

Abstract
Gastrointestinal (GI) anthrax, caused by the bacterial infection of Bacillus anthracis, posts a significant bioterrorism threat by its relatively high mortality rate in humans. Different from inhalational anthrax by the route of infection, accumulating evidence indicates the bypass of vegetative bacteria across GI epithelium is required to initiate GI anthrax. Previously, we reported that purified anthrolysin O (ALO), instead of tripartite anthrax edema and lethal toxins, is capable of disrupting gut epithelial tight junctions and barrier function in cultured cells. Here, we show that ALO can disrupt intestinal tissue barrier function in an ex vivo mouse model. To explore the effects of ALO in a cell culture model of B. anthracis infection, we showed that anthrax bacteria can effectively reduce the monolayer integrity of human Caco-2 brush-border expressor (C2BBE) cells based on the reduced transepithelial resistance and the increased leakage of fluorescent dye. This disruption is likely caused by tight junction dysfunction observed by the reorganization of the tight junction protein occludin. Consequently, we observe significant passage of vegetative anthrax bacteria across C2BBE cells. This barrier disruption and bacterial crossover requires ALO since ALO-deficient B. anthracis strains fail to induce monolayer dysfunction and allow the passage of anthrax bacteria. Together these findings point to a pivotal role for ALO within the establishment of GI anthrax infection and the initial bypass of the epithelial barrier.
Keywords: Gastrointestinal anthrax, anthrolysin O, cholesterol-dependent cytolysin, epithelial barrier disruption, anthrax infection
Introduction
Bacillus anthracis, the etiologic agent of anthrax is a potent bioterrorism agent [1; 2; 3; 4]. Anthrax infection initiates when B. anthracis spores enter the host through one of three routes (skin, lung, or gastrointestinal tract) and result in cutaneous, inhalational, or gastrointestinal (GI) anthrax, respectively. All three routes of infection can lead to systemic infection and are ultimately lethal if untreated. Both inhalational and GI anthrax post a significant bioterrorism threat by their high mortality rate. The first line of defense against GI anthrax infection is mediated by gastrointestinal epithelial cells, which form a barrier to separate luminal contents and the underlying tissue compartment [5; 6]. This barrier is controlled by blockage of paracellular movement through regulation of fluid and electrolyte diffusion across tight junctions. Tight junctions are formed by discrete multi-protein complexes that connect neighboring cells by associating with the cytoskeleton. Gut epithelial barrier disruption occurs in certain bacterial infections and in inflammation [5]. GI anthrax differs from inhalational anthrax as the spores are ingested and germinate on or within the epithelium of the GI tract [1] in order to gain access to the underlying lymphatics, including the Peyer’s Patch [7]. The mechanism by which GI anthrax is initiated is poorly understood. However, a route of passage through the GI epithelial layers is likely required, whether in the form of a primary lesion [7] or other means to circumvent this barrier.
Anthrax bacteria secrete over 5% of its gene products [8]. Of those, anthrolysin O (ALO) is capable of disrupting GI epithelial barrier function in cultured cells [9]. ALO is homologous to the cholesterol dependent cytolysin (CDC) family, which is required for maximal virulence in several bacterial species of Streptococcus, Listeria, and Clostridium [10; 11; 12; 13]. Bacterial CDC is best known to facilitate the escape of intracellular bacteria from lysosomal degradation exemplified by listeriolysin O (LLO), perfringolysin O (PFO), and ALO [12; 13; 14]. However, growing evidence indicates that bacterial CDCs exhibit diverse extracellular functions as well. CDCs can modulate intracellular calcium [9; 15], reorganize the actin cytoskeleton [16], disrupt the epithelial tight junctions [9; 17], alter histone modifications [18], activate toll-like receptors [19; 20], and trigger cytokine production [21]. At high concentrations, CDCs are also known to be hemolytic and cytolytic [9; 10; 12; 22; 23].
While purified ALO can cause barrier disruption of cultured gut epithelial cells, the relevance of ALO in tissue or in bacterial infection is unclear. Initial reports found that anthrax bacteria are only weakly hemolytic and suggest that ALO production is relatively low [24]. However, later studies show that ALO is produced at biologically relevant amounts in culture and during infection [22; 23; 25]. Additionally, anti-ALO monoclonal antibodies have been shown to protect against B. anthracis infection in mice [26]. In this study, we used several models to more clearly define the role of ALO during infection. We showed that purified ALO alters the permeability of intestinal tissues using an ex vivo mouse intestinal loop model. Additionally, we found that B. anthracis infection of human Caco-2 brush border expressor (C2BBE) cells specifically alters tight junction architecture and allows the movement of anthrax bacteria across the epithelial barrier in an ALO-dependent manner. These results highlight the potential of ALO to influence the development of anthrax infection within the GI tract.
Methods and Materials
Bacteria, cell culture, and ALO production
C2BBE cells, a subclone of human colonic adenocarcinoma Caco-2 cells, were used at passages 55–80 (33). The cells were cultured in high glucose Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, 4 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10 μg/ml human transferrin (all from Invitrogen, Carlsbad, CA). On the day of experimentation, all B. anthracis strains were sub-cultured into fresh Brain Heart Infusion media (BHI) with appropriate antibiotic for ~6 hours at 37°C until the OD ≈ 0.6. B. anthracis Sterne strain 7702 was grown in antibiotic free media while UT231 (50 μg/ml Kanamycin), BJH250 (50 μg/ml Spectinomycin and Erythromycin) and BJH258 (50 μg/ml Spectinomycin, Kanamycin, and Erythromycin) were grown in the presence of indicated antibiotics. Cells were centrifuged and media was exchanged with C2BBE cell culture media without antibiotics. The recombinant ALO from E. coli was made as previously reported [9].
Ex vivo Intestinal Permeability Assay
To assess intestinal permeability, we utilized a mouse ex vivo intestinal loop model [27]. C57Bl/6 female mice were sacrificed with CO2 followed by cervical dislocation. Positive control mice were interperitoneally injected with 100μg/mouse LPS to stimulate intestinal barrier dysfunction. The abdomen was surgically opened and intestines removed. Two ~2 centimeter longitudinal jejunal segments were cut and each segment lumen was gently rinsed with PBS. One end of the segment was sutured and ~200μl of 1mM FITC-dextran (Invitrogen, Carlsbad, CA) in PBS supplemented with various concentrations of purified ALO (10–250μg/ml) was pipetted into the lumen. The open end of the segment was then tied off with suture to close the loop. Segments were transferred into wells containing PBS and incubated at 37°C for the indicated time. Extra intestinal PBS was sampled at 75 minutes after treatment, which was within the window during which untreated intestinal segments remained intact. The amount of FITC-dextran that transverse the intestine was quantified by fluorescence plate reader at 521nm. Fluorescence was normalized for the length of intestinal segment.
Cell Monolayer Permeability Quantification
C2BBE cells were seeded on 0.4-μm pore polyethylene terephthalate transwells (Corning Costar, Lowell, MA) at 5×104 cells per well, and transepithelial resistance (TER) was monitored using a Millicell-ERS (Millipore, Medford, MA). Monolayers were grown between 10–14 days to produce differentiated, confluent monolayers. Two hours prior to infection, the medium was changed to serum-free medium. Cell monolayers were then apically infected with various B. anthracis strains at a multiplicity of infection (MOI) between 1–1000 bacteria per epithelial cell. The TER and cross monolayer dextran flux were monitored after 21 hours of infection. For the dextran flux assays, 10μg/ml of a 3-kDa fluorescein-labeled dextran (Invitrogen, Carlsbad, CA) was added to the apical transwell chamber during infection. Basolateral samples were taken at various time points, and fluorescence was quantified on a Safire2 fluorometer (Tecan, Research Triangle Park, NC).
Immunofluorescence Microscopy
C2BBE cells were grown within 0.4 μm pore transwell cell culture inserts and infected as previously stated. The infected transwells were washed once in ice-cold HBSS, drained, and fixed in −20°C methanol overnight. Transwells were then fixed with 1X PBS plus 0.1% n-octyl-β-D-glucopyranoside and 1mM CaCl2 (named PBS+) in the presence of 0.1mg/ml bis-(sulfosuccinimidyl) suberate. Fixative was quenched by adding 100mM ethylenediamine. Transwells were washed 3 times with PBS+ and blocked with 1% non-fat powdered milk, 2% teleostean fish gelatin, and 1% BSA in PBS+. Transwells were then incubated for 3 hours with primary antibody, washed with blocking buffer, incubated for 1hr with secondary antibody, and washed again with blocking buffer. Both primary mouse anti-occludin, mouse anti-E-cadherin, or rabbit anti-ZO-1 (1:50) and secondary anti-mouse conjugated Cy5 or anti-rabbit Cy3 (1:1000) antibodies were diluted in blocking buffer (all from Invitrogen, Carlsbad, CA). Membranes were cut from the transwell and mounted with Prolong Gold (Invitrogen, Carlsbad, CA). Slides were visualized by confocal microscopy. The images were processed using Image J and are maximum intensity projections of confocal Z-stacks, which covered the range of fluorescence within the treated and untreated C2BBE cells.
Quantification of Bacterial Crossing of Epithelial Monolayers
C2BBE cells were seeded on 3.0μm pore polyethylene terephthalate transwells (Corning Costar, Lowell, MA) at 5×104 cells per well, and transepithelial resistance was monitored using a Millicell-ERS (Millipore, Medford, MA). Cells were selected for experiments when specific transepithelial resistance windows were reached: between 300–400Ω and >400Ω. Two hours prior to treatment, the medium was changed to serum-free medium. Cell monolayers were then apically infected with various B. anthracis strain at a MOI of 1000 bacteria per epithelial cell. The TER and basolateral chamber bacteria were monitored after 21 hours of infection. The number of bacteria was quantified in basolateral media samples by dilution plating on BHI agar plates.
Results
ALO disruption of mouse intestinal barrier function ex vivo
We have previously shown that purified ALO can cause epithelial barrier disruption of C2BBE human gut epithelial cells. To address whether ALO can disrupt the barrier function of GI tissues, we used an ex vivo mouse intestinal loop model to examine the effect of ALO on the jejunum of C57Bl/6 mice (Figure 1) [27]. After ALO treatment for 75 minutes, we assayed whether intralumenal 3kD FITC-dextran was able to translocate across the intestine into the extralumenal space. This treatment window was chosen for the minimal leakage of intralumenal FITC-dextran of the untreated control segments. As a positive control, mice were interperitoneally injected with lipopolysaccharide (LPS; 100μg/mouse) 2 hours prior to sacrifice, which is known to increase the transepithelial permeability to FITC-dextran (Figure 1). ALO stimulated increased permeability to FITC-dextran in a dose-dependent manner (Figure 1). From these, we concluded that ALO can cause barrier dysfunction of mouse GI tissues.
Figure 1. ALO treatment alters mouse intestine barrier function in ex vivo segments of mouse jejunum.
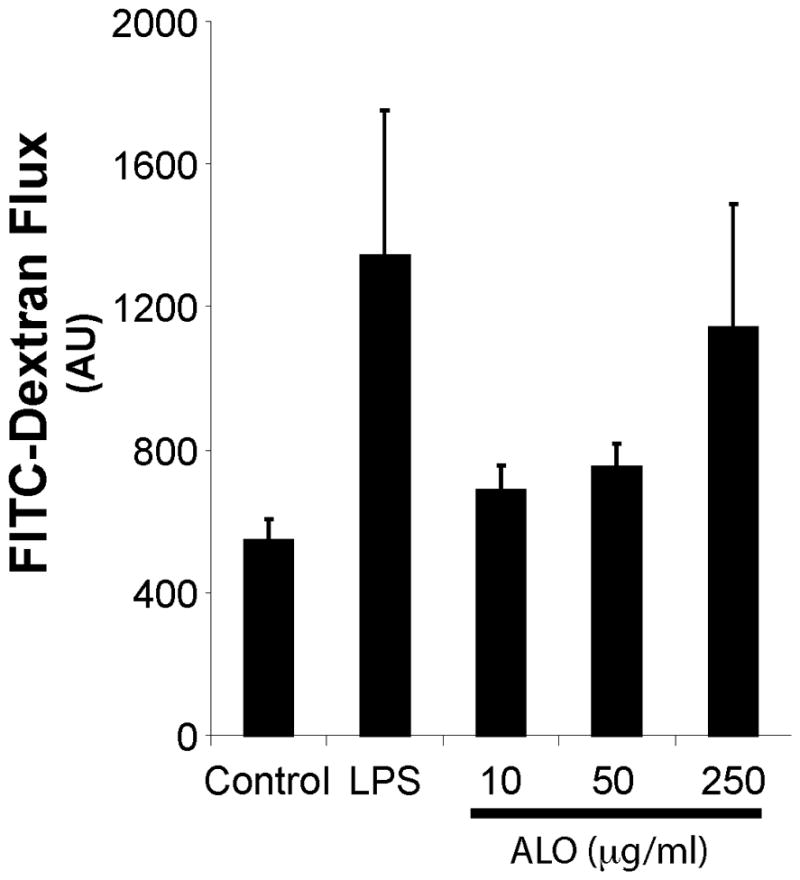
FITC-dextran flux from the lumen of the intestine to the extra intestinal PBS after 75min of the treatment of indicated ALO concentrations or the 2 hour interperitoneal pretreatment of LPS was measured.
B. anthracis induced C2BBE monolayer dysfunction
Our results showed that purified E. coli produced recombinant ALO can lead to GI barrier disruption in human C2BBE cells and in ex vivo mouse intestinal segments. However, it is unclear whether ALO is necessary to cause epithelial barrier disruption during bacterial infection. Therefore, we then examined whether B. anthracis infection will disrupt epithelial barrier function in an ALO-dependent manner. We infected C2BBE epithelial monolayers for up to 21 hours with the mid-log phase growth Sterne strain 7702 and monitored the monolayer permeability to FITC-dextran and transepithelial electrical resistance (TER). We found that vegetative anthrax bacteria can cause increased permeability to FITC-dextran and decreased TER, indicative of epithelial barrier disruption (Figure 2A, B). Such disruption is more severe with increased MOI. Interestingly, we also found that the ALO deficient mutant UT231 failed to cause epithelial barrier disruption (Figure 2C, D). B. anthracis secretes three phospholipases, which are known to mediate ALO activity [14]. Therefore, we assayed the effect of the deletion of these three phospholipases in the presence or absence of ALO gene disruption (Figure 2C, D). We found that BJH250 which is defective in all three phospholipases was still capable of disrupting the barrier function while the ability of phospholipase/ALO knockout mutant (BJH258) to alter monolayer permeability to FITC-dextran and TER was similar to UT231 (Figure 2C, D). Our data indicate that ALO is necessary to cause gut epithelial barrier disruption.
Figure 2. B. anthracis infection produces C2BBE monolayer dysfunction.

(A) Apical to basolateral movement of FITC-dextran and (B) decrease in TER after infection of polarized C2BBE cells with B. anthracis Sterne strain 7702 for 21 hours. The change of (C) FITC-dextran flux and (D) TER after the infection of C2BBE monolayer with three mutant strains of Sterne 7702 (UT231 = ΔALO; BJH250 = ΔPhospholipases; BJH258 = ΔALO ΔPhospholipases).
Monolayer dysfunction correlates to tight junction protein translocation
Epithelial paracellular permeability is controlled by multi-protein complexes, including the tight and adherens junctions. To address the integrity of these two junctions, we used immunofluorescence microscopy to examine the location of the transmembrane protein, occludin, and the membrane associated protein, ZO-1, in the tight junction as well as E-cadherin in the adherens junction. As expected, the staining of these three proteins results in the typical cobblestone appearance of functional tight and adherens junctions (Fig. 3A. D, G). We also found that the infection of B. anthracis Sterne strain 7702 specifically induced the remodeling of the peripheral localization of occludin to a punctate staining within C2BBE cells, indicative of tight junction dysfunction (Figure 3B). Interestingly, the ALO-deficient mutant did not cause such alteration. On the other hand, the infection of Sterne strain 7702 only mildly affects the localization of ZO-1 with the slight enrichment at multicellular junctions (i.e. the convergence of three or more cells onto one point) and the thinning of bicellular junctions (Figure 3E). Again, ALO-deficient strain UT231 had no effect. The adherens junction protein E-cadherin did not change during the infection by all three strains of B. anthracis (Fig. 4G, H, I), pointing to a specificity of B. anthracis to alter the tight junction alone. Our data indicate that ALO is necessary to cause translocation of occludin.
Figure 3. ALO-dependent occludin rearrangement during B. anthracis infection.

C2BBE monolayers were either left uninfected (A–C) or infected with strain 7702 (D–F) or UT231 (G–I). Localization of occludin (A, D, G), ZO-1 (B, E, H), or E-cadherin (C, F, I) is shown as maximum intensity projections of confocal Z-stacks.
Figure 4. Transepithelial resistance dependent bacterial crossing of C2BBE monolayers.

C2BBE monolayers were infected with either B. anthracis strain 7702 or the ALO-negative UT231 at 1000:1 MOI for 21 hours. Movement of bacteria from the apical transwell chamber to the basolateral chamber was calculated by dilution plating of the basolateral media. This is a representative of 4 experiments.
Cross monolayer movement of B. anthracis
As we showed that B. anthracis infection of epithelial cells resulted in a distinct disruption of the epithelial barrier, we then examined whether such disruption could lead to the movement of vegetative bacteria across the C2BBE monolayer by monitoring the number of B. anthracis within the media in basolateral transwell. We first found little or no bacteria in the basolateral transwell after allowing C2BBE cells to fully form their tight junctions (14 days post-seeding, 400–600Ω TER). We then tested whether B. anthracis selectively target monolayers with reduced TERs. This decision was based on our finding that purified ALO appears to target areas of intestinal tissue renewal, such as the villus tips, where epithelial shedding and newly formed tight junctions are prevalent (data not shown). To do so, we assayed the ability of infection to lead to bacterial crossing at two different basal TER ranges from between 300–400Ω and >400Ω. In monolayers with TER between 300–400Ω, 21 hours of infection led to approximately 0.04% (~6.7×104 colony forming units (CFU)) of the initial inoculum to cross from the apical chamber into the basolateral chamber (Figure 4). When a monolayer’s basal TER increased above 400Ω, 200-fold less bacteria were found in the basolateral chamber, which indicates a TER dependence of bacterial crossing. When infected with the ALO deficient strain UT231, no bacteria was seen to cross either 300–400Ω or ≥400Ω monolayers. Thus, B. anthracis strains which produce ALO can more readily cross a monolayer with a lower TER, suggesting that the barrier function of intestinal epithelial cells is important for protection from anthrax infection.
Discussion
Our experiments for the first time show that B. anthracis infection leads to gut epithelial barrier disruption of human C2BBE cells through specific rearrangement of tight junctions and allows anthrax bacteria to pass across the monolayer. We also found that both tight junction disruption and bacterial crossing are ALO-dependent as strains incapable of producing ALO fail to cause the epithelial barrier disruption and are unable to initiate cross monolayer movement of bacteria. Together with our previous finding on the effect of purified ALO on epithelial barrier disruption [9], they establish how the colonization of vegetative anthrax bacteria in the intestinal tract could use ALO to selectively disrupt the paracellular junctions between gut epithelial cells. This opens a pathway for vegetative B. anthracis bacteria to leave the intestinal lumen and enter into the underlying lymphatics, including the Peyer’s patches.
The intestine is a complex organ that has various cell types, differentiation states, and biological functions in each segment. Thus, wide ranges of TER are observed throughout the intestine which are dependent upon the location and physiological state of the tissue [28]. We found that C2BBE monolayers with lower initial TERs between 300–400Ω were required for the passage of anthrax bacteria, lower than the maximal TERs that can be achieved [9; 29; 30]. While it is difficult to correlate the meaning of TER of immortalized C2BBE cells with that of the intestinal epithelial tissues, our data suggests that B. anthracis might preferentially target less differentiated or newly regenerated epithelial cells. This preferential targeting might happen at a segment of GI tract that has lower TER, such as the upper small intestine. Alternatively, such targeting may occur at villus tips where epithelial shedding and other cellular damage occurs or within the villus crypts where tight junctions are less well formed. We also found that high MOI (1000 bacteria to 1 epithelial cell) and a long incubation time (~21 hours) are required to produce these observed alterations. This may in part explain why GI anthrax infection requires high doses of anthrax spores and can be facilitated by experimental manipulations that cause damage in GI tract [1; 7].
The limiting factor for bacterial induced monolayer disruption could be the amount of ALO produced by the bacteria. It is worth noting that other B. anthracis secreted factors, such as the protease InhA which mediates blood coagulation, only function when local concentrations reach threshold levels and require compact spatial localization of the bacteria [31]. Such requirements could be the case for ALO, as sufficiently high titers or spatially constrained vegetative bacteria would lead to high local concentrations of ALO. This localized ALO would affect individual or groups of GI epithelial cells and open gaps within the barrier for the bacteria to cross. It is also possible that accessory secreted proteins are required for optimal ALO function within infection. Future work addressing the complex protein interactions with ALO during infection is needed to define ALO within B. anthracis infection of the GI tract.
Our work also updates the action of ALO and other CDCs. Most reports indicate an intracellular site for CDC activity exemplified by LLO. Previous reports on ALO have indicated similar intracellular activity, which is required for B. anthracis escape from intracellular vesicles within macrophages [14]. Our report is the first to link exogenously produced ALO to specific alterations of gut epithelium. Therefore, this work expands both the potential locations at which ALO can function as well as the potential cell types that ALO can target. Other homologous CDCs can potentially work in a similar manner. For example, pneumolysin is known to disrupt tight junction of lung epithelial cells [11; 17]. Research that specifically addresses the similarity and difference between ALO and other CDC-induced barrier cell dysfunction is needed to address the role as an extracellular toxin.
In conclusion, we have determined that B. anthracis infection targets epithelial cells as portals to bypass the epithelial barrier and that this movement is dependent on the expression of ALO. This work not only shows a role for ALO during the complex interplay between bacterium and host cell within infection, but also expands research on CDCs to include an extracellular function. When put together, this work potentially explains the requirement for high spore doses or other ways to disrupt the intestinal mucosa in the known in vivo models for gastrointestinal anthrax. Future work highlighting in vivo consequences of the expression of ALO are required to fully understand the role of ALO during GI anthrax infection.
Acknowledgments
We thank Dr. Teresa Koehler in providing B. anthracis Sterne strains 7702 and UT231, and Dr. Phil Hanna in supplying the B. anthracis strains BJH250 and BJH258. We also thank Dr. Eugene Chang, Dr. Mark Musch, and Jon Chang for their assistance with developing and implementing the ex vivo intestinal experiments. We thank Dr. Yang-Xin Fu for mice sharing. This work was supported by NIH GM62548, and AI66503, pilot grants from Digestive Disease Research Core Center of the University of Chicago (NIH DK42086) to WJT.
References
- 1.Beatty ME, Ashford DA, Griffin PM, Tauxe RV, Sobel J. Gastrointestinal anthrax: review of the literature. Arch Intern Med. 2003;163:2527–31. doi: 10.1001/archinte.163.20.2527. [DOI] [PubMed] [Google Scholar]
- 2.Dixon TC, Meselson M, Guillemin J, Hanna PC. Anthrax. N Engl J Med. 1999;341:815–26. doi: 10.1056/NEJM199909093411107. [DOI] [PubMed] [Google Scholar]
- 3.Hudson MJ, Beyer W, Böhm R, Fasanella A, Garofolo G, Golinski R, Goossens PL, Hahn U, Hallis B, King A, Mock M, Montecucco C, Ozin A, Tonello F, Kaufmann SHE. Bacillus anthracis: balancing innocent research with dual-use potential. Int J Med Microbiol. 2008;298:345–64. doi: 10.1016/j.ijmm.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inglesby TV, O’Toole T, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Friedlander AM, Gerberding J, Hauer J, Hughes J, McDade J, Osterholm MT, Parker G, Perl TM, Russell PK, Tonat K. Anthrax as a biological weapon, 2002: updated recommendations for management. Jama. 2002;287:2236–52. doi: 10.1001/jama.287.17.2236. [DOI] [PubMed] [Google Scholar]
- 5.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 6.Mitic LL, Anderson JM. Molecular architecture of tight junctions. Annu Rev Physiol. 1998;60:121–42. doi: 10.1146/annurev.physiol.60.1.121. [DOI] [PubMed] [Google Scholar]
- 7.Glomski IJ, Piris-Gimenez A, Huerre M, Mock M, Goossens PL. Primary involvement of pharynx and peyer’s patch in inhalational and intestinal anthrax. PLoS Pathog. 2007;3:e76. doi: 10.1371/journal.ppat.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chitlaru T, Gat O, Grosfeld H, Inbar I, Gozlan Y, Shafferman A. Identification of in vivo-expressed immunogenic proteins by serological proteome analysis of the Bacillus anthracis secretome. Infect Immun. 2007;75:2841–52. doi: 10.1128/IAI.02029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bourdeau RW, Malito E, Chenal A, Bishop BL, Musch MW, Villereal ML, Chang EB, Mosser EM, Rest RF, Tang WJ. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J Biol Chem. 2009;284:14645–56. doi: 10.1074/jbc.M807631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palmer M. The family of thiol-activated, cholesterol-binding cytolysins. Toxicon. 2001;39:1681–9. doi: 10.1016/s0041-0101(01)00155-6. [DOI] [PubMed] [Google Scholar]
- 11.Jedrzejas MJ. Unveiling molecular mechanisms of bacterial surface proteins: Streptococcus pneumoniae as a model organism for structural studies. Cell Mol Life Sci. 2007;64:2799–822. doi: 10.1007/s00018-007-7125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schnupf P, Portnoy DA. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 2007;9:1176–87. doi: 10.1016/j.micinf.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien DK, Melville SB. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect Immun. 2004;72:5204–15. doi: 10.1128/IAI.72.9.5204-5215.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heffernan BJ, Thomason B, Herring-Palmer A, Hanna P. Bacillus anthracis anthrolysin O and three phospholipases C are functionally redundant in a murine model of inhalation anthrax. FEMS Microbiol Lett. 2007;271:98–105. doi: 10.1111/j.1574-6968.2007.00713.x. [DOI] [PubMed] [Google Scholar]
- 15.Gekara NO, Westphal K, Ma B, Rohde M, Groebe L, Weiss S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell Microbiol. 2007;9:2008–21. doi: 10.1111/j.1462-5822.2007.00932.x. [DOI] [PubMed] [Google Scholar]
- 16.Iliev AI, Djannatian JR, Nau R, Mitchell TJ, Wouters FS. Cholesterol-dependent actin remodeling via RhoA and Rac1 activation by the Streptococcus pneumoniae toxin pneumolysin. Proc Natl Acad Sci U S A. 2007;104:2897–902. doi: 10.1073/pnas.0608213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rayner CF, Jackson AD, Rutman A, Dewar A, Mitchell TJ, Andrew PW, Cole PJ, Wilson R. Interaction of pneumolysin-sufficient and -deficient isogenic variants of Streptococcus pneumoniae with human respiratory mucosa. Infect Immun. 1995;63:442–7. doi: 10.1128/iai.63.2.442-447.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamon MA, Batsché E, Régnault B, Tham TN, Seveau S, Muchardt C, Cossart P. Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci USA. 2007;104:13467–72. doi: 10.1073/pnas.0702729104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JM, Ng VH, Maeda S, Rest RF, Karin M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J Exp Med. 2004;200:1647–55. doi: 10.1084/jem.20041215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srivastava A, Henneke P, Visintin A, Morse SC, Martin V, Watkins C, Paton JC, Wessels MR, Golenbock DT, Malley R. The apoptotic response to pneumolysin is Toll-like receptor 4 dependent and protects against pneumococcal disease. Infect Immun. 2005;73:6479–87. doi: 10.1128/IAI.73.10.6479-6487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuchiya K, Kawamura I, Takahashi A, Nomura T, Kohda C, Mitsuyama M. Listeriolysin O-induced membrane permeation mediates persistent interleukin-6 production in Caco-2 cells during Listeria monocytogenes infection in vitro. Infect Immun. 2005;73:3869–77. doi: 10.1128/IAI.73.7.3869-3877.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosser EM, Rest RF. The Bacillus anthracis cholesterol-dependent cytolysin, Anthrolysin O, kills human neutrophils, monocytes and macrophages. BMC Microbiol. 2006;6:56. doi: 10.1186/1471-2180-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shannon JG, Ross CL, Koehler TM, Rest RF. Characterization of anthrolysin O, the Bacillus anthracis cholesterol-dependent cytolysin. Infect Immun. 2003;71:3183–9. doi: 10.1128/IAI.71.6.3183-3189.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drobniewski FA. Bacillus cereus and related species. Clin Microbiol Rev. 1993;6:324–38. doi: 10.1128/cmr.6.4.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross CL, Koehler TM. plcR papR-independent expression of anthrolysin O by Bacillus anthracis. J Bacteriol. 2006;188:7823–9. doi: 10.1128/JB.00525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakouzi A, Rivera J, Rest RF, Casadevall A. Passive administration of monoclonal antibodies to anthrolysin O prolong survival in mice lethally infected with Bacillus anthracis. BMC Microbiol. 2008;8:159. doi: 10.1186/1471-2180-8-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujiya M, Musch MW, Nakagawa Y, Hu S, Alverdy J, Kohgo Y, Schneewind O, Jabri B, Chang EB. The Bacillus subtilis quorum-sensing molecule CSF contributes to intestinal homeostasis via OCTN2, a host cell membrane transporter. Cell Host Microbe. 2007;1:299–308. doi: 10.1016/j.chom.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Madara JL, Trier JS. The functional morphology of the mucosa of the small intestine. In: Johnson LR, editor. Physiology of the gastrointestinal tract. Raven; New York: 1994. pp. 1577–1622. [Google Scholar]
- 29.Turner JR, Angle JM, Black ED, Joyal JL, Sacks DB, Madara JL. PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC kinase. Am J Physiol. 1999;277:C554–62. doi: 10.1152/ajpcell.1999.277.3.C554. [DOI] [PubMed] [Google Scholar]
- 30.Bookstein C, Musch MW, Xie Y, Rao MC, Chang EB. Regulation of intestinal epithelial brush border Na(+)/H(+) exchanger isoforms, NHE2 and NHE3, in C2bbe cells. J Membr Biol. 1999;171:87–95. doi: 10.1007/s002329900561. [DOI] [PubMed] [Google Scholar]
- 31.Kastrup CJ, Boedicker JQ, Pomerantsev AP, Moayeri M, Bian Y, Pompano RR, Kline TR, Sylvestre P, Shen F, Leppla SH, Tang WJ, Ismagilov RF. Spatial localization of bacteria controls coagulation of human blood by ‘quorum acting’. Nat Chem Biol. 2008;4:742–50. doi: 10.1038/nchembio.124. [DOI] [PMC free article] [PubMed] [Google Scholar]