

Abstract
Leukocyte capture on inflamed endothelium is facilitated by a shift in LFA-1 from low to high affinity that supports binding to ICAM-1. LFA-1 bonds help anchor polymorphonuclear leukocytes (PMN) to inflamed endothelium in shear flow, and their redistribution to the leading edge guides pseudopod formation, migration, and extravasation. These events can be disrupted at the plasma membrane by stabilizing LFA-1 in a low- or intermediate-affinity state with allosteric small molecules. We hypothesized that a minimum dimeric bond formation between high-affinity LFA-1 and ICAM-1 under shear stress is necessary to catalyze transmembrane signaling of directed cell migration. Microspheres and substrates were derivatized with monomeric or dimeric ICAM-1 to simulate the surface of inflamed endothelium under defined ligand valence. Binding to dimeric ICAM-1, and not monomeric ICAM-1, was sufficient to elicit assembly of F-actin and phosphorylation of Src family kinases that colocalized with LFA-1 on adherent PMN. Genetic deletion or small molecule inhibition of Src family kinases disrupted their association with LFA-1 that correlated with diminished polarization of arrested PMN and abrogation of transmigration on inflamed endothelium. We conclude that dimeric bond clusters of LFA-1/ICAM-1 provide a key outside-in signal for orienting cytoskeletal dynamics that direct PMN extravasation at sites of inflammation.
Neutrophil recruitment to inflamed vascular endothelium is initiated by selectins that facilitate cell rolling and activation of β2 integrins (CD11/CD18) that mediate stable adhesion and migration across the blood vessel wall (1). Critical to a transition from cell rolling to arrest is a shift in β2 integrins (CD18) from a low- to high-affinity state primed for binding ICAM-1, which is up-regulated on endothelium in response to cytokine stimulation (2). A step following arrest is adhesion strengthening and contact-mediated guidance that correlates with membrane redistribution of LFA-1 into high-density clusters concentrated at the uropod and lamellipod along the major axis of a polarizing neutrophil (3–5). Supporting the hypothesis that LFA-1 participates in guidance of motility is the observation that allosterically locking LFA-1 in an intermediate- or low-affinity conformation with small molecules abrogated neutrophil polarization and transendothelial migration (4).
Published data also support the notion that homodimerization of ICAM-1 is a dynamic event on the membrane of inflamed endothelium that can serve to increase leukocyte adhesion efficiency and transmigration (6 – 8). We have reported that high-affinity LFA-1 will remain bound 10-fold longer to dimeric ICAM-1 than to monomeric ICAM-1 (9). Moreover, neutrophil capture of beads expressing dimer in sheared suspension was 100% more efficient than capture of monomeric ICAM-1 beads, thereby demonstrating a pivotal role of bond valence in adhesion stability under shear flow. For more than a decade it has been known that native ICAM-1 up-regulated on inflamed endothelium exists in equilibrium between the monomeric and dimeric states (10, 11). More recent data suggest that structural rearrangement of the Ig domain supports a transition to dimeric ICAM-1 that may optimally orient the D1 binding site for adhesion via leukocyte LFA-1 (12). Taken together, these data suggest that both LFA-1 conformation and valence in binding ICAM-1 under shear stress can dramatically alter the adhesive dynamics. However, it remains ill-defined how binding to dimeric ICAM-1 regulates the adhesive and migratory phenotype of polymorphonuclear leukocytes (PMN)3 beyond prolonging bond lifetime.
Induction and stability of high-affinity LFA-1 requires ligation of ICAM-1, suggesting a causal link between stable adhesion and the cell’s activation status, including F-actin polymerization (13, 14). For instance, chemokine activation of PMN can rapidly induce a shift in LFA-1 affinity that is mediated by linkage to the cytoskeleton (4, 5). Releasing LFA-1 from its actin cytoskeletal restraints promotes a conformational shift in LFA-1 to a high-affinity state and leads to directed cell movement (15). Membrane-associated nonreceptor Src family kinases (SFK) can directly interact with the integrin cytoplasmic tail and regulate integrin affinity and outside-in signaling, which together link a variety of extracellular cues to cytoskeletal functions such as cell spreading (16, 17). For instance, the SFK member Fyn rapidly recruits to sites of LFA-1 binding and lymphocyte migration on ICAM-1 (18, 19). More recently, it was shown that another member, Syk, is implicated in outside-in signaling through integrins (20, 21). Slow rolling on E-selectin and ICAM-1 was abolished by blockade of Syk and was absent in Syk−/− bone marrow chimeric mice, indicating an important role of Syk in mediating downstream signaling leading to actin polarization (20, 22). Furthermore, co-clustering of high-affinity CD18 and F-actin at the site of PMN homotypic aggregation is blocked by inhibitors of SFK activity (23). Taken together, these results demonstrate that SFK regulate conformational shifts and increased affinity of LFA-1 that are accompanied by F-actin polymerization and leukocyte motility. However, the role of SFK phosphorylation in LFA-1/ICAM-1-mediated neutrophil arrest and migration on inflamed endothelium have not been established.
In the present study, we tested the hypothesis that PMN adhesion to surfaces bearing dimeric ICAM-1 provides a critical outside-in signal via high-affinity LFA-1 that results in F-actin formation, macromolecular assembly with phospho-SFK, and directed cell migration. Inhibition or genetic deletion of SFK disrupted the redistribution of high-affinity LFA-1 along the major axis of polarized PMN, a requisite step for migration and diapedesis on inflamed endothelium in shear flow. We conclude that the cooperative tuning of LFA-1 affinity on the PMN and ICAM-1 valence on the endothelium serves to spatially coordinate and integrate signals from selectins, chemokines, and integrins as they are ligated on a decelerating PMN at vascular sites of inflammation.
Materials and Methods
Abs, small molecules, and reagents
Abs were used at 10 μg/ml or per manufacturer’s suggestion. Reagents used were nonblocking mAb CL23.4, which was a kind gift from the Luscinskas Laboratory (Brigham and Women’s Hospital, Boston, MA), anti-ICAM-1 (BBIG-I1; R&D Systems), anti-Mac-1 (2LPM19c, Dako), goat anti-rabbit IgG fluorescein (Antibodies Incorporated), anti-IL-8 (BioSource International), Ab to phospho-Src family (Tyr416) (Cell Signaling Technology), normal rabbit IgG (Upstate Biotechnology), rat anti-mouse CD11b (5C6, Serotec), anti-CD45 Ab, rhodamine-conjugated donkey anti-rabbit IgG secondary Ab (Invitrogen), small-molecule lovastatin (Calbiochem), recombinant human IL-8, IL-1β, monomeric ICAM-1 (R&D Systems), dimeric ICAM-1/Ig, anti-LFA-1 (TS2/4), anti-CD18 (240Q Fab), and anti-CD18 (327C) (ICOS), Alexa Fluor 488 labeling kit, fluorescein phalloidin (Molecular Probes), Src kinase inhibitor II (EMD Biosciences), and MIP-1α(PeproTech).
ICAM-1 bead assembly
Carboxylate microspheres (diameter of 10 μm) were purchased from Polysciences. Five hundred microliters of beads was washed twice in 1.5 ml of MES buffer (pH 5.0) (Sigma-Aldrich), resuspended in 200 μl of MES, and sonicated for 15 min. 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide, hydrochloride (Molecular Probes) was added at 1 mM, and beads were incubated for 5 min at room temperature and centrifuged at 500 rpm. Monomeric ICAM-1 (172.4 μg/ml) or dimeric ICAM-1/Ig (86.2 μg/ml) was mixed with the beads for 1 h at room temperature at 500 rpm, except for beads that also had anti-IL-8, where both the anti-IL-8 (20 μg/ml) and ICAM-1 were coincubated. Glycine (Sigma-Aldrich) was added (10 mM) and beads were mixed for 30 min at room temperature at 500 rpm. ICAM-1 beads were washed in PBS and resuspended in 1.5 ml of PBS. Monomer- and dimer-coated beads (150 μl) were added to an equivalent volume of Coomassie reagent (Pierce). Samples were vortexed for 30 s and incubated for 10 min at room temperature. Absorbance was measured at or near 595 nm on a NanoDrop ND-1000 spectrophotometer and compared with BSA standard to obtain baseline. Samples were then washed with 5% trypsin-EDTA 10× (Invitrogen) and incubated at room temperature to detect bound ICAM-1 concentration. Samples were centrifuged at 500 rpm, and supernatant was stained with Coomassie reagent and analyzed by a NanoDrop ND-1000 spectrophotometer. Basal trypsin levels were subtracted from ICAM-1 concentrations and the average concentrations of monomeric and dimeric ICAM-1 were found to be 54.35 and 63.43 μg/ml, respectively. This confirmed approximately equal site densities of ICAM-1 on the beads.
Site density on beads was determined by comparison with bead standards containing a known number of binding sites (Quantum Simply Cellular beads, Bangs Laboratories) and prescribed to be ~6000 sites/μm2 as identified by anti-CD54 and analyzed on a FACScan flow cytometer. This is consistent with the ICAM-1 site density observed on activated endothelium (24). Beads with anti-IL-8 were incubated with 50 nM IL-8 for 15 min and then washed immediately before use.
Human PMN isolation
Whole blood was drawn from healthy subjects by venipuncture into sterile syringes with heparin (10 U/ml blood, Elkins-sinn) as described previously (4). PMN were isolated from whole blood using a density gradient media (Matrix/Thermo Scientific). PMN were washed once with HEPES buffer (10 mM KCl, 110 mM NaCl, 10 mM glucose, 1 mM MgCl2, and 30 mM HEPES (pH 7.4)) and were maintained at room temperature in HEPES buffer, human serum albumin (HSA) (0.1%), and CaCl2 (1.5 mM) for not more than 10 min before use.
Bead/PMN assay of F-actin content
PMN suspensions were prepared in HEPES buffer (with 0.1% HSA and 1.5 mM CaCl2). To focus this study predominantly on LFA-1-mediated adhesion, Mac-1 was blocked with 10 μg/ml 2LPM19c for 15 min at 37°C just before assaying bead binding and activation. In conditions where soluble lovastatin (1 mM), Src kinase inhibitor (10 μM), DMSO, or TS2/4 were used, these reagents were also simultaneously incubated with the 2LPM19c. PMN (105/ml) were then mixed for 2 min with beads (106/ml) coated with either ICAM-1 or Ab. PMN/bead mixtures were then washed and resuspended in 0.1 mg/ml lysophosphatidylcholine (Avanti Polar Lipids) in buffered formalin (Fisher Scientific) for 10 min on ice to fix and permeabilize the cells. Phalloidin-FITC (0.165 μM) or anti-CD18 (327C)-Alexa Fluor 488 (all 10 μg/ml) was mixed with the cells at 37°C for 20 min and then washed and resuspended in HEPES buffer. F-actin content on single aggregates of PMN beads was analyzed on a FACScan flow cytometer (BD Biosciences) by first gating this population on a forward vs side scatter plot and then assessing mean fluorescence intensity (MFI). Statistical analysis was performed using GraphPad Prism software using the two-tailed t test or the Newman-Keuls multiple comparison test.
Mouse PMN isolations
Murine bone marrow PMN were isolated using a one-step 62% density gradient medium as previously described (25). Cells were kept at 4°C in a Ca2+/Mg2+-free HBSS. CaCl2 and MgCl2 were added to the buffer at 1 mM final concentration before analysis. PMN were isolated by dextran sedimentation of RBCs, followed by pelleting in Ficoll-Paque Plus (Amersham).
Soluble ICAM-1 binding to mouse PMN
Mouse PMN were isolated from 10-wk-old male C57BL/6 wild-type and hck−/−fgr−/− lyn−/− knockout mice (26). Experiments were run in HEPES buffer with 1.5 mM CaCl2 and 0.1% HSA at room temperature for not longer than 4 h during the experiment. PMN (2 × 105 cells/100 μl) were incubated with 10 μg/ml rat anti-mouse blocking CD11b Ab for 10 min at 37°C, 450 rpm. Without wash, PMN were either left inactivated or activated (as indicated) with 100 ng MIP-1α and allowed to bind soluble dimeric ICAM-1-Alexa Fluor 488 (10 μg/ml) for 10 min. Samples were read by fluorescence flow cytometry.
PMN shape change on ICAM-1 substrates
Monomeric (75 μg/ml) or dimeric (35 μg/ml) ICAM-1 was titrated to achieve equal coating density by incubation on polystyrene for 2 h at room temperature in TBS (pH 8.5). The substrate was washed with TBS, and then HSA (4%) was incubated in TBS on the substrate overnight to block nonspecific interactions. Substrates were then washed in HHB (0.1% HSA and 1.5 mM CaCl2) and kept hydrated until use. Neutrophils were allowed to settle for 1 min while images were taken every second. IL-8 (10 nM) was then added and lightly mixed. Imaging continued for another 6 min after IL-8 addition. Images of adhered neutrophils were analyzed with Image-Pro Plus 5.1 (Media Cybernetics). The ratio of the maximum length to minimum width through the PMN centroid was analyzed and plotted.
Immunoprecipitation and Western blotting
Following incubation of PMN (2 × 107) on monomeric or dimeric ICAM-1 for 7 min, the cells were frozen in liquid nitrogen. The cells were then resuspended in 250 μl relaxation buffer (10 mM HEPES (pH 7.4), 100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin) and then sonicated for 6 s. Nuclei, granules, and unbroken cells were removed by centrifugation at 860 × g for 10 min at 4°C. Supernatants were ultracentrifuged at 100,000 × g for 30 min at 4°C to separate cytosol and membrane fractions. Supernatants were collected (cytosol), or pellets (membrane) were resuspended in 80 μl 1% n-octyl-glucoside (Boehringer Mannheim). Total protein in both fractions was quantified with Bio-Rad protein assay reagent using BSA as a standard. Membrane or cytosol fractions were added to 4× sample buffer and boiled for 10 min. Proteins were subjected to SDS-PAGE and Western blotting.
For immunoprecipitation, PMN were solubilized in lysis buffer (1% Nonidet P-40 in 50 mM NaCl, 50 mM Tris-HCl (pH 7.4)) containing 5 mM EDTA, 1 mM NaF, leupeptin (1 μg/ml), aprotinin (10 μg/ml), pepstatin A (1 μg/ml), 2 mM sodium orthovanadate, and PMSF (100 μg/ml). Lysates were vortexed every 5 min and incubated on ice for 30 min and then centrifuged at 19,000 × g at 4°C for 20 min. The lysates were precleared by incubation/gentle rocking with 50 μl protein G-agarose beads (Invitrogen) at 4°C for 30 min. Immunoprecipitations were conducted by adding rat anti-LFA-1 (BioLegend), incubating at 4°C overnight, followed by addition of protein G-agarose beads and further incubation for 90 min. Immune complexes were washed with the same lysis buffer three times, resolved by SDS-PAGE, and transferred onto nitrocellulose membranes. Immunoblots were blocked with 5% milk in PBST (PBS (Sigma-Aldrich) 1 tablet/200 ml, 0.1% Tween 20) for 1 h and then incubated overnight with primary Abs for phospho-SFK (Tyr416) (Cell Signaling Technology) in 5% milk PBST, followed by Alexa Fluor 680-labeled anti-rat or anti-mouse secondary Abs (Molecular Probes). Immunoblots were scanned by using the LI-COR machine system.
Brain endothelial (bEND) cells-PMN adhesion/migration assay
Bone marrow PMN were isolated from hck−/− fgr−/− lyn−/− and strain-matched wild-type mice. To observe PMN interaction with a physiological substrate, we have developed a microfluidic device capable of generating a defined shear stress within leukocyte-containing fluid. We assembled this device on top of a confluent monolayer of bEND.3 endothelial cells stimulated 5 h before the experiment with 300 U/ml murine TNF-α. To promote adhesion, PMN were initially perfused over the monolayer at a concentration of 4 × 106 PMN/ml and allowed to settle for 1 min. Following static adhesion, we perfused PMN through the microfluidic channels at an average wall shear stress of 1 dyne/cm2 and recorded the resulting PMN-endothelial interactions by phase-contrast videomicroscopy for the subsequent 8 min in multiple regions of the bEND.3 monolayer. PMN interacting with the substrate were readily identified by their phase-bright appearance, and arrested and rolling PMN were discriminated as previously described (4). Arrested PMN were classified as “polarized” if the ratio of length to width of the phase-bright cell image was >1.5. PMN undergoing transendothelial migration (TEM) were identified by a transition from phase-bright to phase-dark appearance as the cells spread to the basal side of the endothelial monolayer. All bEND.3 adhesion data are representative of differences between wild-type and knockout mice (n =2) and six experimental runs.
HUVEC-PMN adhesion/migration assay
HUVEC monolayers at passages 5– 6 were grown to confluency over 2–3 days on gelatin-coated coverslips. We assembled a polydimethyl-siloxane microfluidic device (described above) on the HUVEC monolayers stimulated for 4 h with 30 U/ml TNF-α. PMN were incubated at 37°C for 30 min in 1 μM SFK inhibitor or 0.3% DMSO control, and then perfused over the inflamed monolayers at a concentration of 1 × 106/ml and an average wall shear stress of 2 dyne/cm2. Arrest, polarization, and TEM were determined as described above. Adhesion and polarization data are representative of at least four experimental runs for both control and inhibited PMN.
Real-time fluorescent imaging
In certain flow experiments, control and SFK inhibited PMN at a concentration of 1 × 106/ml were prelabeled with 20 μg/ml 327C or TS2/4 directly conjugated to Alexa 546 fluorophore for 10 min at room temperature. To image a robust polarization response, microfluidic devices were assembled atop confluent HUVEC monolayers stimulated with 5 ng/ml IL-1βfor 4 h. Labeled PMN were perfused at 2 dyne/cm2 average wall shear stress over this inflamed monolayer for 3 min, at which time the inlet reservoir was washed with unlabeled HHB with 1.5 mM Ca and 0.1% HSA to reduce background fluorescence. Sequences of bright-field and 546 nM excitation fluorescence were captured from random fields of arrested PMN for 6 min after the initial wash. High-density clusters of 327C or TS2/4 signal were defined as regions of a PMN with 546 nM pixel intensity 3 SDs higher than the mean pixel intensity of the cell. Polarized PMN that expressed a cluster within 1 μm of the uropod and at least one additional distinct cluster >3 μm from the uropod were quantified as having biaxial integrin distribution. Data are representative of two experimental runs for each condition. Experiments were also performed to image ICAM-1 conformation on stimulated and resting HUVEC monolayers. HUVEC grown to confluence were stimulated with IL-1β (5 ng/ml) for 4 h and subsequently labeled with mAb BBIG-I1 (R&D Systems) that recognizes an epitope common to all ICAM-1, followed with a secondary IgG Alexa Fluor 546 conjugate. Another ICAM-1 mAb specific to domain 4 accessible on dimeric ICAM-1 was also applied (clone CL23.4). HUVEC were then fixed with 2% PFA for 15 min, washed with PBS, and immunofluorescence imaging was used to quantify expression levels on cell body and at junctions.
Total internal reflection fluorescence microscope imaging
PMN (5 × 106/ml in 125 μl) in HBSS with Ca2+/Mg2+ (1 mM) were plated on coverslips coated with monomeric (75 μg/ml) or dimeric (15 μg/ml) ICAM-1 at 37°C for 7 min and then fixed in 1% paraformaldehyde for 10 min (at 37°C). The cells were washed twice with HBSS followed by blocking with 10% FCS for 30 min and stained with Alexa Fluor 488-conjugated anti-human LFA-1 (TS2/4) mAb at 4°C in the dark for 1 h. The cells were washed in PBS and permeabilized with PBS containing 5% FCS and 0.2% Triton X-100 (blocking buffer) at room temperature for 30 min. The cells were then stained with anti-phospho-SFK at 4°C in dark for 2 h. Cells were then washed with blocking buffer five times followed by staining with rhodamine-conjugated donkey anti-rabbit IgG secondary Ab for 1 h at 4°C in the dark. The cells were then washed five times with blocking buffer and immediately imaged. Image acquisition was achieved by Zeiss Axiovert 200M with a 100×/1.45 oil objective and a 1.6× Optovar. As controls, secondary Ab staining alone and staining of hck−/−fgr−/−lyn−/− PMN was performed.
Results
Imaging neutrophils arresting and transmigrating on inflamed endothelium reveals that most transmigrate at cell-cell junctions where β2 integrins engage endothelial ICAM-1 (27, 28). ICAM-1 was labeled on unstimulated and IL-1β-stimulated HUVEC using Ab CL23.4 that specifically recognizes ICAM-1 homodimers, and it was compared with mAb BBIG-I1 that recognizes a common epitope on all ICAM-1. A low, albeit significant, level of BBIG-I1 was observed to bind to unstimulated HUVEC as compared with virtually no binding with CL23.4, suggesting that a low density of monomeric ICAM-1 sites dominated the expression (Fig. 1). IL-1β stimulation induced a comparable 3-fold increase in total and homodimeric ICAM-1 on the surface, and ~75% of Abs recognizing either form of ICAM-1 were proximal to intracellular junctions (Fig. 1b). These data support the conclusion that ICAM-1 up-regulated during inflammatory stimulation with IL-1β rapidly forms dimers that concentrate at intracellular junctions.
FIGURE 1.

ICAM-1 expression on resting and cytokine-stimulated endothelium. HUVEC monolayers were grown to confluence and stimulated with IL-1β for 4 h. Immunofluorescence imaging of ICAM-1 expression and distribution was performed with BBIG-I1, a mAb specific to an epitope recognizing monomeric and dimeric ICAM-1, and mAb CL23.4 that reportedly identifies homodimer via recognition of the Ig domain 4. Cells were fixed with 2% PFA for 15 min and imaged by microscopy. a, Representative images of ICAM-1 expressed on stimulated and unstimulated HUVEC (n = 50 cells analyzed). b, ICAM-1 MFI located on endothelial periphery within 1 μm of intercellular junctions and that on remaining cell body. Data are means ± SEM from three independent experiments for observation of at least 50 cells for each condition. **, p < 0.001.
Activation of PMN through LFA-1 binding to ICAM-1
To assess the capacity for adhesive contact-mediated activation of PMN bound to beads, the extent of F-actin polymerization was measured on cell suspensions by flow cytometry and confirmed on single PMN-bead aggregates by immunofluorescence microscopy (Fig. 2). A number of bead chemistries were assembled to establish localized activation of PMN with a specified ICAM-1 valence. The beads were mixed in shear suspensions with PMN while inducing or blocking the LFA-1 high-affinity state. Monomeric or dimeric ICAM-1 was coupled to the bead surface at equivalent site density. In some experiments beads were coated only with mAb TS2/4 to assess the contribution of bivalent ligation of LFA-1 in the absence of ICAM-1. This Ab recognizes the CD11a subunit of LFA-1 and does not block its recognition of domain 1 of ICAM-1 (9, 29). Neutrophils adherent to dimeric ICAM-1 beads in the absence of chemokine stimulation up-regulated F-actin by ~100% compared with those binding to beads presenting TS2/4 or monomeric ICAM-1. This activation was associated with a conformational upshift in LFA-1 affinity as F-actin localization was blocked to baseline levels by addition of lovastatin, which stabilizes LFA-1 in the low-affinity state. Addition of soluble IL-8 to PMN bound to monomeric ICAM-1 or TS2/4 beads increased activation by ~100%, and this effect was augmented for dimeric ICAM-1 beads to a ~200% increase in F-actin (Fig. 2a).
FIGURE 2.

Increase in F-actin expression upon binding of beads coated with ICAM-1. Whole-cell F-actin in response to PMN capture of beads coated with anti-LFA-1 TS2/4, monomeric ICAM-1, or dimeric ICAM-1 as measured by phalloidin-FITC binding and detected by flow cytometry in the presence of (a) soluble IL-8 (5 nM) or lovastatin (1 mM). Data are expressed as MFI from three to five separate experiments. *, p < 0.01 and **, p < 0.001. b, Immunofluorescence images depict, from left to right: phalloidin signal from dimeric ICAM-1 beads; dimeric ICAM-1 coexpressed with IL-8; monomeric ICAM-1; monomeric ICAM-1 with IL-8. c, In the presence of soluble anti-CD18 (240Q Fab, 10 μg/ml) or anti-LFA-1 (TS2/4, 10 μg/ml). *, p < 0.01. Statistical analysis also reported significance in sTS2/4-mediated activation of PMN bound to both dimeric and monomeric ICAM-1 (p < 0.05).
Direct imaging of phalloidin at the PMN-bead interface confirmed that F-actin formation was a proportional response to G protein-coupled receptor (GPCR) and integrin-mediated signaling. PMN were directly activated via CXCR1/2 by coimmobilizing recombinant IL-8 along with ICAM-1 on beads. As shown in the representative images, F-actin was up-regulated to a greater extent on PMNs bound to dimeric as compared with monomeric ICAM-1 beads (Fig. 2b). Ab 240Q binds to the β (CD18) subunit of LFA-1 and allosterically induces a high-affinity conformation for binding ICAM-1 (5, 30, 31). Addition of univalent Fab fragments of 240Q to PMN bound to dimer ICAM-1 beads increased F-actin production by 115% and 134% over those bound to TS2/4 and monomeric ICAM-1 beads, respectively (Fig. 2c). Addition of soluble bivalent TS2/4 to cross-link LFA-1 bound to either monomer or dimer ICAM-1 activated significantly more F-actin than for PMN bound and unstimulated. Taken together, these data suggest that neutrophil adhesion to dimeric ICAM-1 through LFA-1 is not only sufficient to initiate F-actin production, but is synergistic with stimulation via chemokine or allosteric Ab and is necessary to drive maximal cytoskeletal activation.
SFK-mediated signaling
SFK have been shown to function in transduction of outside-in signaling via high-affinity CD18 and inside-out signaling upon ligation of the chemokine receptor CXCR1 (32). We assessed the role of SFK in cytoskeletal activation via LFA-1 when bound to dimeric ICAM-1 beads. PMN were preincubated with Src kinase inhibitor II (a derivative of 2-thioxo-2,3-dihydro-1H-thieno[2,3-d]pyrimidin-4-one) (33) and then incubated with dimeric ICAM-1 or control IgG-coated beads in the presence or absence of coexpressed IL-8 on the bead surface under conditions of fluid shear stress. Pretreatment of PMN with Src kinase inhibitor II decreased F-actin by ~25% in PMN bound to ICAM-1 beads (Fig. 3a). However, activation was recovered by stimulation with IL-8, thereby suggesting a dominant role for SFK in outside-in signaling of cytoskeletal activation following ligation of ICAM-1. This was confirmed by examining the capacity of Src kinase inhibitor to alter activation of CD18 to the high-affinity conformation upon binding ICAM-1 dimer. Binding of the allosteric high-affinity reporter mAb 327C to PMN enabled detection of activation in the presence of beads coated with dimeric ICAM-1 with and without coexpression of IL-8. Stabilization of high-affinity CD18 bound to dimeric ICAM-1 or IL-8 was not significantly decreased by SFK inhibition (Fig. 3b). However, the increased activation co-signaled in the presence of IL-8 and ICAM-1 was inhibited ~70% toward the baseline with ICAM-1 alone (Fig. 3b). Thus, SFK do not appear to be involved in stabilization of high-affinity LFA-1 during inside-out signaling via CXCR, but are required for the synergistic activation that presumably involves outside-in signaling via LFA-1 binding to dimeric ICAM-1.
FIGURE 3.

SFK regulation of CD18 affinity and outside-in signaling. Expression of F-actin and high-affinity CD18 were assayed using flow cytometry of PMN/bead complexes. a, Total cellular F-actin formation after treatment with Src kinase inhibitor II (10 μM) during PMN binding to beads coated with dimeric ICAM-1, IL-8, or beads expressing both dimeric ICAM-1 and IL-8. Data are expressed as the percentage of the phalloidin MFI of DMSO-treated control PMN binding to the respective bead type from three to five separate experiments. **, p < 0.05 for difference from 100%. b, Expression of high-affinity CD18 on DMSO control and SFK inhibitor (10 μM)-treated PMN as detected by mAb 327C. PMN were agitated with beads coated with IgG control, dimeric ICAM-1, and dimeric ICAM-1 and IL-8 during three to five separate experiments. **, p < 0.001. c, Soluble dimeric ICAM-1 was conjugated to Alexa Fluor 488 and its binding to mouse wild-type (WT) and lyn−/− hck−/− fgr−/− (SFK KO) PMN in the presence and absence of MIP-1α (100 ng) was quantified by MFI from three separate experiments. *, p < 0.01 and +, p < 0.05. MIP-1α enhanced ICAM-1 binding in SFK KO with a significance level of p = 0.05.
To confirm whether SFK are indeed involved in conversion and signaling via high-affinity CD18, we assayed the binding of soluble dimeric ICAM-1 on neutrophils isolated from a murine knockout deficient in all three SFK members (hck, fgr, and lyn) (34). The ICAM-1 binding capacity of bone marrow-isolated PMN was compared between wild-type and the triple knockout mouse in the presence of anti-Mac-1 to ensure that LFA-1 activation was assessed. PMN from SFK-deficient mice bound ~50% less ICAM-1 when compared with wild-type mice, suggesting that the PMN isolation procedure alone activated a portion of LFA-1 in a SFK-dependent manner. Addition of MIP-1α, which stimulates via CXCR2, increased ICAM-1 binding by 1-fold in wild-type mice, and this was decreased by 30% in the SFK knockout mice (Fig. 3c). These data confirmed that SFK are necessary for full activation of LFA-1 binding capacity, but that PMN retain the capacity to activate high-affinity LFA-1 binding to ICAM-1 via GPCR signaling in the complete absence of SFK.
We have recently reported that adhesion via high-affinity LFA-1 is a critical step in the process of neutrophil polarization and transmigration on inflamed HUVEC (4). High-affinity LFA-1 was observed to assemble at the uropod and base of newly formed pseudopods during contact-mediated activation that precedes transmigration on inflamed endothelium (4). Here, we examined the role of SFK in the integration of signaling through high-affinity LFA-1 bound to a substrate bearing dimeric ICAM-1. Human PMN became polarized only after addition of soluble IL-8 subsequent to PMN in contact with the ICAM-1 substrate. To examine the role of LFA-1 cross-linking on cytoskeletal activation and the extent of PMN polarization, soluble TS2/4 was added during adhesion on ICAM-1. Cross-linking of LFA-1 with TS2/4 augmented PMN polarization on dimeric as compared with monomeric ICAM-1 substrate (Fig. 4). Pretreatment with Src kinase inhibitor II, but not DMSO alone, significantly reduced the extent of PMN polarization to a baseline value. These data suggest that during contact-mediated activation via dimeric LFA-1/ICAM-1 bonds, SFK are involved in outside-in signaling that leads to PMN polarization.
FIGURE 4.
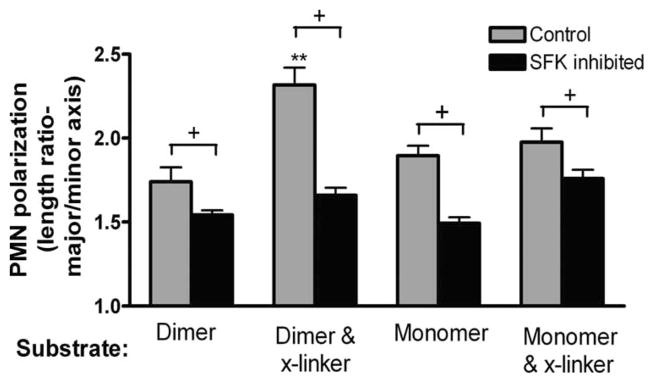
PMN shape change on ICAM-1 substrates. PMN settled on polystyrene coated with dimeric or monomeric ICAM-1. After 1 min, IL-8 was added and the adherent cells were imaged 6 min after stimulation. All PMN were preincubated with Mac-1-blocking Ab 2LPM19c. Indicated samples were treated with an anti-LFA-1 crosslinker (mAb TS2/4) to induce dimerization of LFA-1 and/or treated with Src kinase inhibitor II (10 μM). The average length/width ratio of the PMN was measured based on the visible PMN outline under phase-contrast illumination. +, p < 0.05 and **, p < 0.05 (n = 26–67 cells analyzed per condition from three independent experiments).
Activated SFK and LFA-1 colocalize in macromolecular clusters on ICAM-1
SFK inhibitor effectively suppressed the capacity of LFA-1 to participate in contact-mediated activation and polarization during PMN arrest on dimeric ICAM-1. This would suggest that activated SFK may physically associate with LFA-1 and participate in outside-in signaling. To test this hypothesis, we examined colocalization of LFA-1 with phospho-SFK in the plane of the membrane at sites of adhesive contact with ICAM-1. Substrates of monomeric and dimeric ICAM-1 of equal site density were assembled and adherent PMN were analyzed using total internal reflection fluorescence microscopy. This technique was applied to excite fluorophore-conjugated Abs to LFA-1 and phospho-SFK diffusing on adjacent sides of the plasma membrane and assembling within ~100 nm of the substrate. In this manner, we could detect assembly of LFA-1 clusters and activated SFK in the plane of the membrane bound to a glass slide derivatized with either monomer or dimer ICAM-1. Dimeric ICAM-1 was 75% more effective than monomeric ICAM-1 at initiating macromolecular clusters of LFA-1 on spread PMN (Fig. 5, a and b). Although PMN adhesion was observed on both the monomeric and dimeric ICAM-1 substrates in this static assay, the latter supported a greater extent of LFA-1 activation and PMN polarization. Discrete clusters of activated SFK within the cortical membrane were imaged on PMN spread on dimeric ICAM-1 using an Ab that reports on the phosphorylation of Tyr416, which is in the activation loop of the catalytic domain of Src kinase. These clusters were 3-fold more likely to colocalize with LFA-1 than Ab CD45 and 6-fold more probable than that due to random receptor association in the membrane (Fig. 5, c and d). Moreover, colocalization of Tyr416 and LFA-1 in wild-type PMN was specific, as its signal was absent in PMN isolated from triple SFK knockout mice (data not shown).
FIGURE 5.

LFA-1 clusters are associated with activated SFK. PMN were allowed to settle on ICAM-1-bearing glass coverslips and then fixed, stained, and imaged for LFA-1 and phospho-SFK distribution. a, Representative images of LFA-1 distribution on settled neutrophils were enhanced using gamma adjustment to demonstrate the distribution of high-density clusters. b, The number of clusters per neutrophil was quantified by directly counting the number of distinct regions with fluorescent pixel intensity 2.5 SDs higher than the average. *, p < 0.01. c, PMN were plated on ICAM-1 and then fixed and stained with Alexa Fluor 488-conjugated anti-LFA-1 Ab followed by permeabilization and staining with anti-phospho-SFK (Tyr416) as in a. PMN were imaged by total internal reflection fluorescence microscopy, and colocalization of LFA-1 and Tyr416 or of CD45 and Tyr416 was quantified by rendering clusters of LFA-1, CD45, and Tyr416 into pixilated bimodal images as shown. d, Colocalization was defined as the percentage of Tyr416 pixels that coincided with LFA-1 or CD45 pixels, with “+” denoting significance between monomeric and dimeric ICAM-1 and LFA-1 and CD45 at p < 0.05.
We next determined whether LFA-1 and SFK become physically associated within these focal clusters during adhesion on ICAM-1. Whole-cell lysates and membrane fractions of murine PMN adherent on monomeric vs dimeric ICAM-1 were isolated by differential centrifugation. In whole-cell lysates, Tyr416 detection was not significantly different between PMN on monomeric or dimeric ICAM-1 (Fig. 6a). This baseline level of Tyr416 signal was attributed to autophosphorylated SFK and was associated with intrinsic kinase activity equally abundant on PMN adherent to monomer or dimer. Adhesion of PMN on ICAM-1 resulted in a mobilization of phospho-SFK to the membrane and physical association with LFA-1. This is substantiated by the lower level of Tyr416 associated with LFA-1 in the membrane fraction as compared with the whole-cell lysate (Fig. 6a). Furthermore, a 1-fold greater level of Tyr416 was isolated from the membrane fraction of PMN adherent to dimeric as compared with monomeric ICAM-1 (Fig. 6b). The two distinct bands observed in Fig. 6 were attributed to Lyn, which migrates as a doublet of 55 and 58 kDa. This was substantiated by a lack of signal observed in immunoblots conducted with the lysate from PMN obtained from mice genetically deficient in Lyn (data not shown). Taken together, these data indicate that PMN bound to dimeric ICAM-1 effectively recruit the phosphorylated subunit of Lyn to membrane clusters of LFA-1 within minutes of adhesion to dimeric ICAM-1.
FIGURE 6.

Phospho-SFK (Tyr416) associates with LFA-1 upon adhesion to ICAM-1. Murine neutrophils (2 × 107) were allowed to adhere on monomeric or dimeric ICAM-1 and incubated for 7 min before cell lysis. LFA-1 was immunoprecipitated (IP) with polyclonal antisera and subjected to immunoblotting (IB) with antiphospho-SFK. Murine neutrophils (2 × 107) adherent to monomer or dimer were then frozen in liquid nitrogen. Cell membrane preparations were prepared as described in Materials and Methods. Samples of each preparation were immunoprecipitated with anti-LFA. a, Phospho-SFK (Tyr416) associated with LFA-1 on monomer and dimer in whole-cell lysate and membrane fraction (upper blot). The two bands coincide with 55- and 58-kDa markers and represent the SFK kinase Lyn, which migrates as a doublet at this molecular mass. b, Quantitative image analysis of the immunoprecipitated Tyr416 was performed for the membrane fraction of neutrophils adherent to monomeric vs dimeric ICAM-1. Bars represent normalized intensity of phospho-SFK signal associating with PMN membrane adherent to monomer vs dimer ICAM-1 from three experiments. +, p < 0.05.
To further define the role of SFK in a model of PMN recruitment to inflamed endothelium under shear flow, we observed cell capture, rolling, and subsequent transmigration in a custom-made parallel plate microfluidic flow channel (35). Human PMN pre-treated with soluble SFK inhibitor, and mouse PMN isolated from the bone marrow of wild-type and SFK knockout, were observed during recruitment under a shear of 2 dyne/cm2 on inflamed HUVEC and mouse bEND cells, respectively. Inhibition or deletion of SFK did not impair PMN capture and rolling on the endothelium; however, the fraction of PMN that was arrested was diminished by ~50% on both mouse and human endothelium (Fig. 7). Within seconds of arrest, virtually all PMN in wild-type mice and human controls exhibited shape polarization, and a fraction of these transmigrated to a position beneath the inflamed endothelium, where they are easily detected by their phase-dark appearance. Under conditions in which SFK were inhibited or genetically deleted there was a 90% reduction in the capacity for PMN to become polarized, and transmigration was abrogated.
FIGURE 7.

SFK play a critical role in PMN arrest, polarization, and transmigration across inflamed endothelium. a, PMN in the presence of 1 mM Src kinase inhibitor II or 0.3% DMSO control were perfused through a microfluidic flow chamber at an average wall shear stress of 2 dyne/cm2 and observed interacting with inflamed HUVEC monolayers. Neutrophils were imaged over the course of 6 min and assayed for firm arrest, polarization, and TEM. +, p < 0.05 and **, p < 0.001. b, Wild-type (WT) or lyn−/−hck−/− fgr−/− (SFK KO) murine bone PMN were perfused over inflamed bEND.3 endothelial cells at an average wall shear stress of 1 dyne/cm2. As in a, stable arrest, polarization, and TEM were identified optically. Data are means ± SEM from three independent experiments on murine replicates. *, p < 0.01.
We next examined whether the lack of polarization and migration observed in the SFK-deficient PMN would also manifest in a defect in the redistribution of high-affinity LFA-1 to the leading and trailing membrane of polarized and migrating cells. Human PMN perfused over IL-1β stimulated HUVEC monolayers at 2 dyne/cm2 underwent cell polarization and redistribution of the β2 integrin activation reporter 327C to the uropod and pseudopod, as previously reported (Fig. 8a). Pretreatment of PMN with soluble SFK inhibitor decreased, but did not abrogate, the extent of high-affinity CD18 in polarized cells. Most evident was that LFA-1 remained clustered at the uropod and did not redistribute into high-affinity clusters at the base of forming pseudopods (Fig. 8b). Taken together, the data suggest that binding to dimeric ICAM-1 facilitates cellular cytoskeletal activation and conversion of LFA-1 into a high-affinity state. This promotes LFA-1 colocalization with phospho-SFK, which is necessary for contact-mediated cell polarization and migration across inflamed endothelium.
FIGURE 8.

Integrin redistribution and PMN polarization is disrupted by inhibiting SFK. Neutrophils pretreated with SFK inhibitor or vehicle control were labeled with 20 mg/ml anti-CD11a (TS2/4) or anti-high-affinity CD18 (327C) conjugated to Alexa 546 fluorophore for 10 min at room temperature. Neutrophils were then perfused over IL-1β stimulated HUVEC monolayers at 2 dyne/cm2 shear stress. Sequences of brightfield and Alexa 546 emission were captured from random fields of arrested PMN from 3 to 6 min following the onset of perfusion. a, Images of LFA-1 or active CD18 are representative of >30 neutrophils and were enhanced by both gamma adjustment to identify cluster location and traced to clarify the neutrophil border. Arrows indicate the location of high-density integrin clusters. b, Polarized PMN that expressed a cluster within 1 μm of the uropod, and at least one additional distinct cluster >3 μm from the uropod, were defined as having biaxial integrin distribution (n = 30–35 neutrophils from three independent experiments; +, p < 0.05).
Discussion
Leukocyte binding to the multivalent form of ICAM-1 induces clustering of high affinity LFA-1, which promotes shear-resistant cell adhesion and contact-mediated guidance during transmigration across inflamed endothelium (4, 36). It is well established that ICAM-1 is cooperative in this process, as it reorganizes on the surface of inflamed endothelium into high-density clusters that support multimeric binding of β2 integrins during migration (36–38). Membrane redistribution of focal clusters of high-affinity LFA-1 is observed on PMN following arrest as they proceed to a polarized and migratory phenotype on inflamed endothelium (4). Based on these observations and the fact that small-molecule allosteric antagonists that retain LFA-1 in low or intermediate affinity effectively abrogate PMN polarization and transmigration, we focused this study on the process underlying LFA-1-dependent guidance of PMN and discovered that: 1) PMN binding to dimeric ICAM-1, but not monomeric ICAM-1, initiated cytoskeletal activation synergistically with stimulation by either chemokine or allosteric Ab; 2) SFK became activated and associated with high-affinity focal clusters of LFA-1 on the plasma membrane upon contact with dimeric ICAM-1, which concentrated at intracellular junctions on inflamed endothelium; and 3) SFK activation was required for efficient PMN arrest, polarization, and transmigration on inflamed endothelium in shear flow. These data suggest that mutual coordination of LFA-1 affinity on the leukocyte and membrane redistribution of ICAM-1 homodimers are pivotal and cooperative events in the multistep process of leukocyte recruitment during inflammation.
Integrin affinity and ICAM-1 valence regulate the level of cytoskeletal activation
The physiological importance of dimeric ICAM-1 as compared with monomeric ICAM-1 expression in promoting a migratory phenotype of PMN on endothelial cell surfaces was evident. Fluorescent imaging of ICAM-1 on stimulated HUVEC revealed dimeric ICAM-1 to be the predominant conformation observed on the membrane surface (10). In employing beads presenting ICAM-1 at defined site density and reagents that regulate the affinity and valence of CD18, a hierarchy in the activation state of PMN bound to beads derivatized with various IgGs was clearly evident. Surface expression of dimeric ICAM-1 on beads was sufficient to accumulate high-affinity CD18 and activate F-actin formation at the site of contact. This was attributed to the generation of outside-in signals specifically via high-affinity LFA-1 since Ab blocking of Mac-1 receptors that also bind tightly to ICAM-1 did not diminish PMN activation, yet stabilizing LFA-1 in a low-affinity state with lovastatin effectively blocked signaling. We discovered that at a minimum, formation of LFA-1 dimers formed by binding to ICAM-1 dimers, or cross-linked by mAb TS2/4, was sufficient to initiate outside-in signaling. These data led us to speculate that the distance (e.g., ~100 Å) between respective ICAM-1 molecules presented on the recombinant IgG construct attached to our beads, as well as the native homodimer up-regulated on in-flamed endothelium (12, 39), is a strategic one for conducting the outside-in signal via LFA-1. This spatial acuity between ligated pairs of high-affinity LFA-1 may facilitate linkage with accessory molecules such as talin (e.g., cross-sectional area of ~60 nm) that exist as elongated flexible antiparallel dimers with binding sites for several β integrin cytodomains (40, 41). Dimerization of LFA-1 through ICAM-1 binding may allow a single talin or other accessory molecule to engage with multiple LFA-1 tails within the same spatial region of the submembrane cytoplasm through multimeric interaction. These macromolecular complexes may also provide a scaffold for concentrating phospho-SFK and other integrin-associated signaling molecules through transient interaction, increasing the likelihood of cross-phosphorylation and the successful assembly of a signaling complex that effects outside-in signaling. For example, it has been observed that binding of LFA-1 cytoplasmic domains to talin results in uncoupling of the α and β subunits and subsequent outside-in signaling, implying that proximity between integrins may catalyze efficient uncoupling and signaling (42).
SFK as a transducer of outside-in signaling
Published data support a role for SFK phosphorylation in regulation of adhesion molecules during leukocyte arrest and transmigration on inflamed endothelium. For instance, SFK-induced cortactin phosphorylation coordinates the clustering of E-selectin and ICAM-1 at the site of contact between a leukocyte and inflamed endothelium (38). We observed that redistribution of bound LFA-1 and phospho-SFK upon PMN adhesion provided critical guidance cues for efficient transmigration, as genetic deletion or soluble inhibitors of SFK abrogated TEM. Others have reported that SFK members Hck and Lyn are activated upon ligand binding of high-affinity CD18 and their activity strengthens adhesion by concentrating F-actin and CD18 at sites of adhesion (23). A more refined model of the role of SFK in the multistep process of PMN recruitment is emerging. Imaging adhesion kinetics in our microfluidic channels revealed that PMN capture and rolling was unaffected in the absence of SFK, whereas arrest was inhibited by ~50% and cell polarization and transmigration were virtually abrogated in SFK-deficient mouse PMN or in human PMN treated with soluble inhibitor. PMN adherent to dimeric ICAM-1 initiated SFK phosphorylation, which coincided with its focal clustering with high-affinity LFA-1 in the cortical membrane. Consistent with previous reports, the mechanism underlying the defect induced by inhibiting SFK correlated with an inability to cluster high-affinity LFA-1 along the major axis of shape polarization and to polymerize F-actin after adhesion. We conclude that although SFK are necessary for signal transduction at sites of adhesion, they are particularly instrumental in signaling via focal clusters of high-affinity LFA-1 and in linking this complex to cytoskeletal rearrangement that facilitates guidance to sites of transmigration. In the context of contact-mediated guidance, platelet spreading on fibrinogen via α(IIb)β integrin (3) is linked to protein kinase C β activation and is mediated by the adaptor/scaffolding protein receptor for activated C kinase 1 (RACK1), which can directly modulate interaction with the SFK (43). Rearrangement of LFA-1 by ICAM-1 is known to transduce specific cytoplasmic changes in integrin tails and stabilization in response to GPCR-mediated Rap-1 activation (44). In this regard, in separate studies we observed focal clustering of high-affinity LFA-1 associated with Rap-1 and talin on PMN adherent to dimeric, but not monomeric, ICAM-1 (N. Dixit and S. I. Simon, unpublished data). This is consistent with formation of a macromolecular complex of LFA-1/ICAM-1/Rap-1 with SFK that directs leukocyte adhesion strengthening and migration into sites of inflammation, a process that is deficient in leukocyte adhesion deficiency-III patients who are lacking these complexes (45).
Sequential regulation of outside-in signaling and PMN diapedesis
We propose the following stepwise model for contact-mediated guidance via high-avidity mulitvalent clusters of LFA-1/ICAM-1: Conversion of a single LFA-1 to an intermediate or high-affinity state through inside-out signaling (e.g., chemokine, or clustering of E-selectin ligands) promotes binding to a single ICAM-1 of a homodimer, which facilitates more efficient binding of proximal LFA-1 to the second ICAM-1. A critical geometry is achieved by this specific pattern of clustered LFA-1 bound to oligomerized ICAM-1, which leads to cytoskeletal formation of F-actin and clustering of SFK and other adaptor molecules at focal clusters. These clusters take up the membrane forces anchoring PMN in shear flow, and this may increase the likelihood of converting additional LFA-1 to high affinity, thereby increasing the bond lifetime of the LFA-1/ICAM-1 complex. Supporting the idea of a signaling complex requiring more than a single LFA-1 is the finding that the threshold for PMA to activate nonclustered LFA-1 in mutant K562 cells is higher than in cells retaining the capacity to cluster LFA-1 (46). The implication is that mutant, nonclustered LFA-1 cannot form an effective outside-in signaling complex. Recruitment of F-actin to clustered high-affinity LFA-1 may provide a scaffold for SFK phosphorylation leading to downstream signaling through RACK1 and protein kinase C, which in turn amplifies the number of high-affinity integrins in a positive feedback process that serves to coordinate cell polarization and directed migration at focal sites of contact. This is supported by our observation that SFK were more involved in outside-in signaling upon dimeric ICAM-1 binding and LFA-1 clustering than in conversion of LFA-1 to high affinity in response to GPCR signaling via MIP-1α or IL-8 receptor ligation in the absence of ICAM-1. Disruption of outside-in signaling was evident in SFK knockouts and in the presence of soluble SFK inhibitors where neutrophil recruitment stalled after arrest and polarization on inflamed endothelium, likely due to an inability of LFA-1 to properly distribute into biaxial clusters.
We conclude that an upshift in LFA-1 affinity and formation of multivalent bonds with ICAM-1 homodimer is a dynamic process providing a key navigational step in the transition to a migratory phenotype. In this manner, both the endothelium and leukocyte cooperate in regulating the efficiency and site of leukocyte recruitment during inflammation.
Acknowledgments
We thank the scientists who worked at ICOS, in particular Donald Staunton, who provided the 327C and 240Q used in these studies.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants AI47294 (to S.I.S.), AI65495, and AI68150 (to C.L.) and an NIH training grant to U.Y.S. (AI060555).
Abbreviations used in this paper: PMN, polymorphonuclear leukocytes; bEND, brain endothelial; GPCR, G protein-coupled receptor; HSA, human serum albumin; MFI, mean fluorescence intensity; SFK, Src family kinase; TEM, transendothelial migration.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity β2-integrin on neutrophils. J Immunol. 2004;172:7780–7790. doi: 10.4049/jimmunol.172.12.7780. [DOI] [PubMed] [Google Scholar]
- 2.McEver RP, Zhu C. A catch to integrin activation. Nat Immunol. 2007;8:1035–1037. doi: 10.1038/ni1007-1035. [DOI] [PubMed] [Google Scholar]
- 3.Salas A, Shimaoka M, Kogan AN, Harwood C, Von Andrian UH, Springer TA. Rolling adhesion through an extended conformation of integrin αLβ2 and relation to αI and βI-like domain interaction. Immunity. 2004;20:393–406. doi: 10.1016/s1074-7613(04)00082-2. [DOI] [PubMed] [Google Scholar]
- 4.Green CE, Schaff UY, Sarantos MR, Lum AF, Staunton DE, Simon SI. Dynamic shifts in LFA-1 affinity regulate neutrophil rolling, arrest, and transmigration on inflamed endothelium. Blood. 2006;107:2101–2111. doi: 10.1182/blood-2005-06-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lum AF, Green CE, Lee GR, Staunton DE, Simon SI. Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. J Biol Chem. 2002;277:20660–20670. doi: 10.1074/jbc.M202223200. [DOI] [PubMed] [Google Scholar]
- 6.Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-α-activated vascular endothelium under flow. Blood. 2005;106:584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woska JR, Jr, Morelock MM, Jeanfavre DD, Bormann BJ. Characterization of molecular interactions between intercellular adhesion molecule-1 and leukocyte function-associated antigen-1. J Immunol. 1996;156:4680–4685. [PubMed] [Google Scholar]
- 8.Yang Y, Jun CD, Liu JH, Zhang R, Joachimiak A, Springer TA, Wang JH. Structural basis for dimerization of ICAM-1 on the cell surface. Mol Cell. 2004;14:269–276. doi: 10.1016/s1097-2765(04)00204-7. [DOI] [PubMed] [Google Scholar]
- 9.Sarantos MR, Raychaudhuri S, Lum AF, Staunton DE, Simon SI. Leukocyte function-associated antigen 1-mediated adhesion stability is dynamically regulated through affinity and valency during bond formation with intercellular adhesion molecule-1. J Biol Chem. 2005;280:28290–28298. doi: 10.1074/jbc.M501662200. [DOI] [PubMed] [Google Scholar]
- 10.Miller J, Knorr R, Ferrone M, Houdei R, Carron CP, Dustin ML. Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function associated-1. J Exp Med. 1995;182:1231–1241. doi: 10.1084/jem.182.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reilly PL, Woska JR, Jr, Jeanfavre DD, McNally E, Rothlein R, Bormann BJ. The native structure of intercellular adhesion molecule-1 (ICAM-1) is a dimer: correlation with binding to LFA-1. J Immunol. 1995;155:529–532. [PubMed] [Google Scholar]
- 12.Chen X, Kim TD, Carman CV, Mi LZ, Song G, Springer TA. Structural plasticity in Ig superfamily domain 4 of ICAM-1 mediates cell surface dimerization. Proc Natl Acad Sci USA. 2007;104:15358–15363. doi: 10.1073/pnas.0707406104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dustin ML, Bivona TG, Philips MR. Membranes as messengers in T cell adhesion signaling. Nat Immunol. 2004;5:363–372. doi: 10.1038/ni1057. [DOI] [PubMed] [Google Scholar]
- 14.Porter JC, Bracke M, Smith A, Davies D, Hogg N. Signaling through integrin LFA-1 leads to filamentous actin polymerization and remodeling, resulting in enhanced T cell adhesion. J Immunol. 2002;168:6330–6335. doi: 10.4049/jimmunol.168.12.6330. [DOI] [PubMed] [Google Scholar]
- 15.Lub M, van Kooyk Y, van Vliet SJ, Figdor CG. Dual role of the actin cytoskeleton in regulating cell adhesion mediated by the integrin lymphocyte function-associated molecule-1. Mol Biol Cell. 1997;8:341–351. doi: 10.1091/mbc.8.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arias-Salgado EG, Lizano S, Shattil SJ, Ginsberg MH. Specification of the direction of adhesive signaling by the integrin β cytoplasmic domain. J Biol Chem. 2005;280:29699–29707. doi: 10.1074/jbc.M503508200. [DOI] [PubMed] [Google Scholar]
- 17.Roskoski R., Jr Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun. 2004;324:1155–1164. doi: 10.1016/j.bbrc.2004.09.171. [DOI] [PubMed] [Google Scholar]
- 18.Totani L, Piccoli A, Manarini S, Federico L, Pecce R, Martelli N, Cerletti C, Piccardoni P, Lowell CA, Smyth SS, et al. Src-family kinases mediate an outside-in signal necessary for β2-integrins to achieve full activation and sustain firm adhesion of polymorphonuclear leukocytes tethered on E-selectin. Biochem J. 2006;396:89–98. doi: 10.1042/BJ20051924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernardini G, Kim JY, Gismondi A, Butcher EC, Santoni A. Chemoattractant induces LFA-1 associated PI3K activity and cell migration that are dependent on Fyn signaling. FASEB J. 2005;19:1305–1307. doi: 10.1096/fj.04-3352fje. [DOI] [PubMed] [Google Scholar]
- 20.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced αLβ2 integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26:773–783. doi: 10.1016/j.immuni.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mocsai A, Zhang H, Jakus Z, Kitaura J, Kawakami T, Lowell CA. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood. 2003;101:4155–4163. doi: 10.1182/blood-2002-07-2346. [DOI] [PubMed] [Google Scholar]
- 22.Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16:547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 23.Piccardoni P, Manarini S, Federico L, Bagoly Z, Pecce R, Martelli N, Piccoli A, Totani L, Cerletti C, Evangelista V. SRC-dependent outside-in signalling is a key step in the process of autoregulation of β2 integrins in polymorphonuclear cells. Biochem J. 2004;380:57–65. doi: 10.1042/BJ20040151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lomakina EB, Waugh RE. Micromechanical tests of adhesion dynamics between neutrophils and immobilized ICAM-1. Biophys J. 2004;86:1223–1233. doi: 10.1016/S0006-3495(04)74196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira S, Lowell C. The Lyn tyrosine kinase negatively regulates neutrophil integrin signaling. J Immunol. 2003;171:1319–1327. doi: 10.4049/jimmunol.171.3.1319. [DOI] [PubMed] [Google Scholar]
- 26.Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alcaide P, Auerbach S, Luscinskas FW. Neutrophil recruitment under shear flow: it’s all about endothelial cell rings and gaps. Microcirculation. 2008;19:1. doi: 10.1080/10739680802273892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gopalan PK, Burns AR, Simon SI, Sparks S, McIntire LV, Smith CW. Preferential sites for stationary adhesion of neutrophils to cytokine-stimulated HUVEC under flow conditions. J Leukocyte Biol. 2000;68:47–57. [PubMed] [Google Scholar]
- 29.Huang C, Springer TA. A binding interface on the I domain of lymphocyte function-associated antigen-1 (LFA-1) required for specific interaction with intercellular adhesion molecule 1 (ICAM-1) J Biol Chem. 1995;270:19008–19016. doi: 10.1074/jbc.270.32.19008. [DOI] [PubMed] [Google Scholar]
- 30.Lupher ML, Jr, Harris EA, Beals CR, Sui LM, Liddington RC, Staunton DE. Cellular activation of leukocyte function-associated antigen-1 and its affinity are regulated at the I domain allosteric site. J Immunol. 2001;167:1431–1439. doi: 10.4049/jimmunol.167.3.1431. [DOI] [PubMed] [Google Scholar]
- 31.Huth JR, Olejniczak ET, Mendoza R, Liang H, Harris EA, Lupher ML, Jr, Wilson AE, Fesik SW, Staunton DE. NMR and mutagenesis evidence for an I domain allosteric site that regulates lymphocyte function-associated antigen 1 ligand binding. Proc Natl Acad Sci USA. 2000;97:5231–5236. doi: 10.1073/pnas.97.10.5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, Dobransky T, Feldman RD, Ferguson SS, Kelvin DJ. Regulation of tyrosine kinase activation and granule release through β-arrestin by CXCRI. Nat Immunol. 2000;1:227–233. doi: 10.1038/79767. [DOI] [PubMed] [Google Scholar]
- 33.Kilimnik A, Kostjukova MN, Pyatkin IH, Pronin AM, Strelnikova SR, Fedotov YA, Kolesnikov AV. Novel small molecule inhibitors of C-terminal Src kinase (Csk) Cell Mol Biol Lett. 2003;8:588–589. [Google Scholar]
- 34.Meng F, Lowell CA. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J Exp Med. 1997;185:1661–1670. doi: 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaff UY, Xing MM, Lin KK, Pan N, Jeon NL, Simon SI. Vascular mimetics based on microfluidics for imaging the leukocyte–endothelial inflammatory response. Lab Chip. 2007;7:448–456. doi: 10.1039/b617915k. [DOI] [PubMed] [Google Scholar]
- 36.Kim M, Carman CV, Yang W, Salas A, Springer TA. The primacy of affinity over clustering in regulation of adhesiveness of the integrin αLβ2. J Cell Biol. 2004;167:1241–1253. doi: 10.1083/jcb.200404160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Kowalski JR, Zhan X, Thomas SM, Luscinskas FW. Endothelial cell cortactin phosphorylation by Src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ Res. 2006;98:394–402. doi: 10.1161/01.RES.0000201958.59020.1a. [DOI] [PubMed] [Google Scholar]
- 39.Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005;7:151–185. doi: 10.1146/annurev.bioeng.7.060804.100423. [DOI] [PubMed] [Google Scholar]
- 40.Ziegler WH, Gingras AR, Critchley DR, Emsley J. Integrin connections to the cytoskeleton through talin and vinculin. Biochem Soc Trans. 2008;36:235–239. doi: 10.1042/BST0360235. [DOI] [PubMed] [Google Scholar]
- 41.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 42.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 43.Buensuceso CS, Obergfell A, Soriani A, Eto K, Kiosses WB, Arias-Salgado EG, Kawakami T, Shattil SJ. Regulation of outside-in signaling in platelets by integrin-associated protein kinase C β. J Biol Chem. 2005;280:644–653. doi: 10.1074/jbc.M410229200. [DOI] [PubMed] [Google Scholar]
- 44.Kinashi T, Katagiri K. Regulation of lymphocyte adhesion and migration by the small GTPase Rap1 and its effector molecule, RAPL. Immunol Lett. 2004;93:1–5. doi: 10.1016/j.imlet.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 45.Pasvolsky R, Feigelson SW, Kilic SS, Simon AJ, Tal-Lapidot G, Grabovsky V, Crittenden JR, Amariglio N, Safran M, Graybiel AM, et al. A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J Exp Med. 2007;204:1571–1582. doi: 10.1084/jem.20070058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bleijs DA, van Duijnhoven GC, van Vliet SJ, Thijssen JP, Figdor CG, van Kooyk Y. A single amino acid in the cytoplasmic domain of the β2 integrin lymphocyte function-associated antigen-1 regulates avidity-dependent inside-out signaling. J Biol Chem. 2001;276:10338–10346. doi: 10.1074/jbc.M008967200. [DOI] [PubMed] [Google Scholar]