

Abstract
Dual emissive luminescence properties of solid-state difluoroboron β-diketonate-poly(lactic acid) (BF2bdk-PLA) materials have been utilized as biological oxygen sensors. Dyes with red-shifted absorption and emission are important for multiplexing and in vivo imaging, thus hydroxyl-functionalized dinaphthoylmethane initiators and dye-PLA conjugates BF2dnm(X)PLA (X = H, Br, I) with extended conjugation were synthesized. The luminescent materials show red-shifted absorbance (~435 nm) and fluorescence tunability by molecular weight. Fluorescence colors range from yellow (~530 nm) in 10 – 12 kDa polymers to green (~490 nm) in 20 – 30 kDa polymers. Room-temperature phosphorescence (RTP) and thermally activated delayed fluorescence (TADF) are present under a nitrogen atmosphere. For the iodine-substituted derivative, BF2dnm(I)PLA, clearly distinguishable fluorescence (green) and phosphorescence (orange) peaks are present, making it ideal for ratiometric oxygen-sensing and imaging. Bromide and hydrogen analogues with weaker relative phosphorescence intensities and longer phosphorescence lifetimes can be used as highly sensitive, concentration independent, lifetime-based oxygen sensors or for gated emission detection. BF2dnm(I)PLA nanoparticles were taken up by T41 mouse mammary cells and successfully demonstrated differences in vitro ratiometric measurement of oxygen.
Keywords: Difluoroboron β-diketonate complexes, room-temperature phosphorescence (RTP), thermally activated delayed fluorescence (TADF), heavy-atom effect, poly(lactic-acid), oxygen-sensitive material
INTRODUCTION
Boron containing biomaterials have garnered interest in recent years.1–3 New developments include “turn-on” sensors responsive to reactive oxygen species (ROS),4,5 rapidly degrading polymers for stimuli triggered drug release,6 fluorescent ion sensors,7,8 near-IR probes,9,10 and bioorthogonal fluorescent labels.11,12 These fluorescent dyes use the difluoroboron (BF2) unit to restrict intramolecular rotation and vibrational freedom of the β-diketonate ligand, producing bright and efficient fluorescence.13,14 Various types of BF2 fluorescent materials have been prepared, including β-diketiminate (NBN),15,16 β-ketoiminate (NBO),17–21 and β-diketonate (OBO) complexes.22–24 Difluoroboron can also act as an electron-accepting group, inducing a strong dipole upon excitation, making the dyes sensitive to solvent and media polarity.25–27 Difluoroboron β-diketonates (BF2bdks) are particularly interesting because of their exceptional luminescent properties in both solution and solid states. The Stokes shifts are large enough so that the dyes do not completely self-quench when concentrated, and the medium,28 molecular packing,29–32 or dye design33 can modulate the solid-state luminescence. The ligand framework also provides a versatile scaffold for functionalization by mesogens,34 polymers,35–37 heavy atoms,38 or chelators.39,40 Furthermore, BF2 chelates have demonstrated the ability to restrict intramolecular twisting of the aromatic-carbonyl moiety to produce rare, metal-free phosphorescence in rigid environments.20,41–44
A common application of phosphorescent dyes is optical oxygen sensing.45 Quantifying oxygen via phosphorescence can be achieved by lifetime or intensity-based sensing.46 Lifetime sensing is often deemed more reliable because phosphorescence decay is independent of phosphor concentration and is highly sensitive to collisional quenching by oxygen.47–49 These methods can be achieved with fluorescence and phosphorescence lifetime imaging microscopy (FLIM and PLIM) techniques,50 or even with high-speed cameras.51 Intensity based measurements are more easily performed with common instrumentation (e.g. fluorescence microscopes or color cameras). Pure intensity techniques are dependent on phosphor concentration, which can change throughout experimentation depending on dye stability or material heterogeneity.
Difluoroboron dibenzoylmethane-poly(lactic acid) (BF2dbmPLA) is a multi-emissive material that produces both intense fluorescence and long-lived phosphorescence upon single or multiphoton excitation (Figure 1).52 Using boron dyes as initiators for lactide ring-opening polymerization (ROP) creates a metal-free, biodegradable, single component oxygen sensor, minimizing problems associated with dye leaching and heterogeneity. Furthermore, optical properties can be easily tuned by molecular weight (MW).53,54 These materials are readily fabricated into nanoparticles suitable for cellular uptake and both in vitro and in vivo sensing.55–57 Iodine-substituted difluoroboron dibenzoylmethane poly(lactic acid) (BF2dbm(I)PLA) nanoparticles have enabled tumor hypoxia imaging by ratiometric techniques.58 This polymer, shows fluorescence in the range of 440–485 nm, and phosphorescence at about 530 nm. Blue and green wavelengths like these are easily absorbed or scattered in vivo by absorbers in tissues, which limits BF2dbm(I)PLA imaging primarily to surfaces.59,60 Thus, red-shifted analogues could potentially increase tissue penetration by decreasing the amount of scattered light.61 Additionally, obtaining a palette of colors could make these materials more versatile for multiplexing biological sensing applications.62 Previous model studies involving anthracene- and naphthylene-derived dyes, blended but not conjugated with PLA, showed red-shifted spectral properties.63 Also, recently reported naphthyl-phenyl based BF2bdkPLA conjugates were synthesized with red-shifted dual-emissive optical properties.33
Figure 1.
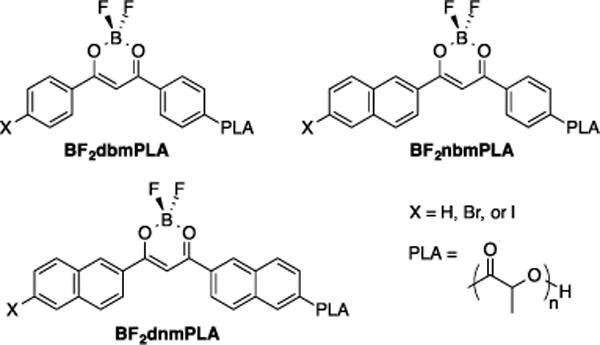
Difluoroboron β-diketonate poly(lactic acid) materials.
This study extends our exploration of boron β-diketonate materials by increasing dye conjugation using dinaphthoylmethane (dnm). Previously, dnm ligands have been utilized as mechanochromic materials,64 two-photon excitable energy donors,65,66 in optoelectronic lanthanide complexes67 and in visible light absorbers for temperature sensitive europium complexes68 The red-shifted absorption and increased π–conjugation of this ligand make it a good candidate for incorporation into oxygen sensing probes. Red-shifted absorbance allows for visible light absorption, which is less phototoxic compared to higher energy UV excitation. Halides, such as bromide and iodide heavy atoms are introduced to increase crossover to the triplet state by enhancing spin-orbit coupling,69,70 for shorter phosphorescence lifetimes and increased intensity of triplet emission at the expense of fluorescence. This study describes the synthesis and optical characterization of BF2dnmPLA materials and probes the effects of halide substitution and polymer molecular weight on the optical properties in dilute solutions, thin films, and nanoparticles. The utility of the iodide derivative, BF2dnm(I)PLA for cellular oxygen sensing is also demonstrated.
EXPERIMENTAL
Materials
3,6-Dimethyl-1,4-dioxane-2,5-dione (D,L -lactide, Aldrich) was recrystallized twice from ethyl acetate and stored under nitrogen. Tin (II) 2-ethylhexanoate (Sn(oct)2, Spectrum), boron trifluoride diethyl etherate (Aldrich, purified, redistilled) and all other reagents and solvents were used as received without further purification. Solvents CH2Cl2 and THF were dried and purified over 3 Å molecular sieves activated at 300 °C according to a previously described method.71 All other chemicals were reagent grade from Sigma Aldrich and were used without further purification. The compounds 1-(6-(2-((tetrahydro-2H-pyran-2-yl)oxy)ethoxy)naphthalen-2-yl)ethanone33 and methyl 6-iodo-2-naphthoate72 were prepared as previously described. Poly(lactic-acid) was synthesized from ethylene glycol as previously described53 (Mn (GPC/MALS) = 13 800 Da, PDI = 1.03).
Methods
1H NMR (300 MHz) spectra were recorded on a Varian UnityInova 300/51 instrument in CDCl3, or D6-DMSO. 1H NMR spectra were referenced to the signals for the residual protiochloroform at 7.26 ppm, protioDMSO at 2.50 ppm, and protioacetone at 2.09 ppm. Coupling constants are given in hertz. High-resolution mass spectra of ligands and dyes were recorded with a Micromass Q-TOF Ultima spectrometer using electrospray ionization (ESI) MS techniques. Polymer molecular weights were determined by gel permeation chromatography (GPC) (THF, 25 °C, 1.0 mL / min) using multi-angle laser light scattering (MALS) (λ = 658 nm, 25 °C) and refractive index (RI) (λ = 658 nm, 25 °C) detection. Polymer Laboratories 5 μm mixed-C columns (guard column plus two columns) along with Wyatt Technology (Optilab T-rEX interferometric refractometer, miniDAWN TREOS multi-angle static light scattering (MALS) detector, ASTRA 6.0 software) and Agilent Technologies instrumentation (series 1260 HPLC with diode array (DAD) detector, ChemStation) were used in GPC analysis. UV-vis spectra were recorded on a Hewlett-Packard 8452A diode-array spectrophotometer. Steady-state fluorescence spectra were recorded on a Horiba Fluorolog-3 Model FL3-22 spectrofluorometer (double-grating excitation and double-grating emission monochromator). Time-correlated single-photon counting (TCSPC) fluorescence lifetime measurements were performed with a NanoLED-370 (λex = 369 nm) excitation source and a DataStation Hub as the SPC controller. Phosphorescence lifetimes were measured with a 1 ms multi-channel scalar (MCS) excited with a pulsed Xenon lamp (λex = 400 nm; duration <1 ms). Lifetime data were analyzed with DataStation v2.4 software from Horiba Jobin Yvon. Fluorescence quantum yields (ΦF) for initiator and polymer samples in CH2Cl2 were calculated versus anthracene as a standard using a previously described method: ΦF anthracene = 0.27,73,74 nD20 EtOH = 1.360, nD20 CH2Cl2 = 1.424.75 Optically dilute CH2Cl2 solutions of the dyes were prepared in 1 cm path length quartz cuvettes with absorbances < 0.1 (a.u.).
Thin films were prepared on the inner wall of vials by dissolving polymers in CH2Cl2 (~2 mg/mL), then evaporating the solvent under a low N2 flow. Blends of dye-PLA conjugates and PLA were prepared by weighing dye-PLA and PLA into vials, dissolving in CH2Cl2 (~2 mg/mL), then evaporating the solvent under low N2 flow (Table S1). The films were dried in vacuo overnight before measurements were taken. Polymers were fabricated into nanoparticles by previously described methods.55 Nanoparticle size was determined by dynamic light scattering (DLS) on a Wyatt Corporation DynaPro Plate Reader II. Ratiometric oxygen sensitivity calibration is performed as previously described.58 Images of films and nanoparticles were taken with a Canon EOS 7D camera with handheld UV lamp excitation (λex = 354 nm).
1-[6-(2-Hydroxyethoxy)-2-napthyl]-3-(2-naphthyl)-propane-1,3-dione (dnmOH) (1)
The aromatic ketone, 1-(6-(2-((tetrahydro-2H-pyran-2-yl)oxy)ethoxy)naphthalen-2-yl)ethanone was prepared as previously described.33 1-(6-(2-((Tetrahydro-2H-pyran-2-yl)oxy)ethoxy)naphthalen-2-yl)ethanone (740 mg, 2.35 mmol) and methyl 2-naphthoate (526 mg, 2.82 mmol) were weighed in a dry 100 mL round-bottom flask and dissolved in anhydrous THF (~ 20 mL). A suspension of anhydrous THF (~10 mL) and NaH (85 mg, 3.5 mmol) was transferred to the flask via cannula. The reaction mixture was heated at 60 °C and monitored by TLC until the limiting reagent (ketone) was consumed. After 1 d, the reaction mixture was cooled to room temperature and quenched by drop-wise addition of saturated NaHCO3 (aq) (10 mL). THF was removed by rotary evaporation and the remaining aqueous layer was acidified with 1M HCl and extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were washed with distilled water (2 × 100 mL) and brine (2 × 100 mL), then dried over Na2SO4, filtered, and concentrated in vacuo. The resulting brown, oily residue was dissolved in THF (50 mL) and water (15 mL), and p-TsOH (50 mg, 0.29 mmol) was added. The reation mixture was heated at 60 °C and monitored by TLC. After 18 h, the reaction mixture was cooled to room temperature and THF was removed by rotary evaporation. The resulting residue was dissolved in CH2Cl2, washed with distilled water (2 × 100 mL), brine (2 × 100 mL), and dried over anhydrous Na2SO4. The solution was filtered, and solvent was removed via rotary evaporation. The tan colored crude product was purified via recrystallization with 1:1 hexanes/EtOAc to give 1 as a tan powder: 490 mg (54%). 1H NMR (300 MHz, CDCl3): δ 17.12 (s, 1H, -OH), 8.58 (s, 1H, 1″-ArH), 8.53 (s, 1H, 1′-ArH), 8.08 (d, J = 3.0, 1H, 8″-ArH), 8.05 (d, J = 3.0, 1H, 8′-ArH), 8.01 (d, J = 7.2, 1H, 4″-ArH), 7.95-7.89 (m, 3H, 4′-ArH, 3″-,5″-ArH), 7.83 (d, J = 8.7, 1H, 3′-ArH), 7.59 (m, broad, 2H, 6″-, 7″-ArH), 7.26 (d, J = 8.7, 1H, 7′-ArH), 7.21 (s, 1H, 5′-ArH), 7.13 (s, 1H, -COCHCO), 4.35 (t, J = 4.2, 2H, -ArOCH2CH2OH), 4.09-4.05 (m, 2H, -ArOCH2CH2OH), 2.05 (s, 1H, -ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd for C25H21O4 385.1440 [M + H]+; found 385.1433.
1-[6-(2-Hydroxyethoxy)-2-napthyl]-3-(6-bromo-2-naphthyl)-propane-1,3-dione (dnm(Br)OH) (2)
The bromine derivative was prepared as described for 1 using methyl 6-bromo 2-naphthoate instead of methyl 2-naphthoate. A tan powder was obtained: 340 mg (46%). 1H NMR (300 MHz, (CD3)2SO): δ 17.34 (s, 1H, -OH), 8.87 (s, 1H, 1″-ArH), 8.81 (s, 1H, 1′-ArH), 8.34 (s, 1H, 5″-ArH), 8.29 (d, J = 8.4, 1H, 8″-ArH), 8.20 (d, J = 8.7, 1H, 8′-ArH), 8.00–8.15 (m, broad, 3H, 3′- ArH, 3″, 4″-ArH), 7.95 (d, J = 8.7, 1H, 7″-ArH), 7.77 (d, J = 8.7, 1H, 7′-ArH), 7.61 (s, 1H, COCHCO), 7.45 (s, 1H, 5′-ArH), 7.25 (d, J = 8.7, 1H, 4′-ArH), 4.96 (t, J = 4.2, 1H, -ArOCH2CH2OH), 4.15 (t, J = 4.2, 2H, -ArOCH2CH2OH), 3.78 (m, 2H, -ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd for C25H20O4Br 463.0545 [M + H]+; found 463.0538.
1-[6-(2-Hydroxyethoxy)-2-napthyl]-3-(6-iodo-2-naphthyl)-propane-1,3-dione (dnm(I)OH) (3)
The iodine derivative was prepared as previously described for 1 with methyl 6-iodo 2-naphthoate instead of methyl 2-naphthoate. The crude dnm(I)OH product 3 was purified by recrystallization with acetone instead of hexanes/EtOAc to yield a tan powder: 98 mg (11%). 1H NMR (300 MHz, (CD3)2SO): δ 17.39 (s, 1H, -OH), 8.84 (s, 1H, 1″-ArH), 8.80 (s, 1H, 1′-ArH), 8.52 (s, 1H, 5″-ArH), 8.27 (d, J = 9.0, 1H, 8″-ArH), 8.18 (d, J = 9.0, 1H, 8′-ArH), 8.05-7.94 (m, broad, 5H, 3′, 4′-ArH, 3″-, 4″-, 7″-ArH), 7.61 (s, 1H, COCHCO), 7.44 (s, 1H, 5′-ArH), 7.29 (d, J = 12, 1H, 7′-ArH), 4.95 (t, J = 6.0, 1H, -ArOCH2CH2OH), 4.15 (t, J = 6.0, 2H, -ArOCH2CH2OH), 3.78 (m, 2H, -ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd for C25H20O4I 511.0406 [M + H]+; found 511.0421.
BF2dnmOH (4)
The ligand dnmOH (1), (250.0 mg, 0.705 mmol) was added to a dry 100 mL round bottom flask under nitrogen, and then dissolved in THF/CH2Cl2 (20/20 mL) to give a deep yellow solution. Boron trifluoride diethyl etherate (120 μL, 0.845 mmol) was added via syringe, turning the solution bright yellow. The reaction was refluxed at 60 °C and monitored by TLC until the ligand substrate was consumed (24 h). Solvents were removed via rotary evaporation, generating a yellow solid. The crude material was purified by recrystallization with 1:1 EtOAc/acetone to yield a yellow-orange powder: 225 mg (74%). 1H NMR (300 MHz, (CD3)2SO): δ 9.12 (s, 1H, 1″-ArH), 9.09 (s, 1H, 1′-ArH), 8.40-8.30 (m, 2H, 8′-ArH, 8″-ArH), 8.01 (d, J = 8.1, 1H, 4″-ArH), 8.16 (m, 3H, 5″-, 3″-ArH, 7′-ArH), 8.06-8.00 (m, 2H, 3′-ArH, COCHCO), 7.75-7.65 (m, 2H, 6″-, 7″-ArH), 7.51 (s, 1H, 5′-ArH), 7.34 (d, J = 8.7, 1H, 4′-ArH), 4.98 (bs, 1H, -ArOCH2CH2OH), 4.19 (t, J = 4.8, 2H, -ArOCH2CH2OH), 4.07 (t, J = 4.8, 2H, -ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd for C25H20BO4F2 433.1423 [M + H]+; found 433.1414.
BF2dnm(Br)OH (5)
The bromide complex 5 was prepared as previously described for 4 using dnm(Br)OH (2) instead of dnm(H)OH. A yellow-orange powder was obtained: 35 mg (31%). 1H NMR (300 MHz, (CD3)2SO): δ 9.12 (s, 1H, 1″-ArH), 9.09 (s, 1H, 1′-ArH), 8.48–8.33 (m, broad, 3 H, 8″-, 5″-ArH, 8′-ArH), 8.18–8.14 (m, broad, 4H, 4″, 3″-ArH, 3′-ArH, -COCHCO-), 8.03 (d, J = 8.7, 1H, 7″-ArH), 7.83 (d, J = 8.7, 1H, 7′-ArH), 7.51 (s, 1H, 5′-ArH), 7.30 (d, J = 8.7, 1H, 4′-ArH), 4.98 (t, J = 4.2, 1H, -ArOCH2CH2OH), 4.19 (t, J = 4.2, 2H, -ArOCH2CH2OH), 3.80 (m, 2H, -ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd C25H18BO4F2BrNa 533.0347 [M + Na]+; found 533.0340.
BF2dnm(I)OH (6)
The iodide complex 6 was prepared as previously described for 4 using dnm(I)OH (3) in place of dnmOH with the following exception: the dnm(I)OH ligand was dissolved in anhydrous THF instead of THF/CH2Cl2. A yellow powder was obtained: 31 mg (44%). 1H NMR (300 MHz, (CD3)2SO): δ 9.09 (s, broad, 2H, 1″-ArH, 1′-ArH), 8.59 (s, 1H, 5″-ArH), 8.41 (d, J = 6.0, 1H, 8″-ArH), 8.35 (d, J = 9.0, 1H, 8′-ArH), 8.17-7.98 (m, broad, 6H, 3″-ArH, 3′-ArH, 4″-ArH, 4′-ArH, 7″-ArH, COCHCO), 7.52 (s, 1H, 5′-ArH), 7.35 (d, J = 12, 1H, 7′-ArH), 4.98 (t, J = 6.0, 1H, -ArOCH2CH2OH), 4.19 (t, J = 6.0, 2H, -ArOCH2CH2OH), 3.79 (m, 2H, -ArOCH2CH2OH). HRMS (ESI, TOF) m/z calcd C25H18BO4FI [M − F]+ 539.0327; found 539.0320.
Polymer Synthesis
Preparative scale reactions to produce BF2dnmPLA (7a–b), BF2dnm(Br)PLA (8a–b), and BF2dnm(I)PLA (9a–b) were conducted as previously described.33 Reagent loadings, reaction times, and monomer conversions are presented in Table 1. At the end of each polymerization, an aliquot was taken to determine monomer conversion by 1H NMR spectroscopy. For polymers with low monomer conversion (< 50%), an additional precipitation into cold methanol was necessary to remove residual monomer. The polymer molecular weights were determined by 1H NMR spectroscopy and GPC (MALS/RI). Molecular weights, polydispersity indices (PDIs), and yields are collected in Table 1. Yields are corrected for monomer conversion.
Table 1.
Polymer Synthesis and Molecular Weight Characterization
| Polymer | Loadinga | Mnb (NMR) |
Mnc (GPC) |
Mwc (GPC) |
PDIc (GPC) |
Time (h) |
Convd (%) |
Yielde (%) |
|
|---|---|---|---|---|---|---|---|---|---|
| BF2dnmPLA | 7a | 300:1/40 | 11,800 | 12,200 | 14,600 | 1.20 | 3.0 | 79 | 68 |
| 7b | 300:1/40 | 38,000 | 31,800 | 41,300 | 1.30 | 14.0 | 85 | 81 | |
| BF2dnm(Br)PLA | 8a | 300:1/40 | 12,500 | 12,500 | 14,900 | 1.19 | 3.0 | 56 | 78 |
| 8b | 300: 1/40 | 26,200 | 26,000 | 31,500 | 1.21 | 10.0 | 75 | 41 | |
| BF2dnm(I)PLA | 9a | 200:1/40 | 13,000 | 12,500 | 14,000 | 1.12 | 2.0 | 71 | 46 |
| 9b | 300:1/40 | 25,500 | 23,200 | 27,600 | 1.19 | 6.0 | 63 | 83 |
Molar ratio of monomer: Sn(oct)2 per 1 equivalent of boron initiator.
Estimated from the relative integration (dye-OCH2CH2-OPLA/PLA-H).77
Molecular weight data determined by GPC with MALS/RI detection.
Percent monomer conversion determined by relative integration (PLA-H/Lactide-H).
Corrected for monomer conversion.
BF2dnmPLA (7a)
The dinaphthyl polymer 7a was obtained as a yellow crystalline foam: 325 mg (68%). Mn (GPC/MALS) = 12,200 Da, PDI = 1.20; Mn (1H NMR) = 11,800 Da. 1H NMR (600 MHz, CDCl3): δ 8.89 (s, 1H, 1′-ArH), 8.77 (s, 1H, 1″-ArH), 8.13 (m, 2H, 8′-ArH, 8″-ArH), 7.96 (m, broad, 6H, 3′-, 4′-, 5′-ArH, 3″-, 7″-ArH), 7.66 (m, broad, 2H, 7′, 6′-ArH), 7.51 (s, 1H, 5″-ArH), 7.34 (d, J = 8.7, 1H, 4″-ArH), 7.19 (s, 1H, -COCHCO-), 5.16 (m, broad, 168 H, PLA-CH-CH3), 4.59 (t, J = 4.8, 2H, -ArOCH2CH2-), 4.35 (t, J = 4.8, 2H, -ArOCH2CH2-), 1.57 (s, broad, 543 H, PLA-CHCH3).
BF2dnmPLA (7b)
The dinaphthyl polymer 7b was obtained as a yellow-green rubbery foam: 972 mg (81%). Mn (GPC/MALS) = 31,100 Da, PDI = 1.30; Mn (1H NMR) = 38,000 Da. 1H NMR (600 MHz, CDCl3): δ 8.89 (s, 1H, 1′-ArH), 8.77 (s, 1H, 1″-ArH), 8.13 (m, 2H, 8′-ArH, 8″-ArH), 7.96 (m, broad, 6H, 3′-, 4′-, 5′-ArH, 3″-, 7″-ArH), 7.66 (m, broad, 2H, 7′, 6′-ArH), 7.51 (s, 1H, 5″-ArH), 7.34 (d, J = 8.7, 1H, 4″-ArH), 7.19 (s, 1H, -COCHCO-), 5.16 (m, broad, 517 H, PLA-CH-CH3), 4.59 (t, J = 4.8, 2H, -ArOCH2CH2-), 4.35 (t, J = 4.8, 2H, -ArOCH2CH2-), 1.57 (s, broad, 1732 H, PLA-CHCH3).
BF2dnm(Br)PLA (8a)
The bromo-dinaphthyl polymer 8a was obtained as yellow crystalline foam: 212 mg (78%). Mn (GPC/MALS) = 12,500 Da, PDI = 1.19; Mn (NMR) = 12,500 Da. 1H NMR (600 MHz, CDCl3): δ 8.77 (m, broad, 2H, 1″ -ArH, 1′ -ArH), 8.17–8.11 (m, broad, 3H, 3″-, 8″, -ArH, 8′ -ArH), 7.92–7.85 (m, broad, 5H, 3′-, 7′, -ArH, 4″-, 5″, -7″-, ArH), 7.70 (d, J = 9, 1H, 4′ -ArH), 7.42 (s, 1H, -COCHCO-), 7.18 (s, 1H, 5′ -ArH), 5.31–5.11 (m, broad, 180 H, PLA -CH-CH3), 4.55 (m, 2H, -ArOCH2CH2-), 4.35 (m, 2H, -ArOCH2CH2-), 1.57 (s, broad, 646 H, PLA -CHCH3).
BF2dnm(Br)PLA (8b)
The bromo-dinaphthyl polymer 8b was obtained as yellow crystalline foam: 693 mg (41%). Mn (GPC/MALS) = 26,000 Da, PDI = 1.21; Mn (NMR) = 26,200 Da. H NMR (600 MHz, CDCl3): δ 8.77 (m, broad, 2H, 1″ -ArH, 1′ -ArH), 8.17-8.11 (m, broad, 3H, 3″-, 8″, -ArH, 8′ -ArH), 7.92-7.85 (m, broad, 5H, 3′-, 7′, -ArH, 4″-, 5″, -7″-, ArH), 7.70 (d, J = 9, 1H, 4′ -ArH), 7.42 (s, 1H, -COCHCO-), 7.18 (s, 1H, 5′ -ArH), 5.31-5.11 (m, broad, 355 H, PLA -CH-CH3), 4.55 (m, 2H, -ArOCH2CH2-), 4.35 (m, 2H, -ArOCH2CH2-), 1.57 (s, broad, 1405 H, PLA -CHCH3).
BF2dnm(I)PLA (9a)
The iodo-dinaphthyl polymer 9a was obtained as yellow-orange foam: 98 mg (46%). Mn (GPC/MALS) = 12,500 Da, PDI = 1.12; (NMR) = 13,000 Da. 1H NMR (600 MHz), (CDCl3): δ 8.77 (s, broad, 2H, 1″-ArH, 1′-ArH), 8.36 (s, 1H, 5″-ArH), 8.19–8.10 (m, 2H, 8″-ArH, 8′-ArH), 7.95 – 7.87 (m, broad, 4H, 3′-, 4′-ArH, 3″-, 4″-ArH), 7.60 (s, 1H, 7″-ArH), 7.43 (s, 1H, COCHCO), 7.33 (s, 1H, 5′-ArH), 7.19 (d, J = 9, 1H, 5′-ArH), 5.30–5.11 (bm, 173H, PLA-CH-CH3), 4.62 – 4.55 (m, 2H, -ArOCH2CH2-), 4.37 – 4.34 (m, 3H, -ArOCH2CH2OH, PLA-OH)), 1.63-1.47 (s, broad, 596H, PLA-CH3).
BF2dnm(I)PLA (9b)
The iodo-dinaphthyl polymer 9a was obtained as yellow-orange foam: 314 mg (83%). Mn (GPC/MALS) = 23,200 Da, PDI = 1.19; (NMR) = 27,600 Da. 1H NMR (600 MHz), (CDCl3): δ 8.77 (s, broad, 2H, 1″-ArH, 1′-ArH), 8.36 (s, 1H, 5″-ArH), 8.19-8.10 (m, 2H, 8″-ArH, 8′-ArH), 7.95 – 7.87 (m, broad, 4H, 3′-, 4′-ArH, 3″-, 4″-ArH), 7.60 (s, 1H, 7″-ArH), 7.43 (s, 1H, COCHCO), 7.33 (s, 1H, 5′-ArH), 7.19 (d, J = 9, 1H, 5′-ArH), 5.30-5.11 (bm, 346 H, PLA-CH-CH3), 4.62 – 4.55 (m, 2H, -ArOCH2CH2-), 4.37 – 4.34 (m, 3H, -ArOCH2CH2OH, PLA-OH)), 1.63-1.47 (s, broad, 1290 H, PLA-CH3).
Cell Culture
4T1 cells (American Type Culture Collection, Manassas, VA) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Life Technologies, Carlsbad, CA) in a humidified incubator with 5% carbon dioxide. Cells were plated at a density of 10,000 cells per well of a 48-well plate. On the following day, cells were loaded with nanoparticles as described.56 Briefly, following media removal, the wells were rinsed with PBS plus calcium and magnesium (PBS+Ca,Mg), and media without serum was added. Nanoparticle solution (50 μg) was incubated with the cells for 1 h at 37 °C. Upon uptake completion, cells were rinsed with PBS+Ca,Mg (3X) to remove any unbound nanoparticles then growth media was replaced. Plates were placed in incubators at either 0.5% oxygen or ambient conditions for 18 h. Hypoxic plates were wrapped in parafilm following the incubation period and were unwrapped immediately before imaging began.
Microscopic Imaging
Fluorescence optical imaging was performed on an inverted Zeiss Axio Observer microscope with a mercury lamp. A QV2 multichannel imager (Photometrics) employed a sequence of beamsplitters to separate the emitted fluorescent light from the sample onto different quadrants on a Hamamatsu Orca Flash4 CMOS camera. The fluorescence and phosphorescence signals were filtered at 505 nm and 565 nm, respectively, with Δλ = 25 nm.
Image Data Analysis
Acquired images were first cropped to produce individual images of the 505 nm fluorescence and 565 nm phosphorescence. Images were corrected to a background reference, regions with low signal were removed with a mask, and then spatially aligned with an affine transformation. Ratios of the 505 nm image relative to the 565 nm image (F/P) were calculated for each time point. The F/P and brightfield images were superimposed for visualization.
RESULTS AND DISCUSSION
Synthesis
Difluoroboron dinaphthoylmethane poly(lactic acid) materials, BF2dnm(X)PLA (X = H, Br, I) (7–9), are generated from BF2dnm(X)OH initiators (4–6) via bulk polymerization of D,L-lactide in the presence of stannous octoate (Sn(oct)2) (Scheme 1). The β-diketonate ligands 1–3 were generated by Claisen condensation using NaH as the base. Corresponding boron initiators 4–6 were prepared by addition of boron trifluoride diethyl etherate in CH2Cl2/THF. Dinaphthyl boron complexes, the iodide-substituted sample in particular, showed limited solubility compared to other β-diketonate derivatives such as the previously reported napthyl phenyl derivative.33 Consequently, typical column chromatographic methods proved ineffective (e.g. silica; EtOAc, THF/hexanes, acetone or THF). Instead, the boron complexes were purified by recrystallization with acetone/EtOAc (BF2dnmOH, 4 and BF2dnm(Br)OH, 5) or pure acetone (BF2dnm(I)OH, 6) and were obtained as yellow-orange powders. Dye-dye interactions in polymer matrices play a key role in fluorescence properties.53,59 Though extended π conjugation can lead to greater π stacking and aggregation, presenting challenges for purification, fortuitously, this feature presents unique benefits for optical tuning.
Scheme 1.
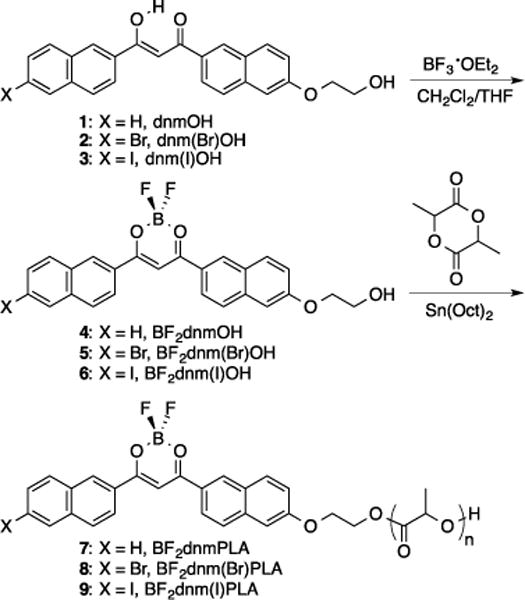
Synthesis of Difluoroboron Dinaphthoylmethane-Poly(lactic acid) Conjugates
Dye-polymer conjugates were synthesized as previously described.33,76 Polymers with various molecular weights (Mn (GPC) = 12 – 30 kDa) were generated by controlling the reaction time and lactide equivalents. The polymers were characterized by size-exclusion chromatography with multi-angle light scattering detection (SEC-MALS) and 1H NMR spectroscopy (Table 1). Unlike previously studied BF2dbmOH (phenyl-phenyl) initiators, these dinaphthyl dyes did not dissolve in lactide melts of low equivalences until after addition of the stannous octoate catalyst and polymerization commenced. Previous studies have shown that initial dissolution of the dye in the lactide melt is not crucial for growing polymers with narrow polydispersities.33 Previous preparations of low molecular weight polymers (5 – 10 kDa) utilized low lactide loadings (i.e. 50 equivalents).53 But under these conditions, dinaphthyl initiators had larger than expected MWs and large PDIs. However, increasing catalyst loading from 1/50 to 1/40 per initiator led to a decrease in PDI (e.g. BF2dnmOH (4), 1/50: PDI = 1.74 versus 1/40: PDI = 1.41). For high lactide equivalences (i.e. >200), BF2dnm(X)OH initiators dissolved in the monomer melts more easily and narrower PDIs were obtained. As previously described,33,53,58 reactions were typically stopped before complete monomer conversion to limit transesterification and thermal depolymerization side reactions.
Optical Properties in Solution
The optical properties of boron dyes and polymers were analyzed in dilute CH2Cl2 (Table 2). Extinction coefficients (ε), quantum yields (ΦF), and fluorescence lifetimes (τF) of initiators 4–6 were determined in dilute solutions to minimize aggregation (~10−6 M and optical density < 0.10). The dye-PLA conjugates (7–9) dissolved easily under these conditions in comparison to dye initiators. Absorption spectra of all dinapthyl initiators and polymers are nearly identical, with a slight red-shift in λabs for heavy atom substituted materials (λabs polymers: hydrogen (4) = 434 nm, bromine (5) = 436 nm, iodine (6) = 439 nm; Figure S1) and expected trends in extinction coefficients for initiators and polymers.33 Luminescence measurements showed green fluorescence for all initiators and polymers (e.g. initiators: λem: H (4) = 518 nm, Br (5) = 520 nm, I (6) = 521 nm). The efficiency of singlet emission varied substantially as evidenced by quantum yields of 0.66, 0.57, and 0.19 for H, Br and I dyes, respectively. Initiators with the heavy atoms Br and I exhibited lower fluorescence quantum yields due to enhanced crossover to the triplet state, followed by phosphorescence, oxygen quenching, or non-radiative decay in the organic solvent.79 Fluorescence lifetimes also became shorter with increasing weight of the heavy atom (τF: H (4) = 2.47 ns, Br (5) = 2.04 ns, I (6) = 0.74 ns).
Table 2.
Optical Properties of Boron Initiators and Polymers in CH2Cl2 Solution
| Sample | λabsa (nm) |
εb (M−1 cm−1) |
λemc (nm) |
τFd (ns) |
ΦFe | |
|---|---|---|---|---|---|---|
| BF2dnmOH | 4 | 434 | 65000 | 518 | 2.47 | 0.66 |
| BF2dnmPLA | 7a | 434 | 45000 | 505 | 2.27 | 0.62 |
| 7b | 434 | 40000 | 503 | 2.26 | 0.59 | |
| BF2dnm(Br)OH | 5 | 436 | 50000 | 520 | 2.17 | 0.57 |
| BF2dnm(Br)PLA | 8a | 436 | 49000 | 516 | 2.04 | 0.51 |
| 8b | 436 | 45000 | 516 | 2.03 | 0.51 | |
| BF2dnm(I)OH | 6 | 439 | 60000 | 521 | 0.74 | 0.19 |
| BF2dnm(I)PLA | 9a | 439 | 51000 | 513 | 0.64 | 0.15 |
| 9b | 439 | 49000 | 512 | 0.60 | 0.15 |
Absorption maxima.
Extinction coefficients calculated at the absorption maxima.
Fluorescence emission maxima excited at 369 nm (xenon lamp).
Fluorescence lifetime excited at 369 nm (LED) monitored at the emission maximum. All fluorescence lifetimes are fitted with single-exponential decay.
Relative quantum yield versus anthracene in EtOH as a standard.78
Optical Properties of Films
Boron polymers were also studied in the solid state as films. Solution-cast films were prepared in vials by dissolving dye-PLA samples in CH2Cl2 followed by slow evaporation under nitrogen. Luminescence properties of the samples are summarized in Table 3. The increased conjugation of the dinaphthyl dyes (Np-Np) has led to red-shifted fluorescence in comparison to naphthyl-phenyl (Np-Ph) and phenyl-phenyl (Ph-Ph) dye–PLA analogues.33,53 For example, for ~12 kDa polymer films without halide heavy atoms emission maxima λem are as follows: BF2dnmPLA (Np-Np) = 523 nm, BF2nbmPLA (Np-Ph) = 461 nm, BF2dbmPLA (Ph-Ph) = 437 nm. For polymers of similar MWs, the presence of halide substituents, Br or I, results in slight redshifts in fluorescence (H (7a): 523 nm Br (8a): 534 nm I (9a): 535 nm).
Table 3.
Optical Properties of Boron Polymer Films
| Sample | Polymer | Fluorescence | Phosphorescence | ||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Mna (kDa) |
Dye Loading b (%) |
λ Fc (nm) |
τpw0d (ns) |
λ Pe (nm) |
τpw0f (ms) |
||
| BF2dnmPLA | 7a | 12.2 | 3.5 | 523 | 11.7 | 541 | 50 |
| 7b | 31.8 | 1.4 | 494 | 7.1 | 509 | 156 | |
| 7c | g | 1.0 | 484 | 3.3 | 499 | 381 | |
| BF2dnm(Br)PLA | 8a | 12.5 | 4.0 | 534 | 6.3 | 567 | 17 |
| 8b | 26.1 | 2.0 | 506 | 4.1 | 555 | 26 | |
| 7c | g | 1.0 | 490 | 3.2 | 564 | 38 | |
| BF2dnm(I)PLA | 9a | 12.5 | 4.5 | 535 | 1.2 | 576 | 4.5 |
| 9b | 23.2 | 2.4 | 503 | 0.9 | 569 | 6.3 | |
| 9c | g | 1.0 | 496 | 0.8 | 566 | 7.0 | |
Number-average molecular weight.
Weight percent dye in the polymer.
Steady-state fluorescence emission maximum under air excited at 400 nm (xenon lamp).
Fluorescence lifetime excited at 369 nm (LED) monitored at the emission maximum. All fluorescence lifetimes are fit with triple-exponential decay.
Delayed emission maxima under N2 excited at 400 nm (xenon flash lamp; 1 ms delay).
Pre-exponential weighted RTP lifetime excited at 400 nm (xenon flash lamp; 1 ms delay). RTP lifetime fit to triple-exponential decay.
Blends prepared from 7a, 8a, 9a and PLA.
The solid-state fluorescence properties of BF2bdks are highly dependent on the molecular weight (MW). Changing dye loading in this way is facile method for tuning fluorescence. Previously reported dbm (Ph-Ph)53,58 and nbm (Np-Ph)33 substituted PLA polymers exhibited red-shifted emission for low MW polymers due to increase dye-dye interactions stabilizing excited state dipoles. Longer MW polymers decreased these dye-dye interactions resulting in blue-shifted emission in the solid-state. This same trend was observed in this study with dnm dyes (e.g. BF2dnmPLA: ~12.2 kDa (7a) = 523 nm and ~31.8 kDa (7b) = 494 nm. Fluorescence lifetime (τF) is also influenced by polymer MW. In short polymers, neighboring fluorophores can stabilize the excited state, producing longer fluorescence lifetimes than longer polymers (e.g. BF2dnmPLA: 7a: τF = 11.7 ns, 7b: τF = 7.1 ns). The halide can also influence the fluorescence lifetime. Heavy atoms increase the rate of intersystem crossing (ISC) to the triplet state by enhancing spin-orbit coupling.69 This resulted in less intense fluorescence, shown in the images in Figure 2, and shorter fluorescence lifetimes (7a: τF = 11.7 ns, 8a: τF = 6.3 ns, 9a: τF = 1.2 ns).
Figure 2.
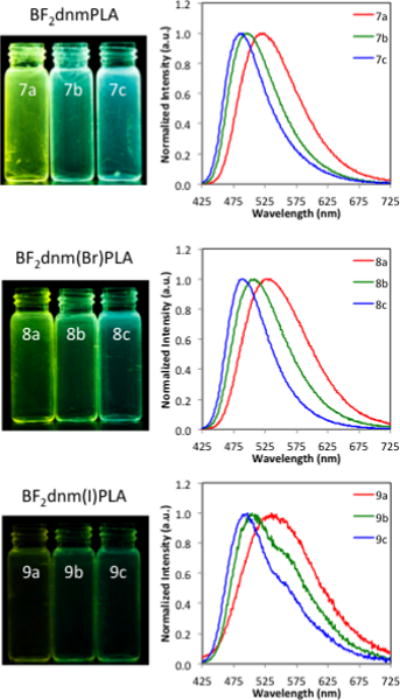
Effects of dye loading on fluorescence. Images of polymer film fluorescence (λex = 354 nm) and fluorescence spectra (λex = 400 nm) for BF2dnmPLA (7a–c), BF2dnm(Br)PLA (8a–c), and BF2dnm(I)PLA (9a–c) under air.
Delayed emission was investigated for the polymers under a nitrogen atmosphere. Room-temperature phosphorescence (RTP) was observed for all BF2dnm(X)PLA materials. Iodide substituted dyes show the expected trends in phosphorescence wavelength (λP). Emission is redshifted with extended conjugation (λP: BF2dnm(I)PLA: 565 nm versus BF2dbm(I)PLA: 526 nm, ~12 kDa). The hydrogen derivative BF2dnm(I)PLA, however, appears anomalous (BF2dbmPLA: 509 nm, BF2nbmPLA 545 nm, BF2dnmPLA (9a): 541 nm, ~12 kDa). This can be explained by the fact that delayed emission maxima are comprised of both RTP and thermally activated delayed fluorescence (TADF), which is commonly observed in BF2bdkPLA materials.52,53,61,63 For example, Klimant et al. recently reported BF2 and aluminum (III) chelates of 9-hydroxyphenalanone in polystyrene and Teflon matrices with TADF and RTP emissive properties.79 With increased dye conjugation the magnitude of the wavelength shift in phosphorescence (ΔλP) is less than the shift in fluorescence (ΔλF). Consequently, the fluorescence to phosphorescence energy gap (ΔE) gets smaller for Np-Np (dnm) systems versus Np-Ph (nbm) and Ph-Ph (dbm) systems. This has important implications for both thermally activated delayed fluorescence and ratiometric sensing with these dual emissive materials. While small singlet triplet energy gaps facilitate intersystem crossing, they also enhance thermal back population to singlet state. As illustrated by BF2dnmPLA (7a), the gap between λF and the delayed emission maximum λDE is very small (λF = 523 nm, λDE = 541 nm). For this sample the delayed emission is dominated by TADF, which accounts for the unexpected blue shift. This hypothesis is confirmed by low temperature measurements where TADF is absent and phosphorescence dominates.80,81 For example, delayed emission for BF2dnmPLA (7b) changes from green (509 nm; delayed fluorescence plus phosphorescence) at RT to red-orange (580 nm; phosphorescence) at 77K (Figures 3 and S2).
Figure 3.
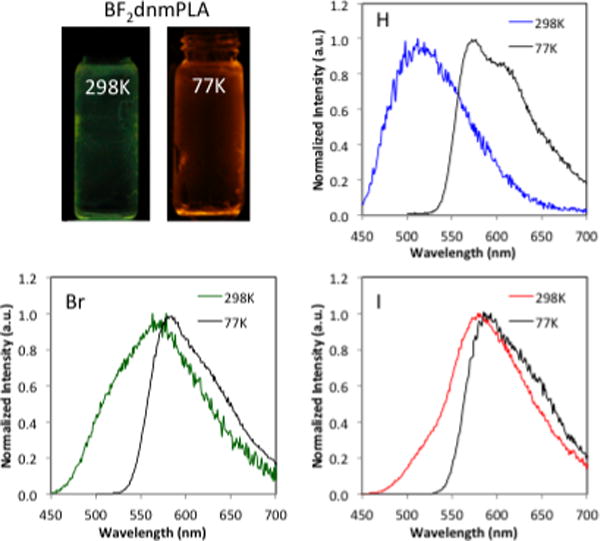
Temperature dependent delayed emission of BF2dnm(X)PLA. Images of BF2dnmPLA (7b) and delayed emission spectra BF2dnmPLA (7b) (H), BF2dnm(Br)PLA (8b) (Br), and BF2dnm(I)PLA (8b) (I), (λex = 400 nm, 2 ms delay; room temperature (298 K) and liquid nitrogen (77K)).
Heavy atoms also influence delayed emission. Like fluorescence λF, the delayed emission maxima for the bromide dye 8a (567 nm) and iodide dye 9a (576 nm) are redshifted compared to the non-halogenated analogue 7a (541 nm) (Figure S3). Also, the singlet triplet energy gaps for the halogenated dyes are greater than for the 7a. Rapid intersystem crossing (ISC) for the heavy atom substituted dyes results in short phosphorescence lifetimes (τP) (hydrogen (7a) 50.1 ms, bromine (8a) 16.9 ms, iodine (9a) 4.5 ms) and increased phosphorescence intensity relative to the hydrogen analogue (Figure 4). Unlike the H dye where TADF dominates at room temperature and longer wavelength phosphorescence is present at low temperature, for bromine (8b) and iodine (9b) dyes have similar delayed emission maxima at room temperature and 77K, however, at low temperature blue region of the band corresponding to TADF disappears and the bandwidth narrows.
Figure 4.
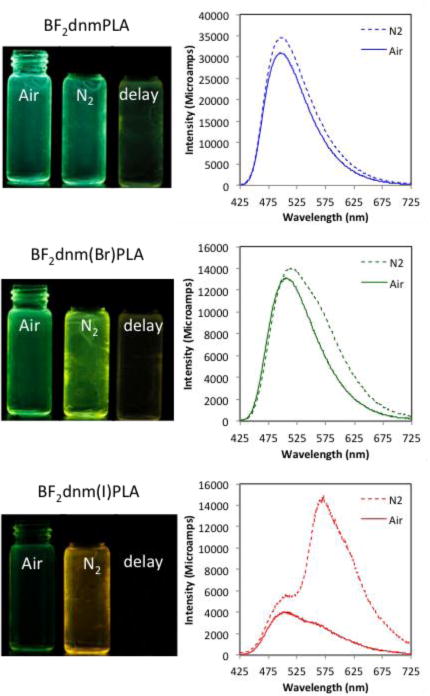
Images and total emission spectra of boron polymers BF2dnmPLA (7b), BF2dnm(Br)PLA (8b), and BF2dnm(I)PLA(9b) in air and N2. Photographs were taken with UV lamp excitation (λmax = 354 nm); delayed emission images (i.e. delay) were captured after the UV lamp was turned off.
While materials with narrow singlet triplet gaps and strong TADF are desirable for OLED,82–85 they are problematic for ratiometric oxygen sensing, given the need for two discrete peaks, namely, stable fluorescence for use as the standard and oxygen sensitive phosphorescence as the sensor. Typical ratiometric sensing materials are multicomponent, comprised of standard and sensor dyes that are combined in a suitable matrix.86–88 Fluorescence and phosphorescence wavelengths and signal intensities are easily controlled by dye selection, and intensities, by dye loading. In dual emissive dyes such as BF2bdkPLAs, different approaches to wavelength and intensity modulation are required. As previously described,33,58 halide heavy atom substitution increases phosphorescence intensity, which is also affected by molecular weight. Dye loading modulates the singlet to triplet energy gap, which for the dinaphthyl materials in this study also helps to addresses the TADF issue.
To adapt these materials for ratiometric sensing and achieve two discrete peaks it is necessary to increase the singlet-triplet energy gap with dye loading. All dye-polymer conjugates, 7a–9a and 7b–9b, exhibit TADF due to considerable overlap in fluorescence and phosphorescence peaks. Emission spectra under nitrogen appear as a single broad band with the exception of longer 23 kDa polymer 9b in which fluorescence appears as a high-energy shoulder on the more intense phosphorescence peak. To test whether fluorescence and phosphorescence peaks can be separated, more dilute dye/polymer samples are required. However, attempts to grow higher molecular weight materials did not yield the targeted products. Broad PDIs and dye degradation were observed for higher loadings and longer reaction times. Higher molecular weights were not achieved. Previously it was demonstrated that spectral properties could be modeled with dye/polymer blends at high dilution.53,63 To avoid phase separation at higher dye loadings, in this study dye-PLA/PLA blends were prepared. First, it was confirmed that spectral properties of 7b (1.4%) could be replicated by blending 7a (3.5%) and PLA. Then samples 7c–9c were prepared in a comparable manner to achieve 1% dye loading. Although 1% loadings were not sufficient to produce peak separation in the H and Br materials, two discrete peaks are evident in the spectrum for BF2dnm(I)PLA/PLA (9c) (Figure S4). Even at 1.0 % dye loading (9c), the phosphorescence was 3.4 times stronger than the fluorescence in air. Further dilution to 0.5 and 0.2% dye loadings shows slightly better peak resolution (Figure 5). However, both fluorescence and phosphorescence blueshift, which may indicate a change from aggregate to monomer emission, and the corresponding drop in intensity with lower dye loadings limits signal detection (e.g. lifetimes), making these blends less practical for lifetime and dual mode lifetime/intensity imaging methods. With the exception of BF2nbm(Br)PLA (26 kDa, 1.9 dye loading) which exhibited a fluorescence to phosphorescence intensity ratio (F/P) of 0.95,33 most boron bdkPLA materials with large singlet triplet energy gaps display weak phosphorescence at low dye loadings (i.e. high MWs). For example, BF2dbm(I)PLA (17.6 kDa, 2.6% dye loading) showed a dramatic decrease in phosphorescence intensity (i.e. F/P = 1.67). The BF2dnm(I)PLA sample 9c with only 1% dye loading and discrete fluorescence and phosphorescence peaks exhibits the most intense relative phosphorescence to fluorescence value to date (F/P = 0.26). Thus, extended conjugation and the propensity of BF2dnm(I) materials to π–stack89 or aggregate58 results in enhanced optical properties. That dual luminescence can be achieved with very low dye loadings, is a promising result for ratiometric oxygen sensing.
Figure 5.
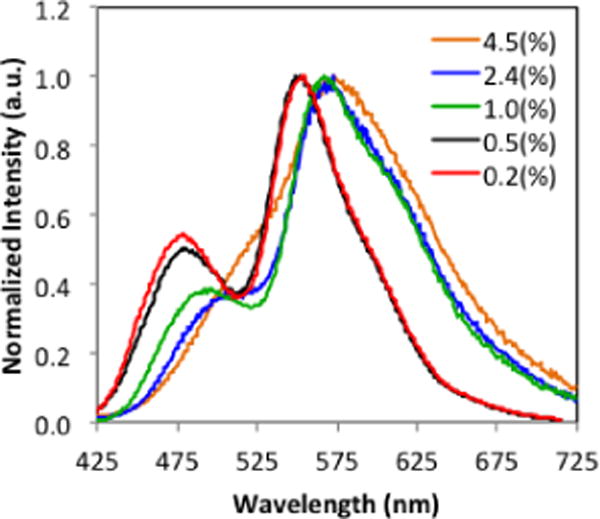
Fluorescence to phosphorescence ratio tuning for BF2dnm(I)PLA/PLA blends at different dye loadings. (See Table S1 for blend preparation.)
Nanoparticles
Polymers were fabricated into nanoparticles to investigate their potential as oxygen nanosensors. Higher MW polymers 7b–9b were selected for study given longer phosphorescence lifetimes that correlate with higher oxygen sensitivity, making all of these materials suitable for lifetime imaging methods. Given peak separation in film samples (Figures 4 and 5), the iodide polymer 9b and blend 9b/PLA served as the focus of ratiometric sensing experiments. Boron nanoparticles (BNPs) were fabricated by nanoprecipitation (i.e. solvent displacement) from DMF/H2O by previously described methods.55
Nanoparticles were characterized with respect to size and optical properties. Nanoparticle size (hydrodynamic radius, RH), polydispersity (PD) and luminescence properties are summarized in Table 4. Dynamic light scattering measurements indicated boron nanoparticle radii ranging from 29 – 47 nm, suitable for cellular uptake,90–92 and polydispersities were also typical for BF2bdkPLA materials.55,56,58 Nanoparticles made from dye-polymer conjugates 7b–9b exhibited no changes in optical properties or DLS measurements after five days. In contrast, for the blend sample 9b/PLA, changes were noted in spectra and DLS data after approximately two hours that became increasing pronounced over time. For example, multimodal distributions were evident in DLS measurements and phosphorescence intensity decreased. Stereoblock formation93,94 and PEGylation95,96 could enhanced blend nanoparticle stability. Absorption properties of BNPs (Table 4) are nearly identical to dye solutions (λmax = 435 – 440 nm), and emission properties mirror those of films (Figure 6).
Table 4.
Boron Nanoparticle Characterization
| Sample | DLSa | Absorbance | Fluorescence | Phosphorescence | |||||
|---|---|---|---|---|---|---|---|---|---|
|
RH (nm) |
PD | λmax (nm) |
λFb (nm) |
τFc (ns) |
λPd (nm) |
τPe (ms) |
|||
| BF2dnmPLA | 7b | NPH | 46.0 | 0.29 | 434 | 493 | 2.87 | 503 | 71.4 |
| BF2dnm(Br)PLA | 8b | NPBr | 36.2 | 0.31 | 436 | 500 | 2.11 | 560 | 12.6 |
| BF2dnm(I)PLA | 9b | NPI | 29.1 | 0.15 | 438 | 498 | 1.20 | 570 | 5.7 |
| BF2dnm(I)PLA | 9b/PLAf | NPI* | 47.0 | 0.35 | g | 479 | 0.80 | 565 | 5.9 |
BNP size and polydispersity determined by DLS (Figure S5)
Fluorescence maximum under air (λex = 400 nm)
Fluorescence lifetime at maximum emission under air (λex = 369 nm LED)
Phosphorescence maxima in delayed emission spectra at room temperature under N2 (λex = 400 nm xenon lamp)
Phosphorescence lifetime at delayed emission spectra maxima under N2 (λex = 400 nm xenon flash lamp)
BNPs made by co-precipitation of polymer 9b and PLA in 1:10 ratio by mass (0.2% loading)
Dye absorbance is negligible (Figure S6)
Figure 6.
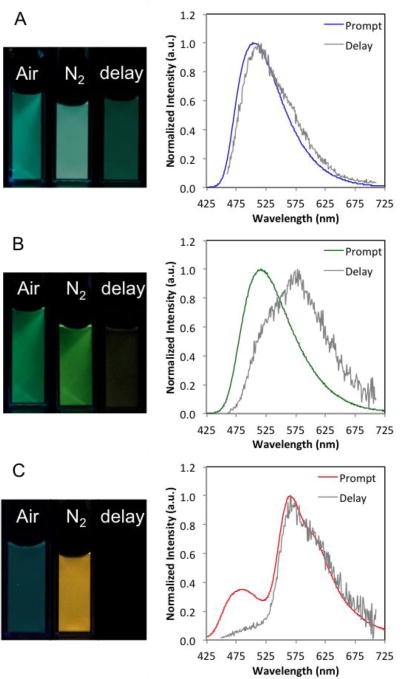
Boron nanoparticles: (A) NPH (7b), (B) NPBr (8b), and (C) NPI* (9b/PLA). Images under air, nitrogen and delayed (i.e. under nitrogen after the UV lamp is turned off.) Total emission spectra under nitrogen (Prompt) and delayed emission spectra under nitrogen (Delay).
Though fluorescence lifetimes are similar for particles and films, phosphorescence lifetimes are more sensitive to environmental changes, with BNPs show decreased delayed emission lifetimes compared to film counterparts (hydrogen 7b: film τP = 156 ms, BNP τP = 71 ms; bromine 8b: film τP = 26 ms, BNP τP = 12 ms; iodine 9b: film τP = 6.3 ms, BNP τP = 5.7 ms).
Oxygen Sensing
Given suitability for ratiometric oxygen sensing and imaging, iodide nanoparticles NPI were calibrated for oxygen sensitivity (Figure 7) and utilized in tumor cell O2 imaging studies. Due to the small ΔE, TADF is evident in NPI as for 9b films. Lifetimes for both delayed fluorescence and phosphorescence are independently measureable (τ497 nm = 7.2 ms and τ570 nm = 5.7 ms). Data are presented as wavelength versus intensity to illustrate this point, rather than the typical wavelength versus normalized fluorescence plots.3,33,58 However, fluorescence changes are minimal compared to phosphorescence and calibration plots are still essentially linear from 0–5.3% O2 (R2 = 0.981).
Figure 7.
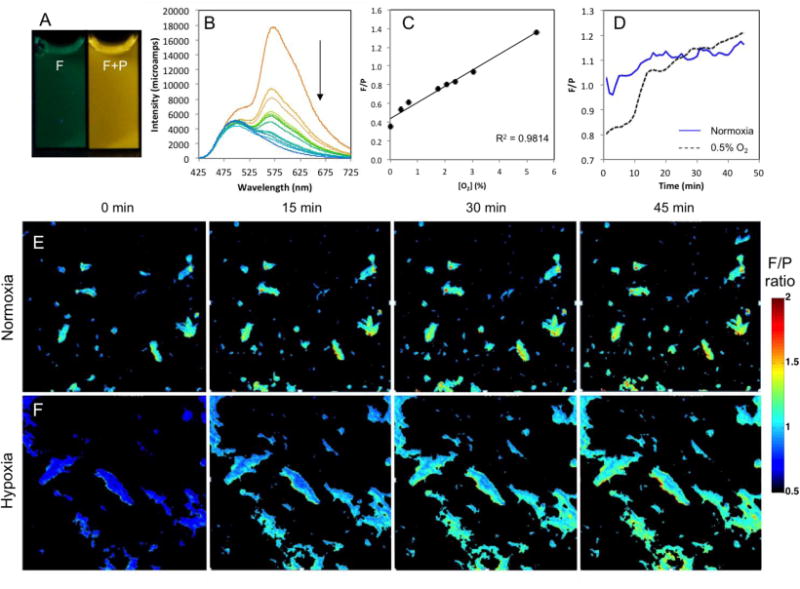
Boron nanoparticle oxygen sensing. (A) Images of BNPs made from BF2dnm(I)PLA 9b (NPI) in air (fluorescence) and N2 (fluorescence + phosphorescence) (λex = 354 nm). (B) Total emission spectra of NPI at O2 levels 0–21%. Arrow indicates decreasing phosphorescence at 570 nm with increasing O2. (C) Fluorescence intensity (505 nm) over phosphorescence intensity (565 nm) (F/P) calibration plot showing a linear fit in the 0 – 5.3% O2 range. (D) QuadView microscope quantification of F/P intensity ratio monitored for 45 min. (E) QuadView microscope images of NPI within 4T1 cells under normoxia (air; ambient). (F) QuadView microscope images of the hypoxia (0.5% O2) to normoxia transition.
Methods for monitoring oxygen levels with boron nanoparticles in in vitro and in vivo contexts have been developed by Dewhirst and Palmer.57,97 BNPs were taken up by 4T1 cells and maintained in two different oxygen environments overnight (~18 hours). The cellular luminescence properties were recorded every minute over a 45 minute time course (Figure 7 and SI Videos). Cells maintained under normoxia conditions showed a relatively steady F/P ratio over this time (~1.1 F/P). BNP-loaded cells incubated under hypoxic conditions were exposed to normoxia (air; ambient) and monitored by microscopy. A robust increase in the F/P ratio was observed as the oxygen levels equilibrated to room air (0.8 – 1.2). These changes occurred very slowly in the first ten minutes, but rapidly in the next ten minutes as the F/P ratios approach those observed under normoxia conditions. Thus, these nanoparticles demonstrated a rapid and dynamic oxygen-sensing activity within cells. Interestingly, nanoparticles under either condition demonstrated an upward drift of the F/P ratio as the imaging proceeded. This may be due to partial dye degradation over the course of the experiment, changes in the dye environment, or other factors that influence polymer and nanoparticle properties. We are currently investigating the nature of this signal drift as boron materials and imaging parameters are further optimized for sensing applications.
CONCLUSION
A series of hydroxyl-functionalized difluoroboron dinaphthoylmethane dyes were synthesized for the bulk polymerization of DL-lactide to yield new dye-PLA conjugates. Dyes showed tunable fluorescence, phosphorescence, and thermally activated delayed fluorescence in the solid state dependent upon dye loading (i.e. PLA molecular weight). At room temperature under N2, thermally activated delayed fluorescence dominated for the non-halogenated material, BF2dnmPLA, whereas phosphorescence was more intense for the bromine and iodine substituted polymers. Given the propensity of dinaphthyl dyes to aggregate, low dye loadings (i.e. higher MW polymers) were required to achieve good spectral separation of fluorescence and phosphorescence signals for ratiometric oxygen sensing. Boron dye polymers were successfully fabricated into nanoparticles and were calibrated for O2 sensing. The nanoparticles can be excited outside the ultraviolet range, which is important for biological imaging applications. The hydrogen and bromine derivatives show long phosphorescence lifetimes and high sensitivity to O2 quenching, making them useful for lifetime based sensing methods. The iodine sample, BF2dnm(I)PLA, on the other hand, displays distinct fluorescence and phosphorescence peaks making it compatible with ratiometric imaging. These nanoparticles demonstrated the ability both to be loaded into cells and to respond to alterations of oxygen levels. Thus, ratiometric imaging with these nanoparticles shows promise in determining intracellular oxygen levels as a determinant for therapeutic action.
Supplementary Material
Acknowledgments
This research was supported by the National Cancer Institute of the National Institutes of Health (R01 CA167250). We gratefully acknowledge the University of Virginia Cancer Center for a fellowship to JSK through the Farrow Fellowship Fund and the NCI Cancer Center Support Grant, P30 CA44579. We also acknowledge the North Carolina Biotechnology Center (2013-MRG-1111) and the Optical Molecular Imaging and Analysis Facility at Duke University for microscopic imaging. We thank Prof. J. N. Demas, Tiandong Liu, and Caroline Kerr for their advice and assistance.
Footnotes
SUPPORTING INFORMATION
UV/vis spectra for boron polymerization initiators (4–6), dye/PLA blend preparation (7c–9c), images and emission spectra of polymers 7a–9a and blends 7c–9c in air and nitrogen, images of polymers at low temperature (77K), DLS and absorption spectra of nanoparticles, 1H NMR spectra of ligands, dyes, and polymers (1–9), and time-lapse videos of 45 minute time courses of both normoxia and normoxia-to-hypoxia experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cheng F, Jakle F. Polym Chem. 2011;2:2122–2132. [Google Scholar]
- 2.Fraser CL, Zhang G. Mater Today. 2009;12:38–40. [Google Scholar]
- 3.Bowers DT, Tanes ML, Das A, Lin Y, Keane NA, Neal RA, Ogle ME, Brayman KL, Fraser CL, Botchwey EA. ACS Nano. 2014;8:12080–12091. doi: 10.1021/nn504332j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickinson BC, Chang CJ. Nat Chem Bio. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickinson BC, Huynh C, Chang CJ. J Am Chem Soc. 2010;132:5906–5915. doi: 10.1021/ja1014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Gracia Lux C, Joshi-Barr S, Nguyen T, Mahmoud E, Schopf E, Fomina N, Almutairi A. J Am Chem Soc. 2012;134:15758–14764. doi: 10.1021/ja303372u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi S, Akiyama S, Tamao K. J Am Chem Soc. 2001;123:11372–11375. doi: 10.1021/ja015957w. [DOI] [PubMed] [Google Scholar]
- 8.Maeda H, Haketa Y, Nakanishi T. J Am Chem Soc. 2007;129:13661–12674. doi: 10.1021/ja074435z. [DOI] [PubMed] [Google Scholar]
- 9.Ran C, Xu X, Raymond SB, Ferrara BJ, Neal K, Bacskai BJ, Medarova Z, Moore A. J Am Chem Soc. 2009;131:15257–15264. doi: 10.1021/ja9047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Tian Y, Li Z, Tian X, Sun H, Liu H, Moore A, Ran C. J Am Chem Soc. 2013;135:16397–16409. doi: 10.1021/ja405239v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson JC, Meimetis LG, Hilderbrand SA, Weissleder R. Angew Chem, Int Ed. 2013;125:7055–7058. doi: 10.1002/anie.201301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devaraj NK, Hilderbrand S, Upadhyay R, Mazitschek R, Weissleder R. Angew Chem, Int Ed. 2010;122:2931–2934. doi: 10.1002/anie.200906120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao D, Li G, Wu D, Qin X, Neuhaus P, Cheng Y, Yang S, Lu Z, Pu X, Long C. Angew Chem, Int Ed. 2013;125:13921–13925. doi: 10.1002/anie.201304824. [DOI] [PubMed] [Google Scholar]
- 14.Loudet A, Burgess K. Chem Rev. 2007;107:4891–4932. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- 15.Perumal K, Garg JA, Blacque O, Saiganesh R, Kabilan S, Balasubramanian KK, Venkatesan K. Chem Asian J. 2012;7:2670–2677. doi: 10.1002/asia.201200477. [DOI] [PubMed] [Google Scholar]
- 16.Araneda JF, Piers WE, Heyne B, Parvez M, McDonald R. Angew Chem, Int Ed. 2011;50:12214–12217. doi: 10.1002/anie.201105228. [DOI] [PubMed] [Google Scholar]
- 17.Yoshii R, Tanaka K, Chujo Y. Macromolecules. 2014;47:2268–2278. [Google Scholar]
- 18.Yoshii R, Nagai A, Tanaka K, Chujo Y. Chem Eur J. 2013;19:4506–4512. doi: 10.1002/chem.201203703. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Lin W, Wang J, Guan X. Org Lett. 2013;15:1768–1771. doi: 10.1021/ol400605x. [DOI] [PubMed] [Google Scholar]
- 20.Koch M, Perumal K, Blacque O, Garg JA, Saiganesh R, Kabilan S, Balasubramanian KK, Venkatesan K. Angew Chem, Int Ed. 2014;126:6496–6500. doi: 10.1002/anie.201402199. [DOI] [PubMed] [Google Scholar]
- 21.Yoshii R, Nagai A, Tanaka K, Chujo Y. Macromol Rapid Commun. 2014;35:1315–1319. doi: 10.1002/marc.201400198. [DOI] [PubMed] [Google Scholar]
- 22.Poon C-T, Lam WH, Wong H-L, Yam VW-W. J Am Chem Soc. 2010;132:13992–13993. doi: 10.1021/ja105537j. [DOI] [PubMed] [Google Scholar]
- 23.Chibani S, Charaf-Eddin A, Mennucci B, Le Guennic B, Jacquemin D. J Chem Theory Comput. 2014;10:805–815. doi: 10.1021/ct4009848. [DOI] [PubMed] [Google Scholar]
- 24.Galer P, Korosec RC, Vidmar M, Sket B. J Am Chem Soc. 2014;136:7383–7394. doi: 10.1021/ja501977a. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Zhang G. Anal Methods. 2012;4:2641–2643. [Google Scholar]
- 26.Reichardt C. Chem Rev. 1994;94:2319–2358. [Google Scholar]
- 27.Kubota Y, Sakuma Y, Funabiki K, Matsui M. J Phys Chem A. 2014;118:8717–8729. doi: 10.1021/jp506680g. [DOI] [PubMed] [Google Scholar]
- 28.Zhang G, Fiore GL, St Clair TL, Fraser CL. Macromolecules. 2009;42:3162–3169. [Google Scholar]
- 29.Zhang G, Lu J, Sabat M, Fraser CL. J Am Chem Soc. 2010;132:2160–2162. doi: 10.1021/ja9097719. [DOI] [PubMed] [Google Scholar]
- 30.Liu T, Chien AD, Lu J, Zhang G, Fraser CL. J Mater Chem. 2011;21:8401–8408. [Google Scholar]
- 31.Nguyen ND, Zhang GQ, Lu JW, Sherman AE, Fraser CL. J Mater Chem. 2011;21:8409–8415. [Google Scholar]
- 32.Sun X, Zhang X, Li X, Liu S, Zhang G. J Mater Chem. 2012;22:17332–17339. [Google Scholar]
- 33.Samonina-Kosicka J, DeRosa CA, Morris WA, Fan Z, Fraser CL. Macromolecules. 2014;47:3736–3746. doi: 10.1021/ma5006606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sánchez I, Nuñez C, Campo JA, Torres MR, Lodeiro C. J Mater Chem C. 2014;2:9653–9665. [Google Scholar]
- 35.Nagai A, Kokado K, Nagata Y, Chujo Y. Macromolecules. 2008;41:8295–8298. [Google Scholar]
- 36.Zhang X, Cui M, Zhou R, Chen C, Zhang G. Macromol Rapid Commun. 2014;35:566–573. doi: 10.1002/marc.201300834. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka K, Tamashima K, Nagai A, Okawa T, Chujo Y. Macromolecules. 2013;46:2969–2975. [Google Scholar]
- 38.Zhang G, Lu J, Fraser CL. Inorg Chem. 2010;49:10747–10749. doi: 10.1021/ic902591s. [DOI] [PubMed] [Google Scholar]
- 39.Xin L, Chen Y-Z, Niu L-Y, Wu L-Z, Tung C-H, Tong Q-X, Yang Q-Z. Org Biomol Chem. 2013;11:3014–3019. doi: 10.1039/c3ob40376a. [DOI] [PubMed] [Google Scholar]
- 40.Smith LF, Blight BA, Park H-J, Wang S. Inorg Chem. 2014;53:8036–8044. doi: 10.1021/ic500944t. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Xie T, Cui M, Yang L, Sun X, Jiang J, Zhang G. ACS Appl Mater Interfaces. 2014;6:2279–2284. doi: 10.1021/am405209w. [DOI] [PubMed] [Google Scholar]
- 42.Bolton O, Lee K, Kim H-J, Lin KY, Kim J. Nat Chem. 2011;3:205–210. doi: 10.1038/nchem.984. [DOI] [PubMed] [Google Scholar]
- 43.Yuan WZ, Shen XY, Zhao H, Lam JW, Tang L, Lu P, Wang C, Liu Y, Wang Z, Zheng Q. J Phys Chem C. 2010;114:6090–6099. [Google Scholar]
- 44.Kwon MS, Lee D, Seo S, Jung J, Kim J. Angew Chem, Int Ed. 2014;53:11177–11181. doi: 10.1002/anie.201404490. [DOI] [PubMed] [Google Scholar]
- 45.Wang X-d, Wolfbeis OS. Chem Soc Rev. 2014;43:3666–3761. doi: 10.1039/c4cs00039k. [DOI] [PubMed] [Google Scholar]
- 46.Demas JN, DeGraff B, Coleman PB. Anal Chem. 1999;71:793A–800A. doi: 10.1021/ac9908546. [DOI] [PubMed] [Google Scholar]
- 47.Meier RJ, Fischer LH, Wolfbeis OS, Schäferling M. Sensor Actuat B-Chem. 2013;177:500–506. [Google Scholar]
- 48.Sakadžić S, Roussakis E, Yaseen MA, Mandeville ET, Srinivasan VJ, Arai K, Ruvinskaya S, Devor A, Lo EH, Vinogradov SA. Nat Methods. 2010;7:755–759. doi: 10.1038/nmeth.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R. Nature. 2014;508:269–273. doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baggaley E, Botchway SW, Haycock JW, Morris H, Sazanovich IV, Williams JG, Weinstein JA. Chem Sci. 2014;5:879–886. [Google Scholar]
- 51.Ehgartner J, Wiltsche H, Borisov SM, Mayr T. Analyst. 2014;139:4924–4933. doi: 10.1039/c4an00783b. [DOI] [PubMed] [Google Scholar]
- 52.Zhang G, Chen J, Payne SJ, Kooi SE, Demas JN, Fraser CL. J Am Chem Soc. 2007;129:8942–8943. doi: 10.1021/ja0720255. [DOI] [PubMed] [Google Scholar]
- 53.Zhang G, Kooi SE, Demas JN, Fraser CL. Adv Mater. 2008;20:2099–2104. [Google Scholar]
- 54.Zhang G, St Clair TL, Fraser CL. Macromolecules. 2009;42:3092–3097. [Google Scholar]
- 55.Pfister A, Zhang G, Zareno J, Horwitz AF, Fraser CL. ACS Nano. 2008;2:1252–1258. doi: 10.1021/nn7003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Contreras J, Xie JS, Chen YJ, Pei H, Zhang GQ, Fraser CL, Hamm-Alvarez SF. ACS Nano. 2010;4:2735–2747. doi: 10.1021/nn901385y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palmer GM, Fontanella AN, Zhang GQ, Hanna G, Fraser CL, Dewhirst MW. J Biomed Opt. 2010;15:066021–066021. doi: 10.1117/1.3523363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang G, Palmer GM, Dewhirst MW, Fraser CL. Nat Mater. 2009;8:747–751. doi: 10.1038/nmat2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Helmchen F, Denk W. Nat Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- 60.Rubart M. Circ Res. 2004;95:1154–1166. doi: 10.1161/01.RES.0000150593.30324.42. [DOI] [PubMed] [Google Scholar]
- 61.Payne SJ, Zhang G, Demas JN, Fraser CL, Degraff BA. Appl Spectrosc. 2011;65:1321–1324. doi: 10.1366/10-06223. [DOI] [PubMed] [Google Scholar]
- 62.Borisov SM, Krause C, Arain S, Wolfbeis OS. Adv Mater. 2006;18:1511–1516. [Google Scholar]
- 63.Xu S, Evans RE, Liu T, Zhang G, Demas JN, Trindle CO, Fraser CL. Inorg Chem. 2013;52:3597–3610. doi: 10.1021/ic300077g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butler T, Morris WM, Samonina-Kosicka J, Fraser CL. Chem Commun. 2015;51:3359–3362. doi: 10.1039/c4cc09439e. [DOI] [PubMed] [Google Scholar]
- 65.Wang X-H, Peng H-S, Ding H, You F-T, Huang S-H, Teng F, Dong B, Song H-W. J Mater Chem. 2012;22:16066–16071. [Google Scholar]
- 66.Cogné-Laage E, Allemand J-F, Ruel O, Baudin J-B, Croquette V, Blanchard-Desce M, Jullien L. Chem Euro J. 2004;10:1445–1455. doi: 10.1002/chem.200305321. [DOI] [PubMed] [Google Scholar]
- 67.McGehee MD, Bergstedt T, Zhang C, Saab AP, O’Regan MB, Bazan GC, Srdanov VI, Heeger AJ. Adv Mater. 1999;11:1349–1354. [Google Scholar]
- 68.Peng H, Stich MI, Yu J, Sun L, Fischer LH, Wolfbeis OS. Adv Mater. 2010;22:716–719. doi: 10.1002/adma.200901614. [DOI] [PubMed] [Google Scholar]
- 69.Lower S, El-Sayed M. Chem Rev. 1966;66:199–241. [Google Scholar]
- 70.McClure DS. J Chem Phys. 1949;17:905–913. [Google Scholar]
- 71.Williams DBG, Lawton M. J Org Chem. 2010;75:8351–8354. doi: 10.1021/jo101589h. [DOI] [PubMed] [Google Scholar]
- 72.Irvine MW, Costa BM, Dlaboga D, Culley GR, Hulse R, Scholefield CL, Atlason P, Fang G, Eaves R, Morley R, Mayo-Martin MB, Amici M, Bortolotto ZA, Donaldson L, Collingridge GL, Molnar E, Monaghan DT, Jane DE. J Med Chem. 2012;55:327–341. doi: 10.1021/jm201230z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heller CA, Henry RA, Mclaughl Ba, Bliss DE. J Chem Eng Data. 1974;19:214–219. [Google Scholar]
- 74.Melhuish WH. J Phys Chem. 1961;65:229–235. [Google Scholar]
- 75.Rioboo RJ, Philipp M, Ramos MA, Kruger JK. Euro Phys J E. 2009;30:19–26. doi: 10.1140/epje/i2009-10496-4. [DOI] [PubMed] [Google Scholar]
- 76.Zhang G, Evans RE, Campbell KA, Fraser CL. Macromolecules. 2009;42:8627–8633. [Google Scholar]
- 77.Chen J, Gorczynski JL, Zhang G, Fraser CL. Macromolecules. 2010;43:4909–4920. [Google Scholar]
- 78.Demas JN, Crosby GA. J Phys Chem. 1971;75:991–1024. [Google Scholar]
- 79.Lehner P, Staudinger C, Borisov SM, Klimant I. Nat Commun. 2014;5:4460. doi: 10.1038/ncomms5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baleizao C, Nagl S, Schäferling M, Berberan-Santos MN, Wolfbeis OS. Anal Chem. 2008;80:6449–6457. doi: 10.1021/ac801034p. [DOI] [PubMed] [Google Scholar]
- 81.Chu C-S, Lin C-A. Sensor Actuat B-Chem. 2014;195:259–265. [Google Scholar]
- 82.Méhes G, Goushi K, Potscavage WJ, Jr, Adachi C. Org Electron. 2014;15:2027–2037. [Google Scholar]
- 83.Zhang Q, Li B, Huang S, Nomura H, Tanaka H, Adachi C. Nat Photonics. 2014;8:326–332. [Google Scholar]
- 84.Tanaka H, Shizu K, Nakanotani H, Adachi C. Chem Mater. 2013;25:3766–3771. [Google Scholar]
- 85.Méhes G, Nomura H, Zhang Q, Nakagawa T, Adachi C. Angew Chem, Int Ed. 2012;51:11311–11315. doi: 10.1002/anie.201206289. [DOI] [PubMed] [Google Scholar]
- 86.Wang X-d, Stolwijk JA, Lang T, Sperber M, Meier RJ, Wegener J, Wolfbeis OS. J Am Chem Soc. 2012;134:17011–17014. doi: 10.1021/ja308830e. [DOI] [PubMed] [Google Scholar]
- 87.Wang X-d, Gorris HH, Stolwijk JA, Meier RJ, Groegel DB, Wegener J, Wolfbeis OS. Chem Sci. 2011;2:901–906. [Google Scholar]
- 88.Borisov SM, Fischer R, Saf R, Klimant I. Adv Funct Mater. 2014;24:6548–6560. doi: 10.1002/adfm.201401754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bukvetskii B, Fedorenko E, Mirochnik A, Beloliptsev AY. J Struct Chem. 2010;51:545–551. [Google Scholar]
- 90.Panyam J, Sahoo SK, Prabha S, Bargar T, Labhasetwar V. Int J Pharm. 2003;262:1–11. doi: 10.1016/s0378-5173(03)00295-3. [DOI] [PubMed] [Google Scholar]
- 91.Petros RA, DeSimone JM. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 92.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 93.Kersey FR, Zhang GQ, Palmer GM, Dewhirst MW, Fraser CL. ACS Nano. 2010;4:4989–4996. doi: 10.1021/nn901873t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kang N, Perron M-È, Prud’homme RE, Zhang Y, Gaucher G, Leroux J-C. Nano Lett. 2005;5:315–319. doi: 10.1021/nl048037v. [DOI] [PubMed] [Google Scholar]
- 95.Agatemor C, Shaver MP. Biomacromolecules. 2013;14:699–708. doi: 10.1021/bm400060x. [DOI] [PubMed] [Google Scholar]
- 96.Thevenaz DC, Monnier CA, Balog S, Fiore GL. Biomacromolecules. 2014;15:3994–4001. doi: 10.1021/bm501058n. [DOI] [PubMed] [Google Scholar]
- 97.Palmer GM, Fontanella AN, Shan S, Hanna G, Zhang G, Fraser CL, Dewhirst MW. Nat Protoc. 2011;6:1355–1366. doi: 10.1038/nprot.2011.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.