genetic causes of disease, working with iPSCs has its own set of challenges. iPSCs reprogrammed from the same parent fibroblast exhibit variability. Some of this variability likely reflects somatic mosaicism in the human tissue sample from which the iPSCs are derived and the clonal nature of iPSC generation (Abyzov et al., 2012). Variability due to random integration of reprogramming factors should be less of an issue with newer reprogramming methods that leave no genetic footprint (Soldner et al., 2009). Though it is possible to differentiate iPSCs into different nervous system cell types, the protocols vary significantly in their efficiency, and some cell types remain difficult or nearly impossible to make. The differentiation protocols require weeks to months to execute, and the time and cost are considerable.
Still, the investment in this field and its trajectory are astonishing. We expect essentially every aspect of the utility of these cells to continue to improve rapidly. The ambitious vision of creating platforms of iPSCs with the requisite genetic diversity to represent significant human populations to enable patient stratification and “clinical trials in a dish” and, eventually, personalized medicine approaches seems fanciful, but it is difficult at this time to see insurmountable obstacles to that goal.
Cell-Based Genetic Screens
Cell-based screens for genetic modifiers have the potential to uncover therapeutic targets that can be moved forward to clinical applications, even before the underlying mechanisms are fully understood (Figure 1). This is critical because complex diseases might otherwise require decades of hypothesis-driven research to produce a sufficient biological understanding to develop a rational intervention. The main cell-based approaches to identify modifiers of biological or pathophysiological processes involve the use of RNA interference (RNAi), cDNA expression, CRISPR/Cas9-mediated gene down or upregulation, or chemigenomics. Chemigenomics is an approach that involves the use of libraries of molecules whose specificity for their target is so well established that it is straightforward to relate a hit from a screen to the biological pathway that mediates the effects.
RNAi screens have been widely used in models of diseases with features that can be assayed in vitro using surrogate endpoints. Surrogate endpoints are phenotypes that the investigator believes are directly related to a pathway or pathogenic process relevant to the disease that the system is designed to model (the reader is referred to several reviews (Falschlehner et al., 2010; Kassner, 2008; Mohr et al., 2010)). Nevertheless, RNAi screens are challenging (Echeverri et al., 2006; Echeverri and Perrimon, 2006; Kaelin, 2012) and have led to reports whose findings are not reproducible or robust enough to pursue therapeutically (Bhinder and Djaballah, 2013; Prinz et al., 2011). Indeed, overlap between siRNA screening results is surprising low, often less than 10% (Neumann et al., 2010).
Each RNAi trigger has varying potency and efficacy against its intended target and may have unwanted effects against numerous other genes. Minimally, transfection conditions should be determined based on efficient knockdown of known pathway genes by a panel of siRNA triggers, resulting in a phenotypic effect and lack of phenotypic effect with a panel of non-targeting siRNAs. Different conditions may be optimal for different phenotypes even within the same basic pathway. Unfortunately, increasing the dose of siRNA often increases knockdown of the genes of interest but reduces specificity. Ideally, siRNA dose should be optimized based on a phenotypic endpoint, preferably the endpoint to be screened. In a high-content image-based screen involving multiple endpoints, a compromise in the siRNA dose may be needed to optimize sensitivity and specificity in as many individual endpoints as possible. Alternatively, one or two primary endpoints might need to be selected for optimizing the assay conditions.
The most common format for siRNA screens in adherent cells remains plating cells in microtiter wells of plastic or glass high-density tissue culture plates. Efforts are underway to identify biomaterials that more closely simulate the in vivo niche in which the cell is found, direct the fate of stem cells, or even pattern tissue in 2D or 3D (Lutolf et al., 2009). To facilitate cell-based RNAi screens (Conrad et al., 2004; Neumann et al., 2006), high-density cellular microarrays have been developed. Arrays are created that contain dried spots with the nucleic acids to be introduced into the cell (e.g., individual members of an RNAi library) along with the transfection reagent to mediate their entry. Cells are plated on top of the microarray, and transfection occurs from below. Since the transfection reagent necessary for a genome-wide siRNA screen can cost as much as the siRNA library, a smaller spot size can be a considerable cost savings. siRNA screens can also be improved through the use of aliquots of frozen cells that are thawed and plated on top of the spotted siRNA/transfection reagent (Swearingen et al., 2010). This removes variability introduced during tissue culture and improves workflow on screening days. Both of these approaches may work better with cell lines than with primary cells, so successful application may depend on the cell type.
The CRISPR-Cas9 system now allows either gene knockout or knockdown/overexpression in cells and organisms (Gilbert et al., 2013; Zhang et al., 2014), depending on whether a nuclease, transcriptional repressor or activator is recruited to a particular genetic locus. These tools are already being applied to large-scale genetic screens (Shalem et al., 2014; Wang et al., 2014), and some of the early results suggest that these approaches may have a lower rate of off-target effects than conventional siRNA. This may prove to be the method of choice in the future. However, most of the available libraries are pooled and would need to be arrayed for use in adherent cells, such as neurons. In addition, gene knockout and knockdown/overexpression screening strategies will likely offer complementary information because knockout of essential genes may be toxic and knockdown of other genes may be inadequate to produce phenotypic changes. With time, the strengths and limitations of these systems will be better understood, and that knowledge can be incorporated into the design of libraries as well as statistical methods for data analysis and interpretation.
With small-molecule (SM) screens, the focus is typically on an individual target, and the assay readout is as proximal to the target as possible. Assaying calcium flux in cells for inhibitors of a calcium channel or substrate phosphorylation for inhibitors of kinase function would be examples. With siRNA screening, the focus is usually on a phenotype or pathway rather than a single target. Most compounds in a SM library are inert in any given assay. In contrast, because all siRNA will have some effect on the cells (largely because of the off-target effects already discussed), distribution of responses is often much broader in a siRNA screen than a SM screen (see also (Birmingham et al., 2009)).
The screens are the easiest and least time-consuming aspect of target discovery and validation, often taking just 4–8 weeks. However, assay development may take 6 months, and confirmations and validation of hits may take a full year. A plan for prosecution of hits after a screen is critical. It should include a flowchart with specific assays and explicit performance criteria to eliminate genes unfit for further study and to ensure commitment to those that are. These criteria may include additional in vitro cell systems measuring the same phenotype, in vitro systems designed to monitor related pathways to avoid, in vivo models of disease, gene expression from normal and diseased human tissues of interest, toxicity liability assessment, and an assessment of the druggability to name a few. Druggability is an assessment of the likelihood of successfully modulating the function of the protein encoded by the gene with a SM. In general, druggable proteins contain structural pockets that can function as receptors for SM ligands, and are typified by classes of proteins, including G-protein-coupled receptors, nuclear hormone receptors, and enzymes, such as proteases and kinases.
Acquiring Images
Most instruments for acquiring images in high-throughput/high-content screening (HT/HCS) are microscope based and designed to capture epifluorescence (Glory and Murphy, 2007). Newer EM-CCD and CMOS cameras complement available CCD technology and expand the options for sensitivity, chip size, throughput and cost. Newer indirect methods of automated focusing are faster, reduce the overall amount of illumination that the cells receive, thereby reducing phototoxicity, and can yield more robust results than image-based approaches.
Labeling technologies have expanded significantly. Quantum dots (Osakada and Cui, 2011; Zhang et al., 2011) are bright fluorophores with narrow excitation and emission spectra, ideal for multiplexing. New cell-permeable SM dyes expand the dynamic physiology that can be measured (Vendrell et al., 2010). Technologies, such as RNA and DNA aptamers, are being developed as fluorescent biosensors with the potential to be tailored to detect nearly any macromolecule (Paige et al., 2012; Tainaka et al., 2010). Genetically encoded fluorescent proteins remain a mainstay because they are non-invasive, can be targeted to subcompartments or expressed selectively in subtypes of cells, and can be engineered to report an array of complex biological phenomena, such as Ca2+ binding, protein-protein interactions, and transition metals in cells (Palmer et al., 2011; Shekhawat and Ghosh, 2011; Vinkenborg et al., 2010). The number and properties of fluorescent proteins continue to increase. Those that are photoconvertible are especially exciting because they offer the potential of developing whole new types of innovative assays, including the measurement of flux of specific proteins and pathways at a single-cell level (Barmada et al., 2014; Chudakov et al., 2007; Chudakov et al., 2010; Hamer et al., 2010; Tsvetkov et al., 2013b). Light-induced stimulations can be incorporated into screens (Levskaya et al., 2009). For applications where the ∼26-kD size of most fluorescent proteins is problematic, polypeptide labeling strategies are available (Choulier and Enander, 2010; Morris, 2010).
HT cell-based screens have been performed most commonly on fixed cells. Typically, a period of time is allowed to elapse after a genetic or SM perturbation, the cells are fixed and immunostained. An automated microscope is instructed to collect images of the fixed cells at the appropriate wavelength(s), and the images are typically analyzed with simple computer programs written in scripting language. The methods for doing these screens are well developed, and data can be acquired at a very high rate. However, there are limitations. In the case of RNAi screens, the appearance of phenotypes depends on sufficient suppression of a gene or protein at the time the assay is performed. If the images are acquired too early, the phenotype may not be evident, resulting in a false negative. If the images are acquired too late, the complexity of the phenotype can increase due to secondary effects of the knockdown of the target. Analogous issues arise with SM screening. To help overcome this issue, live-cell screens are increasingly popular (Neumann et al., 2006; Neumann et al., 2010). Besides capturing information that might be missed altogether with snapshot approaches, live-cell imaging data make it possible to measure critical kinetic features of biological processes, including rapid responses to stimuli.
A major limitation of essentially all commercially available HT/HCS systems is that they are not well equipped to handle cellular heterogeneity. Most are designed to capture snapshots of cell populations and compute aggregate measures from the population. The approach is understandable, given the heavy past reliance on immortalized cell lines, which are assumed to be homogeneous, and historical limitations on the throughput. Under these conditions, cell-to-cell and well-to-well variability are assumed to represent technical rather than biological variation. But as the field increasingly moves to more physiologically relevant systems, such as primary culture or differentiated iPSCs (see above), aggregate population-based measures become more problematic. These cultures are very heterogeneous, varying in maturity, subtype and even whether they are post-mitotic. Averaging measures from heterogeneous cell populations produces a meaningless result, and the inherent variability means the sensitivity of the assay is low.
Recently, a form of automated imaging called robotic microscopy (RM) was invented to overcome these problems (Arrasate and Finkbeiner, 2005). It was originally designed as a hypothesis-testing tool to resolve pathogenic mechanisms, incidental changes, and coping responses (Arrasate et al., 2004) (Figure 2). But its resolving power, particularly with heterogeneous cell systems, led to its application to HT/HCS (Daub et al., 2009; Sharma et al., 2012) (Figure 3). Its fundamental capability is to acquire single-cell data longitudinally, indefinitely and in an HT manner, allowing the investigator to acquire multivariate data from each cell during its lifetime and then relate that information to that cell's fate.
Figure 2. The power of high-throughput longitudinal single-cell analysis.
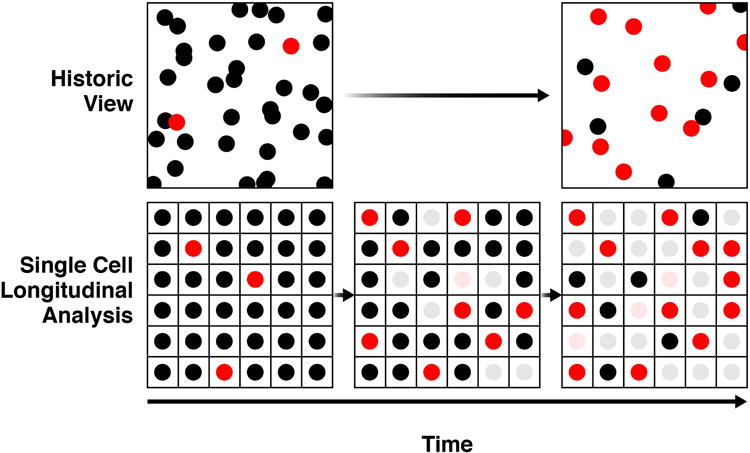
Conventionally, cell-based screening has compared images of two unrelated cell populations (top row). Typically, these populations differ due to the passage of time or to a genetic or pharmacological manipulation of the cells. Assays often involve a measure of cell number (each circle) and a specific biological change/response (shown here as a conversion from a black circle to a red one). With this approach, it is impossible to determine the precise relationship between the observed biological change/response and cell number, and variations in the number of cells present or that exhibit a change or response under nominally identical conditions necessarily must be treated as technical variation, which reduces the overall sensitivity of the system. Longitudinal single-cell analysis overcomes these limitations because each cell is followed over time, effectively allowing it to serve as its own control (bottom row). Whether a biological change or response is associated with a change in cell number can be measured precisely. Indeed, for assays in which the change or response is graded, cell-to-cell variation can be harnessed to uncover exquisitely resolved dose-response relationships. The biological significance of transient or rare changes or responses, which might be highly significant but dismissed with conventional approaches because of their nature, can be elucidated because they can be captured and quantitatively linked to some future fate. For these reasons, the single-cell analysis is 100–1000-fold more sensitive than conventional approaches for measuring effects of perturbations on neuronal survival (Arrasate and Finkbeiner, 2005; Sharma et al., 2012), is well suited to resolve complex pathogenic and coping responses, and is well-suited to enable high-throughput screening of rare, precious or heterogeneous cell populations (e.g., disease models based on patient-derived differentiated human iPSCs).
Figure 3. Analysis of longitudinal single-cell data from screens in primary neurons.
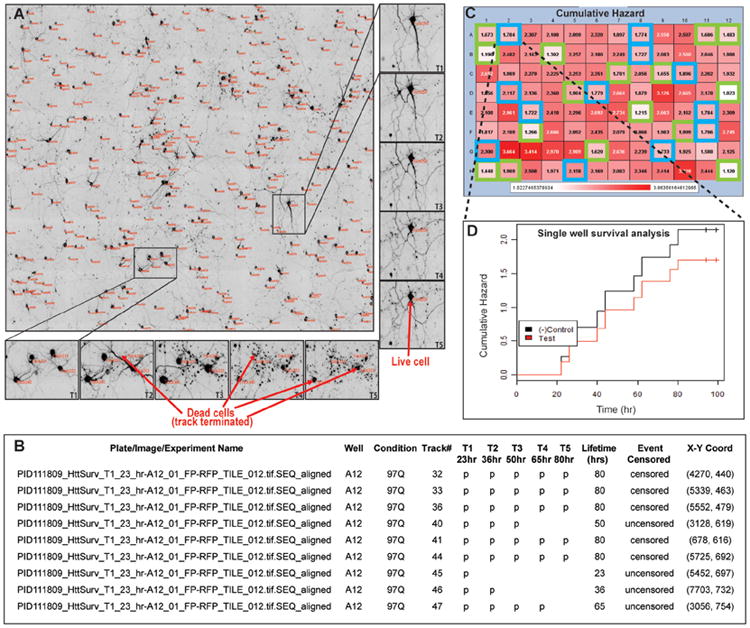
A. Shows a typical montage of microscope fields at the first-time scan after image segmentation. Unique cell-track numbers are shown in red next to each cell. Two regions within this montage have been zoomed in to show cell lifetime detection from the entire time-lapse imaging data set to illustrate the ability to do longitudinal single-cell analysis. B. A typical delimited text file showing the arrangement of data output and feature extraction by the analysis program. C. A representative heat map of a control plate in which a known modifier, the trophic factor brain-derived neurotrophic factor (blue outlined wells) was spotted at different doses throughout the plate in the midst of positive control wells (green outlined wells) and analyzed in a blinded fashion. White shaded wells indicate longer survival and dark red-shaded wells indicate reduced survival. The numbers represent the cumulative death in each well at the end of the experiment. This survival assay had a positive predictive value of 91% and a negative predictive value of 97%, underscoring the highly sensitive and specific nature of robotic microscopy. D. A survival curve from a modifier well showing decreased hazard (increased survival), compared to the negative control wells. Adapted with permission from (Sharma et al., 2012).
The technology has evolved over successive generations. The first generation instrument had the core capability to register the position of a microtiter plate, which is essential to repeatedly find the same individual cells (Arrasate and Finkbeiner, 2005). Image acquisition was automated but delivering plates to the microscope was manual, and the automated focusing was image-based making its performance slow and its accuracy variable. The second generation RM incorporated newer indirect autofocusing methods that were faster, more accurate, and worked on microtiter plates of any well density (Sharma et al., 2012). Importantly, software programs were developed to automatically organize the images that were collected, stitch them together into montages, organize the montages temporally into single files and find and track individual cells from the same microscope field over time. The 3rd generation system was designed to perform fully automated image acquisition 24/7 without human intervention (Video 1). Plates are stored in an automated incubator that contains a robot that delivers the plate to a nest at specific time points prescribed by the software that controls the system. A robotic arm transfers the plate from the nest to the microscope stage, and the automated image acquisition program begins once the plate is in place. A dedicated robotic microscope can capture 72,000 images per day, but usually, investigators capture 9–16 images per well, placing the upper limit of 4,500–8,000 wells per day for assays that require a single image. Because many of the analytical tools for quantifying effects from single-cell analysis are similar to those used for preclinical or clinical trials, the approach to power analysis is similar. Depending on the effect size the investigator wants to detect, the variability of the assay and the number of cells per microscope field, it is possible to calculate the necessary wells and even number of microscope fields the investigator should collect per perturbagen. Most assays are high content and involve capturing images of 2–4 reporter genes at different fluorescence wavelengths. Once an imaging run is finished, the robotic arm transfers the plate back to the incubator. A new 4th generation system has just come on line and achieves higher throughput and content by incorporating fast dual CMOS cameras with a beam splitter and an Andor C1 spinning disc confocal to achieve better spatial resolution and to make create 4D movies. In addition, we are integrating new single-cell transcriptomic technology to investigate the cell-specific phenotypes we observe in greater depth (Actis et al., 2014).
As a practical matter, multiple screens for different collaborators are run simultaneously, and so each of these screens is assigned a fraction of the overall throughput. It is important to point out that, although the throughput of these systems is significant, throughput is not their distinguishing feature. Higher throughput systems exist. The distinguishing feature is the longitudinal single-cell nature of the measurements, which confers a substantial increase in sensitivity and expands the types of biology assays that can be performed. If these features are not critical, then conventional population-based HTS methods will likely be cheaper and faster. Although the technology was invented in the Finkbeiner laboratory, the essential capability to perform longitudinal single-cell analysis has since been established in other academic laboratories (Nagai et al., 2007; Wen et al., 2014).
Image Analysis and Visualization
Analysis of cell-based imaging results from SM or genetic screens involves two steps: 1) extraction of features from the raw images and 2) analysis of those measurements to identify any significant patterns. Currently, image analysis begins by measuring a discrete and narrowly defined phenotype from an image (e.g., neurite length, cell survival, mitochondrial morphology). Analysis programs are written to detect some pattern and/or intensity of the pixels in the image that correspond to a feature of interest and measure it as accurately as possible.
Unfortunately, the number and complexity of features that can be extracted are severely limited. A trained researcher can identify many more features than can be extracted automatically. However, manual annotation is not an option, given the large volumes of data produced. This situation is changing with more computational power. Along with better performance of standard pixel level feature identification, more powerful machine perception software and massive machine resources can now be applied.
Automating feature extraction is a great step forward but also makes analyzing the data much harder just by the simple mass of data that can be rapidly accumulated. To find meaningful correlations and patterns in highly dimensional feature spaces requires complex algorithms and a lot of computational power. Fast and effective visualization tools can help, but as the number of dimensions grows, visualization is not enough. More use of better statistical analysis algorithms that automatically search for interesting patterns in the data are crucial. Also increasingly powerful machine learning techniques can generate models and use them to both make predictions and direct experimentation to test these predictions, thereby improving the models iteratively.
Analysis of longitudinal single-cell data utilizes a suite of statistical tools, called survival analysis, which is used commonly for clinical trials and in time-to-failure studies in engineering (Dekker et al., 2008). Survival analysis and the closely related Cox proportional hazards analysis (Wolfe and Strawderman, 1996) allow predictive statistical models of fate to be built from the variables that are tracked during the experiment. These approaches can be adapted further into Bayesian hierarchical analysis models, which avoid some of the assumptions of frequentist approaches and do a better job handling measurement errors (Miller et al., 2011; Miller et al., 2010). The approach can answer questions that elude conventional approaches (i.e., What is the biological significance for fate of a change I see during the cell's lifetime?). For example, when applied to the study of mechanisms of neurodegenerative disease, it can distinguish thorny and highly interrelated pathogenic processes from coping ones and assign an importance to them based on the quantitative measure of the fate those processes predict.
Single-cell analysis overcomes many of the inherent problems of the population-based approaches described above. Because each cell is followed over time, phenotypes that emerge at any time over the course of the experiment are captured. Even stochastic and informative rare events can be captured at a sufficient level to draw clear conclusions (Barmada et al., 2010). The cell-to-cell variability that reduces the sensitivity of population-based approaches can be harnessed with RM to add additional power to the analysis. For example, the dose of RNAi or other perturbant each cell receives varies from cell to cell, but it can be estimated from the fluorescence of a marker that is co-introduced (Arrasate and Finkbeiner, 2005). Then, after the experiment is done, and the fate of the cell is measured, and the relationship between the dose a cell received and the fate it experienced is quantified.
The abilities of the system to connect the extent of a perturbation to the response on a single-cell level over thousands of cells, to effectively allow each cell to serve as its own control, and to avoid confounds of cell death and cell division make it about 100–1000 times more sensitive for measuring the effects of perturbagens on neuronal survival than commercially available systems that depend on snapshots (Arrasate and Finkbeiner, 2005; Sharma et al., 2012)(Figure 3). The approach is an especially good match for heterogeneous primary cell and differentiated iPSC systems since markers of cell type can be introduced so that different cell types can be identified and their biology can be monitored separately. This likely made it possible to measure disease-relevant phenotypes in iPSC models of HD, ALS, and Parkinson's disease without the need for exogenous stressors, which has been difficult to do with other methods (Bilican et al., 2012; HD iPSC Consortium et al., 2012; Serio et al., 2013; Skibinski et al., 2014). With this high level of sensitivity, conclusions can be drawn about the effects of pharmacological or genetic modifiers with data from as few as eight cells per well, which has made screening with primary cells and iPSCs—even genome-wide siRNA screens and large SM screens—feasible. The imperative for single-cell analysis is increasingly being recognized (Mullassery et al., 2008; Wu et al., 2011), and it has been applied in vivo to do imaging in Caenorhabditis elegans (Murray et al., 2006) and in cell lines to understand the biological significance of variability in signaling (Bendall et al., 2011).
Cell-based imaging screens generate staggeringly large and complex datasets. However, the brain is limited in the number of data dimensions and tradeoffs that can be considered at the same time. Without tools to graphically visualize the results of the analysis and present them quickly, it is difficult to grasp important patterns or trends in the data. Access to both raw data and useful visualizations is critical. Visualization is most helpful in an iterative process where looking at one visualization raises questions that are illuminated by another and so on. The difference between taking a few seconds to present visualization results and a few minutes is often the difference between a system being used frequently or not used at all. While there will never be enough computational power to pre-compute visualizations of all possible interesting aspects of complex data sets, educated guesses can determine which visualizations are most likely to be useful and to improve on those initial guesses by automatically monitoring the usage of the visualization system.
The ready availability of greatly increased computational power should enable a much broader use of statistical investigation. It should become common practice to run basic statistical algorithms to investigate correlations between most pairs of variables, and even some selected tuples, in any data set. This can be done in the background and presented to researchers in a way that makes it easy to check for surprises or anomalies. For repeated sets of experiments, the analysis of the deltas in such correlations between experiments can also yield useful insights. With attribution algorithms, it is even possible to analyze the attribution of changes to important observations and automatically point out the responsible variables/features (Sun and Sundararajan, 2011). Other tools, such as parallel R, and RFAce (https://code.google.com/p/rf-ace/), allow statistical algorithms to be run on large computer clusters.
For statistical algorithms, just like with visualization, computational power cannot grow as quickly as the combinatorics of complex data sets. The possible sets of variables and the possible analyses to be run will always outstrip new increases in computational power. However, with increased computational power, every experiment should have an exploratory computational budget. The question should shift from whether to apply statistical and data mining techniques at all to which variables and techniques to consider, given a specific computational budget. We should move from a world in which more data are considered a hindrance to one in which the ability to throw more related data into the automated analyses we set up for any experiment is seen as a benefit for the potential insights it might engender.
In parallel, better statistical algorithms need to be created. A little-known fact in HTS is that the statistical tests of significance utilize analysis methods that are catastrophically ill-suited to the task. Generally, these tools were designed for data sets in which few hypotheses were tested repeatedly. In HTS, many hypotheses are tested once each. One promising approach is to adapt some of the solutions that have been developed for analysis of microarrays where statisticians have already grappled with similar challenges.
Machine Learning
ML is a technique for building predictive computational models of a phenomenon that are iteratively improved with new facts or evidence. Both supervised and unsupervised learning approaches are promising (Conrad et al., 2004; Glory and Murphy, 2007; Jones et al., 2009; Neumann et al., 2010; Shamir et al., 2010). ML is a rich and rapidly advancing field of computer science, and there are many ML techniques with varying degrees of complexity and computational requirements. At a very high level, an ML system represents entities as sets of features. A feature is something measurable about the entity. All of the features of an entity can be combined into a fingerprint that describes that entity. The ML system builds an internal representation of how various combinations of feature values for an entity (its fingerprint) can predict something about that entity. As the system makes predictions, they are either confirmed or rejected. This feeds back into the ML system and causes it to update its representation. Over many iterations, the system learns to do better and better prediction. Although ML is not a new field, in most situations, the amount of data and computation needed to make it applicable to non-trivial problems are only now becoming generally available.
Besides the insights that these approaches provide for image analysis, they offer an attractive framework for incorporating additional disparate data. For example, it is possible to integrate whole-genome sequencing data and information on the patient's diagnosis, clinical course, and response to medicines with patient-derived iPSC screens. ML has the potential to uncover cellular phenotypes from the imaging data that elude humans and to integrate the additional genetic and clinical data with the phenotypic data in ways that could ultimately help us create a target and therapy discovery platform that is more predictive for clinical trials and lead the way for a personalized medicine approach to diagnosis and to treatment.
ML algorithms often give insight into complex systems when they give predictions that are surprising but correct. The investigation of why non-intuitive predictions are correct often leads to new testable hypotheses. But beyond insight, these systems can be practically applied to solve problems with feedback loops. For example, if an ML system were built to recognize handwriting, and it learned that some complex combination of features predicted that the letter being written was Q, this fact could still be effectively used even if a human couldn't make sense of “why” those features predicted Q. ML systems could potentially also be applied to repetitive tasks, such as growing specific cells or tissues from iPSCs. As long as the ML system effectively and continuously determined what combinations of features led to better yield, it would provide benefit even if the exact reason for its success wasn't immediately understood.
Machine perception is another technology that relies on ML techniques, and it is coming of age as increased computational abilities make it possible to execute the algorithms required. More and more experiments produce results that are images or video that require a non-scaleable application of human effort to interpret. Like all data, each image or video is still represented as a set of bits. With the right feature extraction, it can be fingerprinted, and ML can be applied. If the right representation can be built to predict things about the images based on these features, a level of understanding of the image or video is achieved. More so than in simpler data types, which features to extract becomes critically important. Some recent success has been attained with patterning feature extraction on how the human visual cortex processes input (Le et al., 2012). As we move beyond visualization and apply statistical and ML techniques to large data sets, we can gain new insights into our data and even create practical feedback control loops.
Another under-appreciated aspect of these techniques is that they are free of human bias. This has both an upside and a downside. The upside is that truly novel insight can be gained because these techniques can find patterns in the data that are far outside the scope of where a researcher would normally look. The downside is that these patterns are often not particularly germane to the problem being investigated and often wind up being artifacts of experimental setup or algorithm design. Balancing this upside and downside is an important aspect of working with ML techniques.
The Future
Rapid advances are expected in the near future in all aspects of cell-based screening, including cell-based disease modeling, imaging and analysis. The number of nervous system cell types that are possible to make and the efficiency with which they can be produced inexorably increases. Many existing differentiation protocols have aimed to recapitulate development through the addition of exogenous factors at key time points, which can be slow and inefficient. Increasingly, genetic and small-molecule perturbations are being used, which can create “short cuts” that reduce the time and cost and improve the efficiency of differentiation (Victor et al., 2014; Zhu et al., 2014). Interestingly, ML may have much to offer biologists seeking to solve the unwieldy combinatorial problems involved with refining differentiation protocols. Another focus in the differentiation field is the development of protocols that make subtypes of nervous system cells (Maroof et al., 2013). There is still a widespread concern that even after differentiation, the cell types that are produced are immature and may be unable to recapitulate critical dimensions of disease-related biology that require a certain level of maturity or even aging. Investigators are experimenting with ways to age iPSC cultures naturally or to accelerate the aging process genetically (Miller et al., 2013).
The field has understandably been focused on the development of differentiation methods that produce specific defined cell types. But the nervous system is a complex tissue whose normal function is an emergent property of many cell-cell interactions. For example, the physiology of neurons is very different if they are grown alone or if they are grown with astrocytes, and many critical structures, such as the synapse, the myelinated axon, the neuromuscular junction, require the presence of multiple cell types (Pfrieger and Barres, 1997; Ullian et al., 2001). Clearly, a critical future goal of the field will be to reconstitute nervous system structures that require multiple different cell types. This represents a major challenge to the iPSC field because the protocols to make neurons, astrocytes, microglia-like cells, skeletal muscle and oligodendrocytes are very different. In some cases, it has been possible to differentiate different types of mature cells from the same iPSC line and then recombine them to reconstitute the cell-cell interaction of interest (Serio et al., 2013). A related goal is to model neural circuits with human neurons. Technology to engineer neural circuits in vitro has been developed and needs to be applied to the iPSC field (Garcia et al., 2012; Li et al., 2014; Shein et al., 2009; Shein-Idelson et al., 2011). Notably, as model systems become more complex, the ability to distinguish different cell types and track their biology independently will become even more important for extracting useful data from these heterogeneous systems.
Regardless of the particular screening method or disease, the general concepts governing cell-based screening discussed in the review will likely remain valid. That said, there are likely to be some exciting new directions in cell-based screening. The availability of a fully human iPSC-based platform to evaluate perturbagens by HTS opens up several interesting new possibilities. Human genetics has played a critical role in identifying genes and pathways that are responsible for neuropsychiatric disease. But achieving the statistical power to uncover substantial numbers of interesting genetic variants remains challenging to do because it can be difficult to find enough patients and to afford the costs. In addition, as genetic analyses extend more deeply into non-coding regions of the genome, it becomes even more critical to perform functional validation studies to establish causal relationships between genetic variants and disease. An HT iPSC-based platform could be used to screen putative genetic variants against disease-relevant phenotypes to quickly narrow putative variants, some of which may not have achieved genome-wide significance using human genetic analysis alone, down to a set that have significant functional effects in a fully human system. We could envision a future in which human genetics and HT screening in human CNS models are combined to advance or regress the candidacy of putative disease-modifiers and simultaneously obtain critical functional validation.
With the incorporation of more relevant primary neuron and iPSCs into HTS and the substantial financial investment that entails, the economics of screening will change. It will be increasingly important to extract as much phenotypic information as possible from each screen to justify the expense, and that will likely lead to the design of screens that incorporate multiple biosensors to track independent endpoints. In this context, one of the biggest remaining limitations in image acquisition is the bandwidth available to resolve different fluorophores. The broad excitation and emission spectra of fluorescent proteins make it difficult to use more than three or four simultaneously with conventional filter-based methods to resolve signals. Several approaches to increase the content of screens are under investigation. For example, pixel-by-pixel spectra can be generated, and complex overlapping emissions can be linearly unmixed to distinguish more fluorophores than is possible with conventional filter-based techniques. However, these methods are still too slow for HTS and will require modification. In screens with live-cell imaging, additional terminal assays can be performed after fixing the cells, thus adding content. These can include the selected application of approaches, such as super-resolution microscopy (Bates et al., 2007; Bates et al., 2008; Ding et al., 2009; Grecco and Verveer, 2011) that might be too slow or resource intensive to apply to all the samples in the screen. This can be done through an image-based servo mechanism to direct the instrument to collect the additional data from specific fields of interest identified through automated image analysis.
Predictably, CPU, storage, and bandwidth will all become cheaper, and more computational resources will become available, giving researchers much more powerful techniques for analysis at their disposal. Before any of the techniques above can be used, the nuts and bolts of infrastructure problems of storing, manipulating, moving, and safeguarding data must be solved. Cloud computing solutions will offer simpler alternatives to building everything in the lab, but will potentially complicate other factors like bandwidth and security.
The availability of very large-scale computational power provided by large networked clusters will change the way science is done. We are already seeing the benefits of running existing algorithms in parallel on much larger data sets. Better and faster visualization tools will enhance our ability to understand complex phenomena. The real change will come from applying algorithms that were previously untenable due to computational limitations. These algorithms will, in turn, drive a different approach to investigation.
As techniques, such as ML, machine perception, large-scale modeling and simulation become more common, the way investigators look at problems should change to take advantage of these tools. Adoption of these changes is slow and likely reflects that large-scale approaches are not practical without access to large computational facilities. The problem may be partly overcome by the availability of large-scale computing power by the slice from third-party vendors, such as Google or Amazon. Another reason for the slow adoption of some of these techniques is lack of expertise. ML, simulation, machine perception, visualization, and even the basics of managing large data are all difficult problems that require experts to solve. There is no simple cookbook for applying these techniques to a given problem. The limited supply of people with such experience is dwarfed by the demand. This is a problem that is only fixable by our education system in the long term, but can be ameliorated by collaboration in the short term.
As computational resources become cheaper and more available, researchers will have exciting new options in how they pursue their research. Perhaps the most exciting future directions for cell-based screening will emerge from the effective combination of advances in cell-based modeling, imaging and ML. As cell-based assays become more complex and generate deeper cellular phenotypes, can we use ML to help us prioritize the phenotypic endpoints that will translate best and that should be pursued? The availability of more powerful forms of image analysis, powered by techniques such as deep learning, may lead to a fundamental shift in phenotypic screening, away from simplistic measures of narrow endpoints to more global definitions of cell health. With the ability to produce deep cellular phenotypes in patient-derived iPSCs, will it be possible to use ML to predict features of the clinical course of patients from whom they came? (Figure 4) If so, it could transform approaches to patient stratification, clinical trial design, and improve the reliability of our translational pipeline. We are now entering a period of time where the availability of such computational resources will grow exponentially and so will the development of new algorithms and techniques that take advantage of these resources.
Figure 4. Future applications of high-throughput cell-based screening.

The increasing ability to collect complex cell-based and clinical data and to analyze these data with machine learning approaches is opening up new possibilities for high-throughput cell-based screening. The figure illustrates an initiative being pursued by the authors (S.F. and M.F.) to develop a preclinical pipeline that is more predictive of clinical results. One element of the overall approach, supported by the NIH Common Fund and NINDS, involves the creation of an array of hundreds of biosensors suitable for longitudinal live single-cell imaging and capable of generating a deep phenotypic understanding of a variety of cell structures and functions in brain cells from patient-derived induced pluripotent stem cells (iPSCs). The array of biosensors is referred to as the “Physical Exam of the Cell” and is designed to broadly survey cell physiology, performing a role at a cellular level similar to the role that the physical exam of patients plays in medicine. These large and complex datasets will then be related to similarly deep and complex clinical datasets collected from the patients from whom the iPSCs were generated. Then, with unsupervised machine learning, the two datasets can be examined with the goal of finding features that can be measured from iPSCs that predict one or more clinical features, such as disease progression, of the patients from whom the cells came. New or additional clinical features may be predicted as the iPSC models of disease increase in physiological relevance with improved differentiation protocols or by combining different cell types that lead to emergent properties. A preclinical pipeline that predicts clinical findings could be invaluable for lowering the risk of developing neurotherapeutics by improving the sensitivity of clinical trials through patient stratification and for identifying therapeutic targets and therapies that have better chance of working when tested in patients.
Supplementary Material
Video 1. Third generation robotic microscope. The video illustrates the function and capabilities of a third-generation robotic microscope. An automated imaging acquisition run is initiated by the computer that controls the system and begins when the robot in the incubator is instructed to select a plate of cells for imaging and delivers it to a nest outside the incubator. Then, the robotic arm transfers the plate from the nest to a custom-designed plate holder, equipped with an actuator to minimize variation in the placement of the plate within the holder. The microscope and stage are then directed by the computer to collect an image of a fiduciary mark, which is used by the computer to align the plate, and then images of different fluorescent emissions from different microscope fields is automatically collected. After images are acquired, the plate is returned to its original location in the incubator robotically. The time-lapse series of images of neurons at the end of the movie was made from seven images of the same microscope fields collected on successive days and illustrates the capability of the system to return to precisely the same microscope fields and to follow individual cells for as long and as often as the investigator wishes.
Acknowledgments
We thank members of the Finkbeiner lab and G. Howard for editorial input. S.F. was supported by NIH grants U01 MH1050135, U54 HG008105, R01 NS083390, 3 R01 NS039074, 2 R01 NS045091, U24 NS078370, NSF BRAIN EAGER award, and the Taube/Koret Center for Neurodegenerative Disease. SF is an employee of the J. David Gladstone Institutes, which holds an issued patent and a pending patent for robotic microscopy. MF is an employee of Google. PDK is an employee of Amgen Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L, Ferrandino AF, Rosenberg Belmaker LA, Szekely A, Wilson M, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Actis P, Maalouf MM, Kim HJ, Lohith A, Vilozny B, Seger RA, Pourmand N. Compartmental genomics in living cells revealed by single-cell nanobiopsy. ACS Nano. 2014;8:546–553. doi: 10.1021/nn405097u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ali H, Blackmore M, Bixby JL, Lemmon VP. High content screening with primary neurons. In: Sittampalam GS, Gal-Edd N, Arkin MR, Auld D, Austin C, Bejcek B, Glicksman M, Inglese J, Lemmon V, Li Z, et al., editors. Assay Guidance Manual. Bethesda, MD: Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2013. Internet. [PubMed] [Google Scholar]
- Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, Lipton SA, Ding S. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 2011;9:113–118. doi: 10.1016/j.stem.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Finkbeiner S. Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci USA. 2005;102:3840–3845. doi: 10.1073/pnas.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Aubry L, Bugi A, Lefort N, Rousseau F, Peschanski M, Perrier AL. Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci USA. 2008;105:16707–16712. doi: 10.1073/pnas.0808488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, Tsvetkov A, Pleiss M, Li X, Peisach D, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677–685. doi: 10.1038/nchembio.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Zhuang X. Super-resolution microscopy by nanoscale localization of photo-switchable fluorescent probes. Curr Opin Chem Biol. 2008;12:505–514. doi: 10.1016/j.cbpa.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B, Rothmaier AK, Wedekind F, Zentner J, Feuerstein TJ, Jackisch R. Presynaptic opioid receptors on noradrenergic and serotonergic neurons in the human as compared to the rat neocortex. Br J Pharmacol. 2006;148:795–806. doi: 10.1038/sj.bjp.0706782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhinder B, Djaballah H. Systematic analysis of RNAi reports identifies dismal commonality at gene-level and reveals an unprecedented enrichment in pooled shRNA screens. Comb Chem High Throughput Screen. 2013;16:665–681. doi: 10.2174/13862073113169990045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci USA. 2012;109:5803–5808. doi: 10.1073/pnas.1202922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6:569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow N. Systems biology: Untangling the protein web. Nature. 2009;460:415–418. doi: 10.1038/460415a. [DOI] [PubMed] [Google Scholar]
- Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, et al. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D, Mason M, Garnes J, Dang V, Lievers J, Shoukat-Mumtaz U, et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:355–364. doi: 10.1016/j.mcn.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson EA, McCulloch C, Koganti A, Goodwin SB, Sutter TR, Silkworth JB. Divergent transcriptomic responses to aryl hydrocarbon receptor agonists between rat and human primary hepatocytes. Toxicol Sci. 2009;112:257–272. doi: 10.1093/toxsci/kfp200. [DOI] [PubMed] [Google Scholar]
- Castan I, Valet P, Quideau N, Voisin T, Ambid L, Laburthe M, Lafontan M, Carpene C. Antilipolytic effects of alpha 2-adrenergic agonists, neuropeptide Y, adenosine, and PGE1 in mammal adipocytes. Am J Physiol. 1994;266:R1141–R1147. doi: 10.1152/ajpregu.1994.266.4.R1141. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Mica Y, Lee G, Studer L, Tomishima MJ. Dual-SMAD inhibition/WNT activation-based methods to induce neural crest and derivatives from human pluripotent stem cells. Methods Mol Biol. 2013 doi: 10.1007/7651_2013_59. [DOI] [PubMed] [Google Scholar]
- Choulier L, Enander K. Environmentally sensitive fluorescent sensors based on synthetic peptides. Sensors (Basel) 2010;10:3126–3144. doi: 10.3390/s100403126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Lukyanov S, Lukyanov KA. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc. 2007;2:2024–2032. doi: 10.1038/nprot.2007.291. [DOI] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Churko JM, Burridge PW, Wu JC. Generation of human iPSCs from human peripheral blood mononuclear cells using non-integrative Sendai virus in chemically defined conditions. Methods Mol Biol. 2013;1036:81–88. doi: 10.1007/978-1-62703-511-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke G, Collins RA, Leavitt BR, Andrews DF, Hayden MR, Lumsden CJ, McInnes RR. A one-hit model of cell death in inherited neuronal degenerations. Nature. 2000;406:195–199. doi: 10.1038/35018098. [DOI] [PubMed] [Google Scholar]
- Collinet C, Stöter M, Bradshaw CR, Samusik N, Rink JC, Kenski D, Habermann B, Buchholz F, Henschel R, Mueller MS, et al. Systems survey of endocytosis by multiparametric image analysis. Nature. 2010;464:243–249. doi: 10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- Conrad C, Erfle H, Warnat P, Daigle N, Lörch T, Ellenberg J, Pepperkok R, Eils R. Automatic identification of subcellular phenotypes on human cell arrays. Genome Res. 2004;14:1130–1136. doi: 10.1101/gr.2383804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson's disease. Sci Transl Med. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SJF, Nehme NT, Limmer S, Liegeois S, Pospisilik JA, Schramek D, Leibbrandt A, de Matos Simoes R, Gruber S, Puc U, et al. Genome-wide RNAi screen identifies genes involved in intestinal pathogenic bacterial infection. Science. 2009;325:340–343. doi: 10.1126/science.1173164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook ZR, Housman D. Huntington's disease: Can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis SW, Shi H, Teng C, Korach KS. Promoter and species specific differential estrogen-mediated gene transcription in the uterus and cultured cells using structurally altered agonists. J Mol Endocrinol. 1997;18:203–211. doi: 10.1677/jme.0.0180203. [DOI] [PubMed] [Google Scholar]
- Czepiel M, Balasubramaniyan V, Schaafsma W, Stancic M, Mikkers H, Huisman C, Boddeke E, Copray S. Differentiation of induced pluripotent stem cells into functional oligodendrocytes. Glia. 2011;59:882–892. doi: 10.1002/glia.21159. [DOI] [PubMed] [Google Scholar]
- Daub A, Sharma P, Finkbeiner S. High-content screening of primary neurons: ready for prime time. Curr Opin Neurobiol. 2009;19:537–543. doi: 10.1016/j.conb.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker FW, de Mutsert R, van Dijk PC, Zoccali C, Jager KJ. Survival analysis: Time-dependent effects and time-varying risk factors. Kidney Int. 2008;74:994–997. doi: 10.1038/ki.2008.328. [DOI] [PubMed] [Google Scholar]
- Derian CK, Santulli RJ, Tomko KA, Haertlein BJ, Andrade-Gordon P. Species differences in platelet responses to thrombin and SFLLRN. Receptor-mediated calcium mobilization and aggregation, and regulation by protein kinases. Thromb Res. 1995;78:505–519. doi: 10.1016/0049-3848(95)00084-5. [DOI] [PubMed] [Google Scholar]
- Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, Padmanabhan K, Swoboda JG, Ahmad I, Kondo T, et al. A regenerative approach to the treatment of multiple sclerosis. Nature. 2013;502:327–332. doi: 10.1038/nature12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Takasaki KT, Sabatini BL. Supraresolution imaging in brain slices using stimulated-emission depletion two-photon laser scanning microscopy. Neuron. 2009;63:429–437. doi: 10.1016/j.neuron.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverri CJ, Beachy PA, Baum B, Boutros M, Buchholz F, Chanda SK, Downward J, Ellenberg J, Fraser AG, Hacohen N, et al. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat Methods. 2006;3:777–779. doi: 10.1038/nmeth1006-777. [DOI] [PubMed] [Google Scholar]
- Echeverri CJ, Perrimon N. High-throughput RNAi screening in cultured cells: A user's guide. Nat Rev Genet. 2006;7:373–384. doi: 10.1038/nrg1836. [DOI] [PubMed] [Google Scholar]
- Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med. 2012;4:145ra–104. doi: 10.1126/scitranslmed.3004052. [DOI] [PubMed] [Google Scholar]
- Eglen R, Reisine T. Primary cells and stem cells in drug discovery: Emerging tools for high-throughput screening. Assay Drug Dev Technol. 2011;9:108–124. doi: 10.1089/adt.2010.0305. [DOI] [PubMed] [Google Scholar]
- Emdad L, D'Souza SL, Kothari HP, Qadeer ZA, Germano IM. Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev. 2012;21:404–410. doi: 10.1089/scd.2010.0560. [DOI] [PubMed] [Google Scholar]
- Falschlehner C, Steinbrink S, Erdmann G, Boutros M. High-throughput RNAi screening to dissect cellular pathways: A how-to guide. Biotechnol J. 2010;5:368–376. doi: 10.1002/biot.200900277. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S. Huntington's disease. Cold Spring Harb Perspect Biol. 2011;3:a007476. doi: 10.1101/cshperspect.a007476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino PL, Tavazoie S. The dawn of virtual cell biology. Cell. 2012;150:248–250. doi: 10.1016/j.cell.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia I, Huang L, Ung K, Arenkiel BR. Tracing synaptic connectivity onto embryonic stem cell-derived neurons. Stem Cells. 2012;30:2140–2151. doi: 10.1002/stem.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glory E, Murphy RF. Automated subcellular location determination and high-throughput microscopy. Dev Cell. 2007;12:7–16. doi: 10.1016/j.devcel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Grecco HE, Verveer PJ. FRET in cell biology: Still shining in the age of super-resolution? Chemphyschem. 2011;12:484–490. doi: 10.1002/cphc.201000795. [DOI] [PubMed] [Google Scholar]
- Guo YS, Singh P, Upp JR, Jr, Thompson JC. Species-specific effects of neurotensin on gallbladder contraction in vitro. Dig Dis Sci. 1989;34:21–26. doi: 10.1007/BF01536149. [DOI] [PubMed] [Google Scholar]
- Hamer G, Matilainen O, Holmberg CI. A photoconvertible reporter of the ubiquitin-proteasome system in vivo. Nat Methods. 2010;7:473–478. doi: 10.1038/nmeth.1460. [DOI] [PubMed] [Google Scholar]
- Hayes M, Zavazava N. Strategies to generate induced pluripotent stem cells. Methods Mol Biol. 2013;1029:77–92. doi: 10.1007/978-1-62703-478-4_6. [DOI] [PubMed] [Google Scholar]
- HD iPSC Consortium. Mattis V, Svendsen SP, Ebert A, Svendsen CN, King AR, Casale M, Winokur ST, Batugedara G, Vawter M, et al. Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZB, Takahashi M. Generation of retinal cells from pluripotent stem cells. Prog Brain Res. 2012;201:171–181. doi: 10.1016/B978-0-444-59544-7.00008-1. [DOI] [PubMed] [Google Scholar]
- Jones TR, Carpenter AE, Lamprecht MR, Moffat J, Silver SJ, Grenier JK, Castoreno AB, Eggert US, Root DE, Golland P, et al. Scoring diverse cellular morphologies in image-based screens with iterative feedback and machine learning. Proc Natl Acad Sci USA. 2009;106:1826–1831. doi: 10.1073/pnas.0808843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr Molecular biology. Use and abuse of RNAi to study mammalian gene function. Science. 2012;337:421–422. doi: 10.1126/science.1225787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Jr, Assad-Garcia N, Glass JI, Covert MW. A whole-cell computational model predicts phenotype from genotype. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner PD. Discovery of novel targets with high throughput RNA interference screening. Comb Chem High Throughput Screen. 2008;11:175–184. doi: 10.2174/138620708783877744. [DOI] [PubMed] [Google Scholar]
- Keshavaprasad B, Liu C, Au JD, Kindler CH, Cotten JF, Yost CS. Species-specific differences in response to anesthetics and other modulators by the K2P channel TRESK. Anesth Analg. 2005;101:1042–1049. doi: 10.1213/01.ane.0000168447.87557.5a. [DOI] [PubMed] [Google Scholar]
- Kim DS, Ross PJ, Zaslavsky K, Ellis J. Optimizing neuronal differentiation from induced pluripotent stem cells to model ASD. Front Cell Neurosci. 2014;8:109. doi: 10.3389/fncel.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S, Lipton SA, Zhang K, Ding S. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci USA. 2011;108:7838–7843. doi: 10.1073/pnas.1103113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopin AS, McBride EW, Gordon MC, Quinn SM, Beinborn M. Inter- and intraspecies polymorphisms in the cholecystokinin-B/gastrin receptor alter drug efficacy. Proc Natl Acad Sci USA. 1997;94:11043–11048. doi: 10.1073/pnas.94.20.11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le QV, Ranzato MA, Monga R, Devin M, Chen K, Corrado GS, Dean J, Ng AY. Building high-level features using large scale unsupervised learing. Paper presented at: 29th International Conference on Machine Learning; Edinburgh, Scotland, UK. 2012. [Google Scholar]
- Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu Z, Huang J, Lin X, Luo R, Chen CH, Shi P. NeuroArray: A universal interface for patterning and interrogating neural circuitry with single cell resolution. Sci Rep. 2014;4:4784. doi: 10.1038/srep04784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Matzkies M, Schunkert H, Tang M, Bonnemeier H, Hescheler J, Reppel M. Human and murine embryonic stem cell-derived cardiomyocytes serve together as a valuable model for drug safety screening. Cell Physiol Biochem. 2010;25:459–466. doi: 10.1159/000303051. [DOI] [PubMed] [Google Scholar]
- Lutolf MP, Gilbert PM, Blau HM. Designing materials to direct stem-cell fate. Nature. 2009;462:433–441. doi: 10.1038/nature08602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Rychlik B, You JS, Sisken JE. Sensitivity of cultured human embryonic cerebral cortical neurons to excitatory amino acid-induced calcium influx and neurotoxicity. Brain Res. 1991;542:97–106. doi: 10.1016/0006-8993(91)91003-j. [DOI] [PubMed] [Google Scholar]
- McGonigle P. Animal models of CNS disorders. Biochem Pharmacol. 2014;87:140–149. doi: 10.1016/j.bcp.2013.06.016. [DOI] [PubMed] [Google Scholar]
- McGonigle P, Ruggeri B. Animal models of human disease: Challenges in enabling translation. Biochem Pharmacol. 2014;87:162–171. doi: 10.1016/j.bcp.2013.08.006. [DOI] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7:925–934. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, Finkbeiner S. Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into Huntington's disease molecular pathogenesis. J Neurosci. 2010;30:10541–10550. doi: 10.1523/JNEUROSCI.0146-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S, Bakal C, Perrimon N. Genomic screening with RNAi: Results and challenges. Annu Rev Biochem. 2010;79:37–64. doi: 10.1146/annurev-biochem-060408-092949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MC. Fluorescent biosensors of intracellular targets from genetically encoded reporters to modular polypeptide probes. Cell Biochem Biophys. 2010;56:19–37. doi: 10.1007/s12013-009-9070-7. [DOI] [PubMed] [Google Scholar]
- Mullane K, Williams M. Alzheimer's therapeutics: Continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem Pharmacol. 2013;85:289–305. doi: 10.1016/j.bcp.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Mullassery D, Horton CA, Wood CD, White MRH. Single live-cell imaging for systems biology. Essays Biochem. 2008;45:121–133. doi: 10.1042/BSE0450121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JI, Bao Z, Boyle TJ, Waterston RH. The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nat Protoc. 2006;1:1468–1476. doi: 10.1038/nprot.2006.222. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, Goto Y, Naiki H, Toda T. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- Neumann B, Held M, Liebel U, Erfle H, Rogers P, Pepperkok R, Ellenberg J. High-throughput RNAi screening by time-lapse imaging of live human cells. Nat Methods. 2006;3:385–390. doi: 10.1038/nmeth876. [DOI] [PubMed] [Google Scholar]
- Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Tokumoto Y, Miyake J, Nagamune T. Induction of oligodendrocyte differentiation from adult human fibroblast-derived induced pluripotent stem cells. In Vitro Cell Dev Biol Anim. 2011;47:464–469. doi: 10.1007/s11626-011-9435-2. [DOI] [PubMed] [Google Scholar]
- Okazaki R, Durham SK, Riggs BL, Conover CA. Transforming growth factor-beta and forskolin increase all classes of insulin-like growth factor-I transcripts in normal human osteoblast-like cells. Biochem Biophys Res Commun. 1995;207:963–970. doi: 10.1006/bbrc.1995.1279. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada Y, Cui B. Real-time visualization of axonal transport in neurons. Methods Mol Biol. 2011;670:231–243. doi: 10.1007/978-1-60761-744-0_16. [DOI] [PubMed] [Google Scholar]
- Paige JS, Nguyen-Duc T, Song W, Jaffrey SR. Fluorescence imaging of cellular metabolites with RNA. Science. 2012;335:1194. doi: 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Qin Y, Park JG, McCombs JE. Design and application of genetically encoded biosensors. Trends Biotechnol. 2011;29:144–152. doi: 10.1016/j.tibtech.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penhoat A, Ouali R, Viard I, Langlois D, Saez JM. Regulation of primary response and specific genes in adrenal cells by peptide hormones and growth factors. Steroids. 1996;61:176–183. doi: 10.1016/0039-128x(96)00009-8. [DOI] [PubMed] [Google Scholar]
- Perrin S. Preclinical research: Make mouse studies work. Nature. 2014;507:423–425. doi: 10.1038/507423a. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Pradier L, Habert-Ortoli E, Emile L, Le Guern J, Loquet I, Bock MD, Clot J, Mercken L, Fardin V, Garret C, et al. Molecular determinants of the species selectivity of neurokinin type 1 receptor antagonists. Mol Pharmacol. 1995;47:314–321. [PubMed] [Google Scholar]
- Prinz F, Schlange T, Asadullah K. Believe it or not: How much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- Rasakham K, Liu-Chen LY. Sex differences in kappa opioid pharmacology. Life Sci. 2011;88:2–16. doi: 10.1016/j.lfs.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richert L, Tuschl G, Viollon-Abadie C, Blanchard N, Bonet A, Heyd B, Halkic N, Wimmer E, Dolgos H, Mueller SO. Species differences in the response of liver drug-metabolizing enzymes to (S)-4-O-tolylsulfanyl-2-(4-trifluormethyl-phenoxy)-butyric acid (EMD 392949) in vivo and in vitro. Drug Metab Dispos. 2008;36:702–714. doi: 10.1124/dmd.107.018358. [DOI] [PubMed] [Google Scholar]
- Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura A, et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci USA. 2013;110:4697–4702. doi: 10.1073/pnas.1300398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamir L, Delaney JD, Orlov N, Eckley DM, Goldberg IG. Pattern recognition software and techniques for biological image analysis. PLoS Comput Biol. 2010;6:e1000974. doi: 10.1371/journal.pcbi.1000974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Choi SY, Zhang Y, Nieland TJ, Long S, Li M, Huganir RL. High-throughput genetic screen for synaptogenic factors: Identification of LRP6 as critical for excitatory synapse development. Cell Rep. 2013;5:1330–1341. doi: 10.1016/j.celrep.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Ando DM, Daub A, Kaye JA, Finkbeiner S. High-throughput screening in primary neurons. Methods Enzymol. 2012;506:331–360. doi: 10.1016/B978-0-12-391856-7.00041-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shein M, Greenbaum A, Gabay T, Sorkin R, David-Pur M, Ben-Jacob E, Hanein Y. Engineered neuronal circuits shaped and interfaced with carbon nanotube microelectrode arrays. Biomed Microdevices. 2009;11:495–501. doi: 10.1007/s10544-008-9255-7. [DOI] [PubMed] [Google Scholar]
- Shein-Idelson M, Ben-Jacob E, Hanein Y. Engineered neuronal circuits: a new platform for studying the role of modular topology. Front Neuroeng. 2011;4:10. doi: 10.3389/fneng.2011.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhawat SS, Ghosh I. Split-protein systems: Beyond binary protein-protein interactions. Curr Opin Chem Biol. 2011;15:789–797. doi: 10.1016/j.cbpa.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Scott MA, Ghosh B, Wan D, Wissner-Gross Z, Mazitschek R, Haggarty SJ, Yanik MF. Synapse microarray identification of small molecules that enhance synaptogenesis. Nat Commun. 2011;2:510. doi: 10.1038/ncomms1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Gupta RS. Species-specific differences in the toxicity and mutagenicity of the anticancer drugs mithramycin, chromomycin A3, and olivomycin. Cancer Res. 1985;45:2813–2820. [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci. 2014;34:418–433. doi: 10.1523/JNEUROSCI.2712-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L. Derivation of dopaminergic neurons from pluripotent stem cells. Prog Brain Res. 2012;200:243–263. doi: 10.1016/B978-0-444-59575-1.00011-9. [DOI] [PubMed] [Google Scholar]
- Sun Y, Dong Z, Jin T, Ang KH, Huang M, Haston KM, Peng J, Zhong TP, Finkbeiner S, Weiss WA, et al. Imaging-based chemical screening reveals activity-dependent neural differentiation of pluripotent stem cells. Elife. 2013;2:e00508. doi: 10.7554/eLife.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Sundararajan M. Axiomatic attribution for multilinear functions. 2011:1–20. arXiv.org.
- Sundberg M, Bogetofte H, Lawson T, Jansson J, Smith G, Astradsson A, Moore M, Osborn T, Cooper O, Spealman R, et al. Improved cell therapy protocols for Parkinson's disease based on differentiation efficiency and safety of hESC-, hiPSC-, and non-human primate iPSC-derived dopaminergic neurons. Stem Cells. 2013;31:1548–1562. doi: 10.1002/stem.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swearingen EA, Fajardo F, Wang X, Watson JE, Quon KC, Kassner PD. Use of cryopreserved cell aliquots in the high-throughput screening of small interfering RNA libraries. J Biomol Screen. 2010;15:469–477. doi: 10.1177/1087057110365899. [DOI] [PubMed] [Google Scholar]
- Tainaka K, Sakaguchi R, Hayashi H, Nakano S, Liew FF, Morii T. Design strategies of fluorescent biosensors based on biological macromolecular receptors. Sensors (Basel) 2010;10:1355–1376. doi: 10.3390/s100201355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tsvetkov AS, Arrastate M, Barmada S, Ando DM, Sharma P, Shaby BA, Finkbeiner S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol. 2013b;9:586–592. doi: 10.1038/nchembio.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Vendrell M, Lee JS, Chang YT. Diversity-oriented fluorescence library approaches for probe discovery and development. Curr Opin Chem Biol. 2010;14:383–389. doi: 10.1016/j.cbpa.2010.02.020. [DOI] [PubMed] [Google Scholar]
- Victor MB, Richner M, Hermanstyne TO, Ransdell JL, Sobieski C, Deng PY, Klyachko VA, Nerbonne JM, Yoo AS. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron. 2014;84:311–323. doi: 10.1016/j.neuron.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkenborg JL, Koay MS, Merkx M. Fluorescent imaging of transition metal homeostasis using genetically encoded sensors. Curr Opin Chem Biol. 2010;14:231–237. doi: 10.1016/j.cbpa.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Walter T, Held M, Neumann B, Hériché JK, Conrad C, Pepperkok R, Ellenberg J. Automatic identification and clustering of chromosome phenotypes in a genome wide RNAi screen by time-lapse imaging. J Struct Biol. 2010;170:1–9. doi: 10.1016/j.jsb.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. 2013;12:252–264. doi: 10.1016/j.stem.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014a;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Nguyen HN, Gu Z, Lalli MA, Wang X, Su Y, Kim NS, Yoon KJ, Shin J, Zhang C, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014b;515:414–418. doi: 10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe RA, Strawderman RL. Logical and statistical fallacies in the use of Cox regression models. Am J Kidney Dis. 1996;27:124–129. doi: 10.1016/s0272-6386(96)90039-6. [DOI] [PubMed] [Google Scholar]
- Wray S, Self M, Lewis PA, Taanman JW, Ryan NS, Mahoney CJ, Liang Y, Devine MJ, Sheerin UM, Houlden H, et al. Creation of an open-access, mutation-defined fibroblast resource for neurological disease research. PLoS One. 2012;7:e43099. doi: 10.1371/journal.pone.0043099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Piatkevich KD, Lionnet T, Singer RH, Verkhusha VV. Modern fluorescent proteins and imaging technologies to study gene expression, nuclear localization, and dynamics. Curr Opin Cell Biol. 2011;23:310–317. doi: 10.1016/j.ceb.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- Yang N, Zuchero JB, Ahlenius H, Marro S, Ng YH, Vierbuchen T, Hawkins JS, Geissler R, Barres BA, Wernig M. Generation of oligodendroglial cells by direct lineage conversion. Nat Biotechnol. 2013;31:434–439. doi: 10.1038/nbt.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1220. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress implications and challenges. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu125. [DOI] [PubMed] [Google Scholar]
- Zhang K, Osakada Y, Xie W, Cui B. Automated image analysis for tracking cargo transport in axons. Microsc Res Tech. 2011;74:605–613. doi: 10.1002/jemt.20934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, An MC, Montoro D, Ellerby LM. Characterization of human Huntington's disease cell model from induced pluripotent stem cells. PLoS Curr. 2010;2:RRN1193. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Ambasudhan R, Sun W, Kim HJ, Talantova M, Wang X, Zhang M, Zhang Y, Laurent T, Parker J, et al. Small molecules enable OCT4-mediated direct reprogramming into expandable human neural stem cells. Cell Res. 2014;24:126–129. doi: 10.1038/cr.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Third generation robotic microscope. The video illustrates the function and capabilities of a third-generation robotic microscope. An automated imaging acquisition run is initiated by the computer that controls the system and begins when the robot in the incubator is instructed to select a plate of cells for imaging and delivers it to a nest outside the incubator. Then, the robotic arm transfers the plate from the nest to a custom-designed plate holder, equipped with an actuator to minimize variation in the placement of the plate within the holder. The microscope and stage are then directed by the computer to collect an image of a fiduciary mark, which is used by the computer to align the plate, and then images of different fluorescent emissions from different microscope fields is automatically collected. After images are acquired, the plate is returned to its original location in the incubator robotically. The time-lapse series of images of neurons at the end of the movie was made from seven images of the same microscope fields collected on successive days and illustrates the capability of the system to return to precisely the same microscope fields and to follow individual cells for as long and as often as the investigator wishes.