

Abstract
In mammals, the endocannabinoid signaling pathway provides protective cellular responses to ischemia. Previous work demonstrated increases in long-chain N-acylethanolamines (NAE) in ischemia and suggested a protective role for NAE. Here, a targeted lipidomics approach was used to study comprehensive changes in the molecular composition and quantity of NAE metabolites in a rat model of controlled brain ischemia. Changes of NAE, its precursors, N-acylphosphatidylethanolamines (NAPE), major and minor phospholipids, and free fatty acids (FFA) were quantified in response to ischemia. The effect of intraperitoneal injection of N-palmitoylethanolamine (NAE 16:0) prior to ischemia on NAE metabolite and phospholipid profiles was measured. While ischemia, in general, resulted in elevated levels of N-acyl 16:0 and18:0 NAE, NAPE, and FFA species, pretreatment with NAE 16:0 reduced infarct volume, neurological behavioral deficits in rats, and FFA content in ischemic tissues. Pretreatment with NAE 16:0 did not affect the profiles of other NAE metabolites. These studies demonstrate the utility of a targeted lipidomics approach to measure complex and concomitant metabolic changes in response to ischemia. They suggest that the neuroprotective effects of exogenous NAE 16:0 and the reduction in inflammatory damage may be mediated by factors other than gross changes in brain NAE levels, such as modulation of transcriptional responses.
Supplementary key words: free fatty acid, N-acylphosphatidylethanolamine, lipid signaling, N-palmitoylethanolamine, phospholipids, mass spectrometry
INTRODUCTION
Endocannabinoids are trace lipid signaling molecules that are endogenous ligands of cannabinoid receptors. Several of these ligands are important lipid mediators that regulate a wide range of biological processes in vertebrates [1], invertebrates [2] and plants [3, 4]. Specifically, the endocannabinoid system plays an important role in overall biochemical response to ischemia, which results in nutrient deprivation and accumulation of injurious metabolites that challenge physiological homeostasis in mammals [see reviews [1, 5, 6]]. Previous studies of experimentally induced ischemia in rodent brains both support and in some cases contradict [7, 8] the view that endocannabinoid responses are neuroprotective [reviewed by [9]]. Increased free radical production, over-stimulation of glutamate receptors, and elevated intracellular calcium levels resulting from ischemic damage are thought to be counteracted by endocannabinoid responses [8].
Anandamide, the first endocannabinoid discovered [10], is an arachidonic acid-derived member of the N-acylethanolamines (NAE). NAE are derived from N-acylated phosphatidylethanolamines (PE), which include molecular species with a variety of saturated and unsaturated N-acyl groups, as well as a heterogeneous mix of chains in the diradyl glycerol component [NAPE; reviewed in [11]]. Anandamide formation is thought to originate with the transfer of arachidonic acid from the sn-1 position of precursor phospholipids, such as phosphatidylcholine (PC), to the amine of PE by N-acyltransferases [NAT; [12, 13]]. Free NAE are produced from their NAPE precursors by NAPE-phospholipase D (NAPE-PLD) mediated hydrolysis. Free NAE are rapidly eliminated as signaling sources, likely by transporter-aided cellular reuptake followed by hydrolysis of the NAE; predominantly fatty acid amide hydrolase (FAAH) terminates NAE signaling, yielding free fatty acids (FFA) and ethanolamine [14–16].
Recognition and characterization of the N-acylation-phosphodiesterase pathway, i.e. the endocannabinoid signaling pathway, originated from the observation of sharp increase in concentrations of relatively trace lipids, NAPE and NAE, in ischemic tissues [17–20]. It is now recognized that the role of NAE signaling in response to ischemia is complex; the behavior of NAPE accumulation can differ significantly from NAE release and can vary among acyl groups of varying chain lengths and degrees of saturation [9]. Also, several studies have demonstrated that the damaging effects of ischemia can be reduced by exogenous treatment with NAE [21–24]. Among the various NAE species, NAE 16:0 has received particular attention as having potential neuroprotective effects against ischemic damage [25–29]. NAE 16:0 is among the most abundant of NAE molecular species in a wide range of plants and animals [26, 30, 31], and it is among the most highly up-regulated in response to a variety of stimuli, including hypoxia and ischemia [9].
A comprehensive study of lipid metabolism is required to adequately describe the dynamics of interactions among exogenous addition, storage, release, and degradation of various molecular species that may be of importance in modulation of response to ischemic stress. Targeted lipidomics approaches focusing on effects of altered NAE metabolism in mice [32] and the role of arachidonic acid [13] in signaling have recently been conducted. Here we adopted a lipidomics strategy to measure changes in the molecular composition and quantity of NAE metabolites and describe lipidomic changes associated with ischemia in a transient middle cerebral artery occlusion (MCAO), with and without the exogenous administration of NAE 16:0. Our analyses include detailed measurement of the concentrations of individual acyl groups comprising the NAE family and their NAPE and PE precursors, and other [phosphatidylcholine (PC), phosphatidylserine (PS), sphingomyelin (SM), alk(en)yl,acyl PE (ether-linked PE or ePE), phosphatidylinositol (PI), phosphatidic acid (PA), lysoPE, lysoPC and alk(en)yl,acyl PC (ePC)] phospholipid classes. Finally, we also analyzed the quantity and composition of FFA, since they are important biochemical indicators of metabolic pathways in various pathological conditions, such as membrane disintegration via mechanisms such as deacylation, peroxidation, or free radical reaction [33, 34], and are products of NAE hydrolysis.
MATERIALS AND METHODS
Chemicals
All NAE standards were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Fatty acid methyl ester (FAME) standards were purchased from Sigma-Aldrich (St. Louis, MO, USA). Solvents (HPLC grade) and N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) were purchased from Fisher Scientific (Pittsburgh, PA, USA). Other reagents were of the highest purity commercially available.
Experimental groups
Rats were randomly divided into four experimental groups: (1) sham-operated group (S) that received mock MCAO surgery and treatment with vehicle (ethanol), (2) sham-operated group that received mock MCAO surgery and treatment with 10 mg/kg NAE 16:0 intraperitoneally, (3) ischemia-reperfusion group (I or I/R) that experienced 90 min of MCAO followed by 24 h of reperfusion and treatment with vehicle (ethanol), and (4) I/R group that received 10 mg/kg NAE 16:0 intraperitoneally, concomitantly with MCAO surgery. Brain tissue from ipsilateral (I) and contralateral (C) hemispheres of each of these groups was analyzed separately, yielding lipidomics results for the eight tissue sources summarized in Table I.
Table I.
Identification of experimental groups.
| Surgery | Tissue Sampled | Group Abbreviation | Exogenous Treatment |
|---|---|---|---|
| Ischemia/Reperfusion | Contralateral | IC | Vehicle |
| NAE 16:0 | |||
| Ipsilateral | II | Vehicle | |
| NAE 16:0 | |||
| Sham | Contralateral | SC | Vehicle |
| NAE 16:0 | |||
| Ipsilateral | SI | Vehicle | |
| NAE 16:0 |
Rat MCA transient occlusion – a model of brain ischemia reperfusion injury
Male Sprague Dawley rats (Harlan Laboratories, Inc., Indianapolis, IN, USA) weighing 300–325 g were fed ad libitum and kept in a 22–25°C temperature-controlled room on a 12 h light/dark cycle. All animal-related care and procedures conformed with the Public Health Service Policy on Humane Care and Use of Laboratory animals and were reviewed and approved by the Institutional Animal Care and Use Committee at the University of North Texas Health Science Center.
Brain ischemia reperfusion injury was modeled using transient MCAO as described by us previously [23, 35, 36]. In brief, rats were anesthetized with a ketamine/xylazine cocktail (60 and 10 mg/kg body weight, respectively), and body temperature was maintained at 37 ± 0.5°C throughout surgery. After exposing the left common carotid, the left external carotid, and the left internal carotid artery (ICA), a monofilament nylon suture (3–0; Ethilon; Ethicon Inc., Sommerville, NJ, USA) was introduced through a puncture into the lumen of the ICA. The suture was advanced until adequate resistance was felt to accomplish MCA occlusion. After ninety minutes of MCAO, the suture was withdrawn, followed by 24 h of reperfusion. Sham surgeries were identical except no suture was inserted. After recovery, animals were kept with free access to water and food. Rats were sacrificed by pentobarbital overdose at 24 h after ischemia, death was assured by pneumothorax, and the rats were decapitated. Brains were removed and immediately (within 2 min) flash frozen in liquid nitrogen, and stored at −80°C until extraction.
Measurement of cerebral infarct volume
In order to determine cerebral infarct volume, the brains of identically treated animals were dissected after decapitation. Brains were placed in ice-cold saline for 5 min after removal. Seven coronal slices were then sectioned from each brain at a thickness of 2 mm, and sections were incubated for 15 min at 37°C in a 2% 2,3,5-triphenyltetrazolium chloride (TTC) solution. In the stained sections, infarct area appeared pale-colored and viable areas were colored pink/red. The infarction volume was calculated according to Swanson et al. [37]. In brief, the infarct area in each section was calculated by subtracting the non-infarcted areas of the ipsilateral side from the total corresponding area of the contralateral side that was not affected by the I/R injury. Infarction areas on each section were summed and multiplied by section thickness to compute the total infarction volume, which was expressed as a percentage of total brain volume. Computation and analysis for infarct volume in each brain slice were performed using PCI version 5.3.1 high performance imaging software (Compix Inc., Cranberry Township, PA, USA).
Neurological evaluation
Evaluation for neurological deficits, expressed as neurological deficit scores, was performed on all animals just before euthanasia and scored as described [23, 38] using the following criteria: 0, no observable deficit (normal); 1, failure to extend left forepaw on lifting the whole body by tail (mild); 2, circling towards the contralateral side (moderate); 3, leaning to the contralateral side at rest or no spontaneous motor activity (severe).
Lipid extraction
Rat brain tissues were homogenized in 2 ml hot 2-propanol (70°C) and 1 ml of chloroform. Samples were vortexed and incubated on ice for 30 min prior to overnight extraction at 4°C. Monophasic lipid extracts were partitioned with 2 ml of 1 M KCl. The lower organic phase was washed three additional times with 1 M KCl and subsequently dried to completion under nitrogen. The total lipid content was estimated gravimetrically, and the samples were dissolved in chloroform and stored under nitrogen at −20°C until further analysis.
Phospholipid analysis and quantification
An automated electrospray ionization (ESI)-tandem mass spectrometry approach was used, and sample preparation and data acquisition were carried out as described previously [32], with modifications. Briefly, an aliquot of 2 μl of brain lipid extract, equivalent to 0.6–0.8 mg tissue fresh weight (FW), was analyzed. Precise amounts of internal lipid standards for PC, lysoPC, PE, lysoPE, PA, PS, and PI, as well as solvent, were added.
Mass spectra were acquired on a triple quadrupole MS system (API 4000, Applied Biosystems, Foster City, CA, USA). Sequential precursor and neutral loss scans of the extracts produced a series of spectra, with each spectrum revealing a set of lipid species, PC, PE, PA, PS, or PI, containing a common head group fragment. SM was determined from the same mass spectrum as PC (precursors of m/z 184 in positive mode) [39, 40].
The data were processed as described (32). Peaks corresponding to the target lipids in these spectra were identified, quantified in comparison to the internal standards in the same lipid class, and corrected for instrument/sample background interference and isotopic overlap. SM quantification was by comparison with PC internal standards, using an experimentally determined molar response factor of 0.39 for SM (in comparison with PC). Data were reported as nmoles of each detected lipid metabolite/g tissue FW.
NAPE analysis and quantification
NAPE analysis and quantification were carried out as described previously (32), with modifications. N-17:0 di16:0 PE was synthesized (32) and employed as an internal standard to quantify the NAPE species in the sample extracts. The same rat lipid brain extracts were used for both phospholipid and NAPE analysis. NAPE standard (1.1 nmoles) and solvent were added to an aliquot of extract equivalent to 6–10 mg tissue FW.
Mass spectra were acquired by automated ESI-tandem mass spectrometry on a triple quadrupole MS system (API 4000 QTrap, Applied Biosystems). Sequential neutral loss scans produced a series of spectra, with each spectrum revealing a set of lipid species containing a common ammoniated N-fatty amide head group fragment. N-16:0, N-17:0, N-18:2, N-18:1, N-18:0, N-20:4, N-22:6, and N-22:5 species were detected.
The data were processed as described (32). Peaks corresponding to the target lipids in each N-acyl class (each spectrum) were identified, quantified relative to the N-17:0 di16:0 PE internal standard, and corrected for instrument/sample background interference and isotopic overlap. Due to the interfering presence in the N-20:4 scan of peaks with m/z inconsistent with NAPEs in the m/z range of N-20:4–36:4, N-20:4–36:2, N-20:4–38:6, N-20:4–38:5, N-20:4–40:7, N-20:4–40:5, and N-20:4–40:4 NAPEs, these compounds were not measured. Data were reported as mass spectral signal normalized to N-17:0 di16:0 PE/g tissue FW; the amount of signal produced by 1 nmol of N-17:0 di16:0 PE is 1. Data were evaluated for possible outliers using the Q-test [41] on NAPE lipid class totals; one sham-operated contralateral (SC) rat brain replicate (out of 4) was determined to be an outlier, most likely due to sample mishandling, and was removed from calculations. No response correction factors were employed for potentially different mass spectral responses of various N-acyl chains [32]. NAPE lipidomic data, thus, allow comparison of samples, but may not represent the relative amounts of various molecular species.
NAE extraction and quantification
NAE extraction was conducted as described previously [42]. Each brain hemisphere was removed and homogenized in ice-cold chloroform using a glass tissue grinder. The homogenate was sonicated (60 W) for 2 min to disrupt cells. Approximately 500 mg of tissue was then added to 10 ml of ice-cold chloroform containing deuterated NAE standards (D4-NAE 16:0 and D4-NAE 20:4, 50 ng each). Lipids were extracted by Folch extraction (chloroform/methanol/water, 4:2:1) by the addition of 5 ml cold methanol and 2.5 ml cold PBS buffer and sonication (60 W) in an ice-cold water bath for 10 min, followed by centrifugation. The organic phase was collected for further purification by solid phase extraction (SPE).
Silica SPE cartridges (100 mg, 1.5 ml; Alltech, Deerfield, IL, USA) were conditioned with 2 ml methanol and 4 ml chloroform, and subsequent to loading the sample, the column was washed with 2 ml chloroform and NAE were eluted with 2 ml of ethyl acetate/acetone (1:1). The eluate was collected, evaporated under nitrogen, and derivatized with 50 μl BSTFA and 25 μl dichloromethane for 30 min at 55°C. After derivatization, the samples were again evaporated under nitrogen and reconstituted in 50 μl hexane.
NAE were identified via selective ion monitoring and quantified against the internal deuterated standards (saturated species against deuterated NAE 16:0 and unsaturated species against deuterated NAE 20:4) as trimethylsilyl (TMS)-ether derivatives by GC-MS (Model 6890 GC coupled with a 5973 mass selective detector; Agilent, Wilmington, DE, USA), as described previously [31]. NAE concentration was calculated based on FW.
FFA quantification
Total lipid extracts were combined with 50 μg of heptadecanoic acid (17:0), dried under nitrogen, and derivatized with 400 μl methanol and 6.5 μl 2 M trimethylsilyldiazomethane [43]. The reaction mixture was shaken vigorously for 30 min at room temperature, the reaction was terminated with the addition of 0.2 μl concentrated acetic acid, and the solvent was evaporated under nitrogen. FAME were solubilized in 10 μl acetonitrile, and 2 μl were analyzed by GC (Agilent 6890) using flame ionization detection and a capillary DB-23 column (30 m × 0.25 mm; 0.25 μm film thickness; J&W Scientific, Agilent). Helium was used as a carrier gas at a flow rate of 1 ml/min, and the temperature gradient was as follows: 150°C for 1 min, 150 – 200°C at 8°C /min, 200 – 250°C at 25°C /min and 250°C for 6 min. FAME quantification was based on FAME 17:0 as the internal standard.
Statistical analyses
In all of the lipid analyses experiments, N = 4 (for NAE quantification, N = 6), and the data were expressed as means ± SD. Statistical differences between contra- and ipsilateral, vehicle and treatment, and ischemia and sham tissues were determined by comparing the means, using the two-tailed, unpaired Student’s t-test and significance (P < 0.05) was indicated by ‘a’, ‘b’ and ‘c’ respectively. Histochemistry and neurological assessment data were plotted as the means ± SE. The determination of statistical significance (P < 0.05) was performed with ANOVA and post-hoc Newman Keuls and Bonferroni multiple comparison tests and GraphPad Prism 4 statistical software.
RESULTS
The major metabolites in the endocannabinoid pathway include PE, NAPE, NAE, and FFA. In order to understand ischemia-induced changes in NAE metabolite content and composition, we used a targeted lipidomics approach and conducted comprehensive analyses of total lipid extracts from the contralateral and ipsilateral brain tissues of rats. Ischemia was confirmed in all of the animals that received reperfusion injury, by neurological evaluation (Fig. 1A) and histochemistry (Fig. 1B). Sham-operated rats that had been treated with vehicle or NAE 16:0 showed no ischemic lesions or neurological deficits (data not shown; reported in [23]). The range of neurological deficit scores was between 2 and 3 in vehicle-treated ischemic rats, corresponding to a medium to severe neurological deficit phenotype (Fig. 1A). These animals showed extensive infarction in the cortical and subcortical areas, as determined by vital staining with TTC in coronal brain slices (Fig. 1B). The neurological deficit score of I/R NAE 16:0-treated rats improved significantly to a range of 0–1 (Fig. 1A), and the data were confirmed by significant reduction of ipsilateral infarct volume (Fig. 1B).
Figure 1.
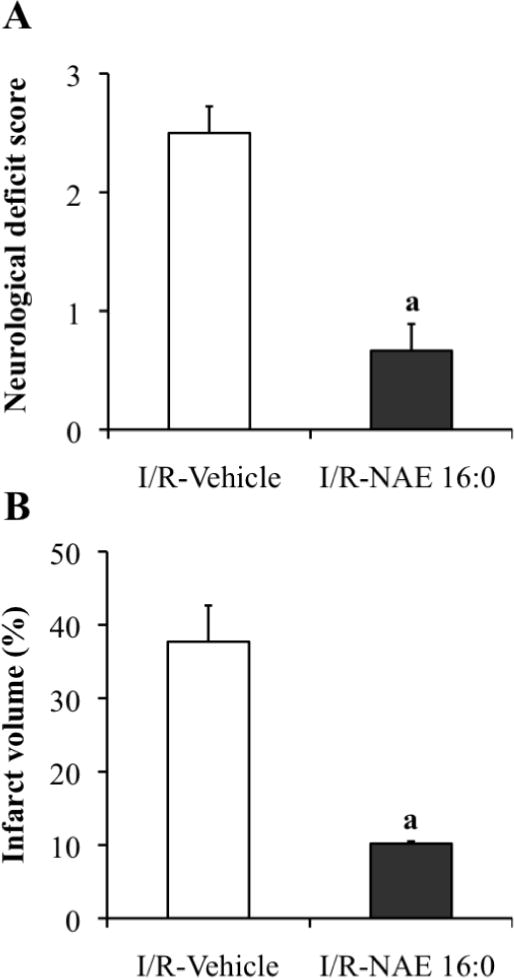
Assessment of neurological deficit scores and histochemistry confirm that NAE 16:0 protects against ischemic brain damage following MCAO I/R.
(A) MCAO-mediated I/R caused moderate to severe neurological deficits, as measured with the standardized neurological deficit score, and treatment with NAE 16:0 reduced neurological deficits significantly (phenotype: none to mild). (B) The size of the ipsilateral ischemic lesion volume (infarct volume) was significantly reduced by NAE 16:0 treatment. Data are shown as mean ± SE. a P < 0.05, compared with I/R vehicle-treated group as determined using one-way ANOVA with Newman Keuls multiple comparison post-hoc test.
Lipids extracted from the contralateral and ipsilateral brain regions of the I/R and sham-operated rats which had been treated with vehicle or NAE 16:0 (Table I) were analyzed for changes in NAE metabolites. Levels of total NAE and their precursor ethanolamine-containing lipid classes, NAPE and PE, and FFA, were determined by summing the analyzed individual molecular species concentrations within each lipid class (Fig. 2). The total amount of PE in contra and ipsilateral tissues showed no significant change associated with ischemia or treatment. In contrast, ischemia with or without treatment resulted in an approximate 3-fold increase in total NAPE and an order of magnitude increase in total NAE, only in the ipsilateral brain tissues (II vs. IC groups, Fig. 2), compared to shams (II vs. SI groups, Fig. 2). Prior administration of NAE 16:0, which resulted in reduced neurological deficit score and infarct volume (Fig. 1), had no effect on endogenous PE, NAPE, or NAE content of ischemic tissues but resulted in a two-fold decline in total FFA content in the ipsilateral tissue (II-NAE 16:0 vs. II-Vehicle, Fig. 2) and an increase in sham-operated contralateral tissue (SC-NAE 16:0 vs. SC-Vehicle, Fig. 2). The total FFA content was two-fold higher in ischemic tissues that did not receive NAE 16:0 treatment, compared to sham-operated tissues (IC- and II-Vehicle groups compared with SC- and SI-Vehicle groups, respectively, Fig. 2).
Figure 2.
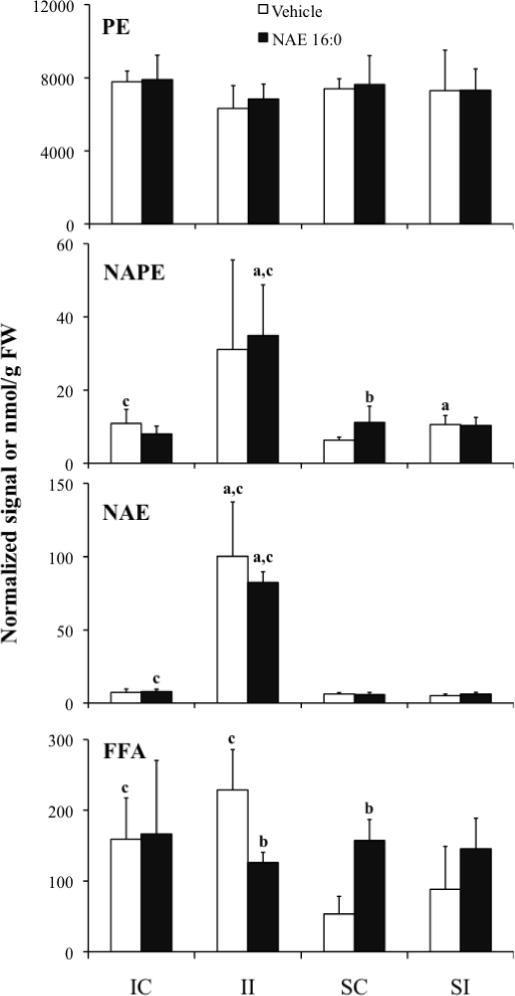
Ischemia/reperfusion- and sham-operation-induced changes in total PE, NAPE, NAE, and FFA content in contralateral (IC and SC) and ispsilateral (II and SI) brain tissues of rats treated with vehicle or exogenous NAE 16:0. Data are shown as mean ± SD. Statistical significance (P < 0.05; N = 4 (for NAE samples N = 6)) between contra- and ipsilateral tissues is indicated by ‘a’, and vehicle and NAE 16:0 treatment by ‘b’, and ischemia and sham tissues by ‘c’ as determined by unpaired Student’s t-test. NAPE values indicate relative normalized mass spectral signal per g of sample FW, where 1 unit of signal indicates the amount of signal that is observed for 1 nmol of the internal standard, N-17:0 di16:0 PE; all other lipid classes are expressed as nmol/g FW.
The changes in acyl-chain composition of NAPE, NAE, and FFA, in response to ischemia, were analyzed (Fig. 3). All three metabolite groups were dominated by the 16:0, 18:0, and 18:1 chains. Arachidonic acid (20:4) represented a larger fraction of FFA than of the N-acyl chains in NAPE or NAE. The dominant acyl groups, 16:0, 18:0, and 18:1, were also the molecular species that primarily contributed to the higher levels in total NAPE and NAE content in ischemic ipsilateral and not contralateral preparations as compared to shams (II vs IC groups and II vs. SI groups in Fig. 2 and Fig. 3). In sham-operated tissues, the content of various molecular species of NAPE and NAE was low and similar to that of ischemic contralateral tissues (IC vs. SC groups, Fig. 3). However, similar to ischemic groups, the major NAPE and NAE species in shams were higher in the ipsilateral tissues compared with contralateral (SI vs. SC groups, Fig. 3). The abundance of FFA 16:0 and 18:0 also was, in general, low in sham-operated tissues of vehicle-treated rats; however, the levels increased by two- to three-fold in rats that were treated with NAE 16:0 (SC- and SI- NAE 16:0 vs. SC- and SI-Vehicle, Fig. 3). Ischemia induced increases in FFA 16:0 and 18:0 content in both contra- and ipsilateral tissues, but these increases tended to be reduced in rats that were pre-treated with NAE 16:0 (II-NAE 16:0 vs II-Vehicle, Fig. 3). Ischemia-related changes observed in the polyunsaturated species, N-linked 18:2, 20:4 and 22:6 of NAPE and NAE and 18:2 and 20:4 of FFA, were not as dramatic as in the saturated and mono-unsaturated species, and their contribution to total accumulation of metabolite was small.
Figure 3.
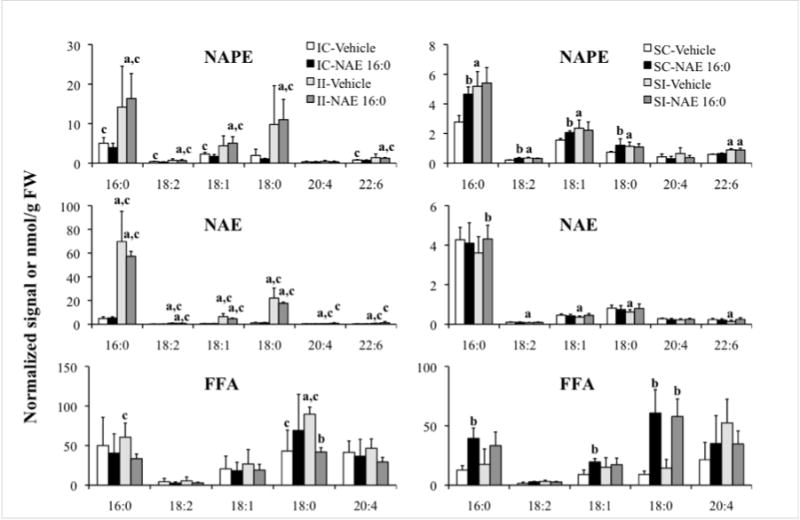
Total lipid extracts from ischemia/reperfusion or sham-operated contralateral (IC and SC) and ipsilateral (II and SI) brain tissues of rats that were treated with vehicle or exogenous NAE 16:0 were analyzed for the composition of NAPE, NAE, and FFA species, differentiated by their acyl chains. Data are shown as mean ± SD. Statistical significance (P < 0.05; N = 4 (for NAE samples N = 6)) between contra- and ipsilateral tissues is indicated by ‘a’, and vehicle and NAE 16:0 treatment by ‘b’, and ischemia and sham tissues by ‘c’ as determined by unpaired Student’s t-test. NAPE values indicate relative normalized mass spectral signal per g of sample FW, where 1 unit of signal indicates the amount of signal that is observed for 1 nmol of the internal standard, N-17:0 di16:0 PE; NAE and FFA lipid classes are expressed as nmol/g FW. Acyl chain designations are indicated by x:y, or carbon chain length:number of double bonds.
In order to further characterize the ischemia-induced changes in NAPE, NAPE sharing a common N-acyl group were characterized in terms of the total acyl carbons and carbon-carbon double bonds in the two chains linked to glycerol. Mass spectral signals for NAPE were assigned to molecular species based on the mass of the intact ion and an N-acyl-containing fragment (major contributing NAPE, Fig. 4; minor contributing NAPE, Fig. 5). The distribution patterns of individual NAPE species with 16:0, 18:0, and 18:1 at the N-position (Fig. 4) and those with 18:2, 20:4, and 22:6 at the N-position (Fig. 5) were all similar and were dominated by diacyl species 40:6, 38:4, 38:6 and 36:1. Once again, as seen in Fig. 3, ischemia-related increases in the dominant NAPE species are evident in the ipsilateral tissue (II vs. IC groups, Fig. 4 and Fig. 5). Molecular species with 1-alk(en)yl,acyl linkages were present only at low amounts in the NAPE profiles (data not shown). There was no apparent effect of exogenous NAE 16:0 treatment on the NAPE profiles.
Figure 4.
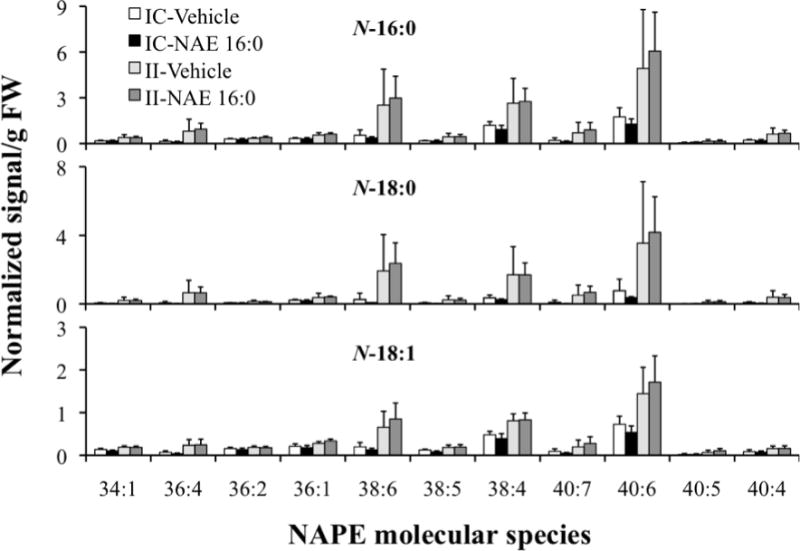
Comprehensive analysis of ischemia/reperfusion-induced levels of NAPE molecular species with acyl chains N-16:0, N-18:0 and N-18:1, in contralateral (IC) and ipsilateral (II) brain tissue of groups treated with vehicle or exogenous NAE 16:0. Data are shown as mean ± SD and N = 4. NAPE molecular species labels indicate the diacyl component (total acyl carbons: total carbon-carbon double bonds) of each detected NAPE species. NAPE species mass spectral signals were normalized against the signal, assigned to be 1 unit, observed for 1 nmol of the internal standard, N-17:0 di16:0 PE.
Figure 5.
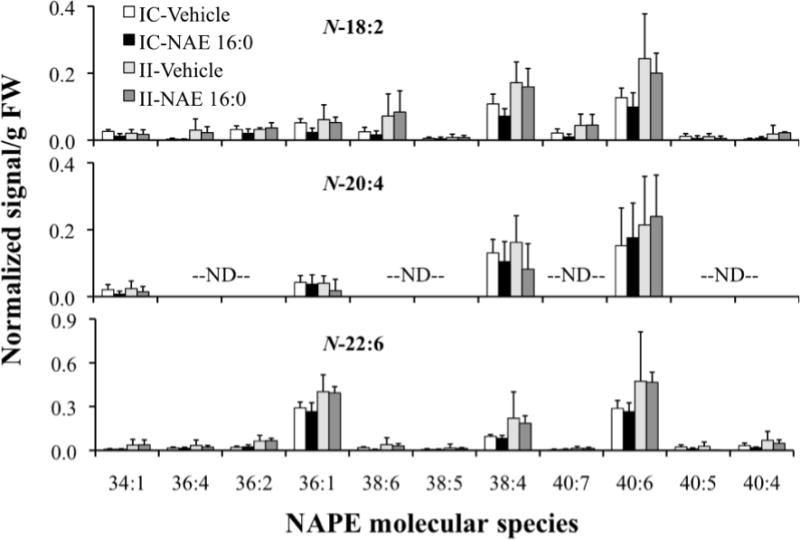
Comprehensive analysis of ischemia/reperfusion-induced levels of NAPE molecular species with acyl chains N-18:2, N-20:4, and N-22:6, in contralateral (IC) and ipsilateral (II) brain tissue of groups treated with vehicle or exogenous NAE 16:0. Data are shown as mean ± SD and N = 4. NAPE molecular species labels indicate the diacyl component (total acyl carbons: total carbon-carbon double bonds) of each detected NAPE species. “ND” indicates molecular species that were not determined (see Experimental Procedures). NAPE species mass spectral signals were normalized against the signal, assigned to be 1 unit, observed for 1 nmol of the internal standard, N-17:0 di16:0 PE.
Finally, major and minor phospholipid class concentrations were compared among the various treatment groups (Fig. 6). Estimates of the total phospholipid content of each treatment group are presented in Table II. Ischemia, with or without pretreatment with NAE 16:0, had no effect on any of the major and minor phospholipids examined, in both contra- and ipsilateral tissues (IC- and II-Vehicle/NAE 16:0 vs. SC- and SI-Vehicle/NAE 16:0 groups, Fig. 6). Absence of an ischemia/reperfusion-induced change in phospholipid patterns may have been due to sampling too early to detect the change or to ischemic damage too small to be reflected in statistically significant changes in phospholipid pattern. Further examination of specific molecular species that contributed to PE indicated a profile similar to that of NAPE molecular species distribution with 40:6, 38:4, 38:6, 40:4 and 36:1 being the predominant diacyl species, which did not change substantially in response to ischemia (Fig. 7). A complete presentation of results for all phospholipid molecular species is provided as Supplemental Data (Table III).
Figure 6.
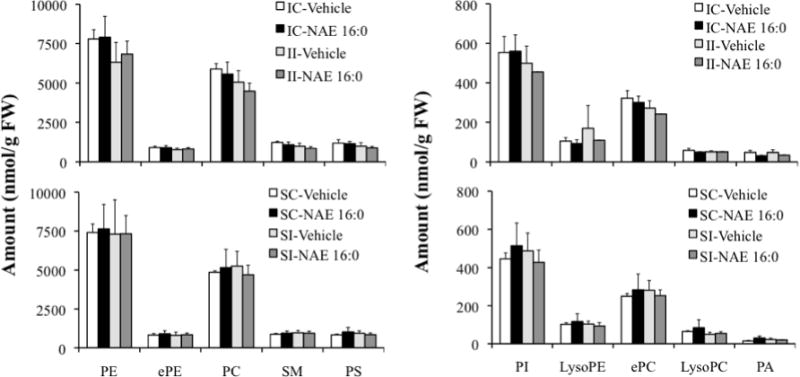
Quantification of major and minor classes of phospholipids in ischemic contralateral and ipsilateral tissues (IC and II) that were treated with vehicle or exogenous NAE 16:0, compared with sham-operated tissues (SC and SI). Data are shown as mean ± SD and N = 4. Abbreviations: phosphatidylethanolamine (PE), alk(en)yl,acyl glycerophosphoethanolamine (ePE), phosphatidylcholine (PC), sphingomyelin (SM) and phosphatidylserine (PS), phosphatidylinositol (PI), lysophosphatidylethanolamine (lysoPE), alk(en)yl,acyl glycerophosphocholine (ePC), lysophosphatidylcholine (lysoPC), and phosphatidic acid (PA).
Table II.
Total phospholipid content in the rat brain tissues of experimental groups.
| Group | Phospholipid (μmol/g FW) Mean ± SD |
|---|---|
| IC-Vehicle | 18.0 ± 1.2 |
| IC-NAE 16:0 | 17.6 ± 2.6 |
| II-Vehicle | 15.1 ± 2.3 |
| II-NAE 16:0 | 14.7 ± 1.6 |
| SC-Vehicle | 15.6 ± 0.7 |
| SC-NAE 16:0 | 16.6 ± 3.6 |
| SI-Vehicle | 16.2 ± 3.5 |
| SI-NAE 16:0 | 15.4 ± 2.1 |
Figure 7.
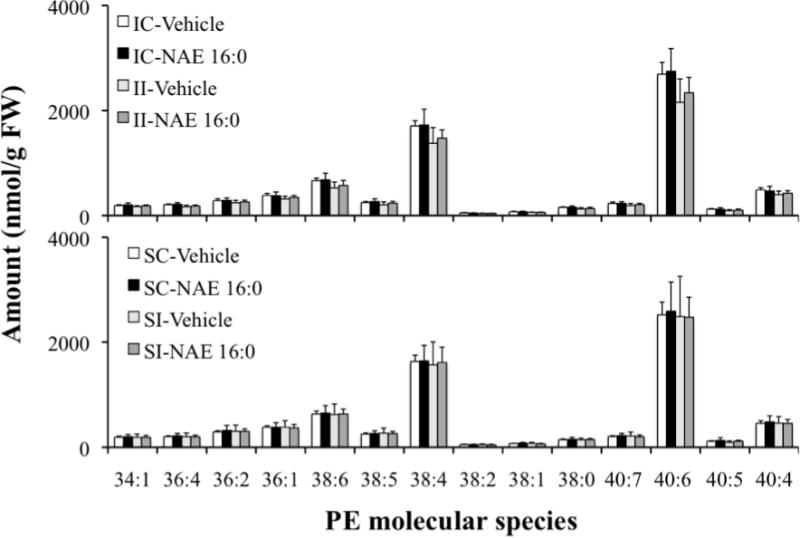
Effect of ischemia/reperfusion injury on levels of PE molecular species in contralateral (IC) and ipsilateral (II) brain tissue of groups treated with vehicle or exogenous NAE 16:0, compared with corresponding sham-operated tissues. Data are shown as mean ± SD and N = 4. PE molecular species labels indicate the diacyl component (total acyl carbons: total carbon-carbon double bonds) of each detected species.
DISCUSSION
In a comprehensive review of available information on the involvement of endocannabinoids in response to cerebral ischemia, Hillard pointed out the potential importance of increased flux of NAE through the biosynthetic pathway and the fact that relatively modest changes reported for anandamide, NAE 20:4, are dwarfed by consistent and much larger increases in saturated and mono-unsaturated members of the NAE family, particularly NAE 16:0 [9]. Our results support this view and offer new insight into the ischemia-related responses of the dominant molecular species of NAE and their NAPE precursors.
As shown previously, MCAO I/R produced a robust ipsilateral lesion as measured with histochemistry and was accompanied by a significant increase in the neurological deficit scores ([23]; Fig. 1). Also paralleling previous studies, NAE 16:0 protected against ischemic brain damage both structurally and functionally by significantly reducing the neurological deficit scores and infarct volume ([23]; Fig. 1). Our results also confirm the accumulation of both NAE and NAPE in cerebral ischemia [20, 29, 44–46], with the relative increase in NAE greatly exceeding that of the NAPE ([47]; Fig. 2). A clear preferential accumulation of species containing saturated and mono-unsaturated N-acyl groups in both NAE and NAPE was observed (Fig. 3) and the molecular species profile of accumulating NAE closely resembled that of the accumulating NAPE (Fig. 4 and 5). This supports the view that PLD hydrolysis of NAPE is non-specific, and that the preferential accumulation of saturated and mono-unsaturated NAE is controlled by the availability of NAPE precursors. NAPE formation via transfer of acyl groups by N-acyltransferases to PE has been suggested to be both the rate-limiting step of NAE formation as well as the source of substrate specificity which is responsible for acyl patterns in both NAE and NAPE [9, 12, 13].
We saw no evidence of the accumulation of anandamide (NAE 20:4) or any of its NAPE precursors after 24 hours of ischemia reperfusion injury (Fig. 3 and 5). Other studies have reported increased anandamide concentrations occurring rapidly, 30 min to 5 hours after MCAO [47–49]. This accumulation has been attributed to evidence that ischemia is accompanied by increased NAPE levels, increased conversion of NAPE to NAE and decreased FAAH expression [9, 47, 48]. These previous studies focused only on occlusion effects and did not include reperfusion in the experimental design. There is evidence that the bulk of anandamide accumulation occurs during reperfusion in both mice [50] and rats [48]. In contrast, although our study measured lipid concentrations after 24 hours of reperfusion after MCAO, we did not observe I/R-related accumulation of anandamide. Degn et al. [29] observed the same result in a study conducted with mice using a time frame similar to ours (24 h post-ischemia), in which neither increases in anandamide or changes in the enzymatic potential for its accumulation were observed.
Despite various discrepancies noted above, it is clear that NAE accumulation in ischemia, as well as after ischemia reperfusion injury, is dominated by saturated and mono-unsaturated species that closely resemble profiles of the precursor NAPE species. N-acyltransferases may be responsible for the observed preferential accumulation of saturated and mono-unsaturated N-acyl groups in NAPE, or perhaps the pool of acyl groups available for transfer is enriched in these species. On the other hand, the lack of specificity of PLD may account for the fact that N-acyl groups of NAE resemble those of NAPE. Relatively high rates of NAPE hydrolysis compared to NAE catabolism may be responsible for the observed accumulation of NAE [9].
One of the goals of our study was to decipher the implications of administering NAE 16:0 as an ischemia protectant [21]. Curiously, neither NAPE nor NAE content was affected by NAE 16:0 treatment; however, the decline noted in the levels of saturated and unsaturated FFA suggests that the protective role of NAE 16:0 is perhaps independent of gross changes in NAE content (Fig. 3). Typically, an ischemic energy crisis accelerates calcium influx into the cytosol, which activates phospholipases and thus the liberation of FFA [34]. On the other hand, accumulation of NAE in ischemic tissues may represent a component of cellular defense mechanisms against the detrimental changes brought about by ischemia and may act as an inhibitor of calcium uptake and electron transport, which has been shown to be concentration dependent [51]. Perhaps the 10-fold increase in NAE content that we reported in response to ischemia was not sufficient to attenuate the increase in FFA. However, it is reasonable to speculate that providing exogenous NAE 16:0, although it did not affect NAE levels at 24 h post-ischemia, may have earlier contributed to the inhibition of FFA formation and thereby offered protection to the tissues by preventing production of reactive oxygen species, destruction of the antioxidant system and inflammation during ischemia, responses associated with increase in FFA content, specifically polyunsaturated FFA [52, 53]. The functional protective effects of NAE 16:0 did not extend significantly into the perfusion period (although structural protection was seen with administration 2 h after occlusion) in an earlier study [23]. A possible mechanism for this protective effect lies in peroxisome proliferator-activated receptor (PPAR)-mediated anti-inflammatory responses [54]. PPAR-alpha is considered a molecular target of NAE 16:0 and 18:1 [55, 56], and its activation has been shown to be critical in cerebral infarct volume reduction produced by exogenous administration of NAE 18:1 in mice [57]. Certainly many other possible mechanistic explanations exist for neuroprotection, including other pathways involving oxidative signals, and this will be resolved only with further experimental evidence.
In conclusion, our targeted, but unbiased lipidomics approach has provided insights and alternatives into understanding the complexity of biochemical events that occur in ischemia and the role of NAE as protectants.
Supplementary Material
Acknowledgments
We would like to thank Mary R. Roth for expert technical assistance. A joint seed grant from the UNT- UNTHSC helped initiate these studies. This work was funded in part by a grant from the U.S. Department of Energy, Office of Science (BES, DE-FG02-05ER15647) to KDC as well as partial support by grants EY014227 from NIH/NEI, RR022570 from NIH/NCRR and AG010485, AG022550 and AG027956 from NIH/NIA and by The Garvey Texas Foundation and the Felix and Carmen Sabates Missouri Endowed Chair in Vision Research (P.K.). Instrument acquisition and method development at the Kansas Lipidomics Research Center were supported by NSF grants MCB 0455318 and DBI 0521587, K-INBRE (NIH Grant P20 RR16475 from the INBRE program of the National Center for Research Resources), and NSF EPSCoR grant EPS-0236913 with matching support from the State of Kansas through Kansas Technology Enterprise Corporation and Kansas State University (R.W.).
Abbreviations
- BSTFA
N,O-Bis(trimethylsilyl)trifluoroacetamide
- ePC
alk(en)yl,acyl glycerophosphocholine
- ePE
alk(en)yl,acyl glycerophosphoethanolamine
- ESI
electrospray ionization
- FAAH
fatty acid amide hydrolase
- FAME
fatty acid methyl ester
- FW
fresh weight
- ICA
internal carotid artery
- I/R
ischemia reperfusion
- MCAO
middle cerebral artery occlusion
- NAE
N-acylethanolamine
- NAPE
N-acyl PE
- NAT
N-acyltransferase
- PA
diacyl glycerophosphate (phosphatidic acid)
- PC
diradyl glycerophosphocholine (phosphatidylcholine)
- PE
diradyl glycerophosphoethanolamine (phosphatidylethanoline)
- PI
diacyl glycerophosphoinositol (phosphatidylinositol)
- PLD
phospholipase D
- PPAR
peroxisome proliferator-activated receptor
- PS
diacyl glycerophosphoserine (phosphatidylserine)
- SM
sphingomyelin
- SPE
solid phase extraction
- TMS
trimethylsilyl
- TTC
2,3,5-triphenyltetrazolium chloride; X:Y designates total fatty acid carbons: carbon-carbon double bonds or total acyl carbons: total carbon-carbon double bonds
References
- 1.De Petrocellis L, Di Marzo V. An introduction to the endocannabinoid system: from the early to the latest concepts. Best Pract Res Clin Endocrinol Metab. 2009;23:1–15. doi: 10.1016/j.beem.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 2.McPartland JM, Matias I, Di Marzo V, Glass M. Evolutionary origins of the endocannabinoid system. Gene. 2006;370:64–74. doi: 10.1016/j.gene.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Chapman KD. Occurrence, metabolism, and prospective functions of N-acylethanolamines in plants. Prog Lipid Res. 2004;43:302–327. doi: 10.1016/j.plipres.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Kilaru A, Blancaflor EB, Venables BJ, Tripathy S, Mysore KS, Chapman KD. The N-acylethanolamine-mediated regulatory pathway in plants. Chem Biodivers. 2007;4:1933–1955. doi: 10.1002/cbdv.200790161. [DOI] [PubMed] [Google Scholar]
- 5.Di Marzo V, Bisogno T, De Petrocellis L. Endocannabinoids: new targets for drug development. Curr Pharm Des. 2000;6:1361–1380. doi: 10.2174/1381612003399365. [DOI] [PubMed] [Google Scholar]
- 6.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 7.Cernak I, Vink R, Natale J, Stoica B, Lea PMt, Movsesyan V, Ahmed F, Knoblach SM, Fricke ST, Faden AI. The “dark side” of endocannabinoids: a neurotoxic role for anandamide. J Cereb Blood Flow Metab. 2004;24:564–578. doi: 10.1097/00004647-200405000-00011. [DOI] [PubMed] [Google Scholar]
- 8.Pellegrini-Giampietro DE, Mannaioni G, Bagetta G. Post-ischemic brain damage: the endocannabinoid system in the mechanisms of neuronal death. FEBS J. 2009;276:2–12. doi: 10.1111/j.1742-4658.2008.06765.x. [DOI] [PubMed] [Google Scholar]
- 9.Hillard CJ. Role of cannabinoids and endocannabinoids in cerebral ischemia. Curr Pharm Des. 2008;14:2347–2361. doi: 10.2174/138161208785740054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 11.Schmid HH, Schmid PC, Natarajan V. The N-acylation-phosphodiesterase pathway and cell signalling. Chem Phys Lipids. 1996;80:133–142. doi: 10.1016/0009-3084(96)02554-6. [DOI] [PubMed] [Google Scholar]
- 12.Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000;61:3–18. doi: 10.1016/s0090-6980(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 13.Astarita G, Piomelli D. Lipidomic analysis of endocannabinoid metabolism in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(26):2755–2767. doi: 10.1016/j.jchromb.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 16.Giang DK, Cravatt BF. Molecular characterization of human and mouse fatty acid amide hydrolases. Proc Natl Acad Sci U S A. 1997;94:2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epps DE, Natarajan V, Schmid PC, Schmid HO. Accumulation of N-acylethanolamine glycerophospholipids in infarcted myocardium. Biochim Biophys Acta. 1980;618:420–430. doi: 10.1016/0005-2760(80)90260-x. [DOI] [PubMed] [Google Scholar]
- 18.Epps DE, Schmid PC, Natarajan V, Schmid HH. N-Acylethanolamine accumulation in infarcted myocardium. Biochem Biophys Res Commun. 1979;90:628–633. doi: 10.1016/0006-291x(79)91281-6. [DOI] [PubMed] [Google Scholar]
- 19.Natarajan V, Reddy PV, Schmid PC, Schmid HH. On the biosynthesis and metabolism of N-acylethanolamine phospholipids in infarcted dog heart. Biochim Biophys Acta. 1981;664:445–448. doi: 10.1016/0005-2760(81)90067-9. [DOI] [PubMed] [Google Scholar]
- 20.Natarajan V, Schmid PC, Schmid HH. N-acylethanolamine phospholipid metabolism in normal and ischemic rat brain. Biochim Biophys Acta. 1986;878:32–41. doi: 10.1016/0005-2760(86)90341-3. [DOI] [PubMed] [Google Scholar]
- 21.Koulen P, Chapman KD. Modulation of intracellular calcium signaling by N-acylethanolamines. Office UP. 2006 ed. Vol 2006-0142395A1, U.S.A. [Google Scholar]
- 22.Duncan RS, Chapman KD, Koulen P. The neuroprotective properties of palmitoylethanolamine against oxidative stress in a neuronal cell line. Mol Neurodegener. 2009;4:50. doi: 10.1186/1750-1326-4-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garg P, Duncan RS, Kaja S, Koulen P. Intracellular mechanisms of N-acylethanolamine-mediated neuroprotection in a rat model of stroke. Neuropharmacology. 2010;166:252–262. doi: 10.1016/j.neuroscience.2009.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schomacher M, Muller HD, Sommer C, Schwab S, Schabitz WR. Endocannabinoids mediate neuroprotection after transient focal cerebral ischemia. Brain Res. 2008;1240:213–220. doi: 10.1016/j.brainres.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 25.Lambert DM, Di Marzo V. The palmitoylethanolamide and oleamide enigmas : are these two fatty acid amides cannabimimetic? Curr Med Chem. 1999;6:757–773. [PubMed] [Google Scholar]
- 26.Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem. 2002;9:663–674. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- 27.Jonsson KO, Vandevoorde S, Lambert DM, Tiger G, Fowler CJ. Effects of homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabinoid anandamide. Br J Pharmacol. 2001;133:1263–1275. doi: 10.1038/sj.bjp.0704199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandevoorde S, Lambert DM. The multiple pathways of endocannabinoid metabolism: a zoom out. Chem Biodivers. 2007;4:1858–1881. doi: 10.1002/cbdv.200790156. [DOI] [PubMed] [Google Scholar]
- 29.Degn M, Lambertsen KL, Petersen G, Meldgaard M, Artmann A, Clausen BH, Hansen SH, Finsen B, Hansen HS, Lund TM. Changes in brain levels of N-acylethanolamines and 2-arachidonoylglycerol in focal cerebral ischemia in mice. J Neurochem. 2007;103:1907–1916. doi: 10.1111/j.1471-4159.2007.04892.x. [DOI] [PubMed] [Google Scholar]
- 30.Sepe N, De Petrocellis L, Montanaro F, Cimino G, Di Marzo V. Bioactive long chain N-acylethanolamines in five species of edible bivalve molluscs. Possible implications for mollusc physiology and sea food industry. Biochim Biophys Acta. 1998;1389:101–111. doi: 10.1016/s0005-2760(97)00132-x. [DOI] [PubMed] [Google Scholar]
- 31.Venables BJ, Waggoner CA, Chapman KD. N-acylethanolamines in seeds of selected legumes. Phytochemistry. 2005;66:1913–1918. doi: 10.1016/j.phytochem.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Kilaru A, Isaac G, Tamura P, Baxter D, Duncan S, Venables B, Welti R, Koulen P, Chapman K. Lipid Profiling Reveals Tissue-Specific Differences for Ethanolamide Lipids in Mice Lacking Fatty Acid Amide Hydrolase. Lipids. 2010:1–13. doi: 10.1007/s11745-010-3457-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bazan NG., Jr Effects of ischemia and electroconvulsive shock on free fatty acid pool in the brain. Biochim Biophys Acta. 1970;218:1–10. doi: 10.1016/0005-2760(70)90086-x. [DOI] [PubMed] [Google Scholar]
- 34.Rehncrona S, Westerberg E, Akesson B, Siesjo BK. Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. J Neurochem. 1982;38:84–93. doi: 10.1111/j.1471-4159.1982.tb10857.x. [DOI] [PubMed] [Google Scholar]
- 35.Wen Y, Yang S, Liu R, Perez E, Yi K, Koulen P, Simpkins J. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain research. 2004;1008:147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 36.Wen Y, Yang S, Liu R, Perez E, Brun-Zinkernagel A, Koulen P, Simpkins J. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2007;1772:473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 37.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 38.Huang Z, Huang P, Panahian N, Dalkara T, Fishman M, Moskowitz M. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 39.Brügger B, Sandhoff R, Wegehingel S, Gorgas K, Malsam J, Helms J, Lehmann W, Nickel W, Wieland F. Evidence for segregation of sphingomyelin and cholesterol during formation of COPI-coated vesicles. The Journal of Cell Biology. 2000;151:507. doi: 10.1083/jcb.151.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liebisch G, Lieser B, Rathenberg J, Drobnik W, Schmitz G. High-throughput quantification of phosphatidylcholine and sphingomyelin by electrospray ionization tandem mass spectrometry coupled with isotope correction algorithm. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2004;1686:108–117. doi: 10.1016/j.bbalip.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Shoemaker D, Garland C, Steinfeld J, Nibler J. Experiments in Physical Chemistry. McGraw Hill, Inc; New, York: 1981. Treatment of experimental data; pp. 25–52. [Google Scholar]
- 42.Muccioli GG, Stella N. An optimized GC-MS method detects nanomolar amounts of anandamide in mouse brain. Anal Biochem. 2008;373:220–228. doi: 10.1016/j.ab.2007.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brondz I. Development of fatty acid analysis by high-performance liquid chromatography, gas chromatography, and related techniques. Analytica Chimica Acta. 2002;465:1–37. [Google Scholar]
- 44.Felder CC, Nielsen A, Briley EM, Palkovits M, Priller J, Axelrod J, Nguyen DN, Richardson JM, Riggin RM, Koppel GA, Paul SM, Becker GW. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996;393:231–235. doi: 10.1016/0014-5793(96)00891-5. [DOI] [PubMed] [Google Scholar]
- 45.Kempe K, Hsu FF, Bohrer A, Turk J. Isotope dilution mass spectrometric measurements indicate that arachidonylethanolamide, the proposed endogenous ligand of the cannabinoid receptor, accumulates in rat brain tissue post mortem but is contained at low levels in or is absent from fresh tissue. J Biol Chem. 1996;271:17287–17295. doi: 10.1074/jbc.271.29.17287. [DOI] [PubMed] [Google Scholar]
- 46.Schmid PC, Krebsbach RJ, Perry SR, Dettmer TM, Maasson JL, Schmid HH. Occurrence and postmortem generation of anandamide and other long-chain N-acylethanolamines in mammalian brain. FEBS Lett. 1995;375:117–120. doi: 10.1016/0014-5793(95)01194-j. [DOI] [PubMed] [Google Scholar]
- 47.Berger C, Schmid PC, Schabitz WR, Wolf M, Schwab S, Schmid HH. Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem. 2004;88:1159–1167. doi: 10.1046/j.1471-4159.2003.02244.x. [DOI] [PubMed] [Google Scholar]
- 48.Amantea D, Spagnuolo P, Bari M, Fezza F, Mazzei C, Tassorelli C, Morrone LA, Corasaniti MT, Maccarrone M, Bagetta G. Modulation of the endocannabinoid system by focal brain ischemia in the rat is involved in neuroprotection afforded by 17beta-estradiol. FEBS J. 2007;274:4464–4775. doi: 10.1111/j.1742-4658.2007.05975.x. [DOI] [PubMed] [Google Scholar]
- 49.Muthian S, Rademacher DJ, Roelke CT, Gross GJ, Hillard CJ. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience. 2004;129:743–750. doi: 10.1016/j.neuroscience.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 50.Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci. 2003;23:7767–7775. doi: 10.1523/JNEUROSCI.23-21-07767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Epps DE, Palmer JW, Schmid HH, Pfeiffer DR. Inhibition of permeability-dependent Ca2+ release from mitochondria by N-acylethanolamines, a class of lipids synthesized in ischemic heart tissue. J Biol Chem. 1982;257:1383–1391. [PubMed] [Google Scholar]
- 52.Kinouchi H, Imaizumi S, Yoshimoto T, Motomiya M. Phenytoin affects metabolism of free fatty acids and nucleotides in rat cerebral ischemia. Stroke. 1990;21:1326–1332. doi: 10.1161/01.str.21.9.1326. [DOI] [PubMed] [Google Scholar]
- 53.Paik MJ, Li WY, Ahn YH, Lee PH, Choi S, Kim KR, Kim YM, Bang OY, Lee G. The free fatty acid metabolome in cerebral ischemia following human mesenchymal stem cell transplantation in rats. Clin Chim Acta. 2009;402:25–30. doi: 10.1016/j.cca.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 54.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 55.LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D. The search for the palmitoylethanolamide receptor. Life Sci. 2005;77:1685–1698. doi: 10.1016/j.lfs.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Guzman M, Lo Verme J, Fu J, Oveisi F, Blazquez C, Piomelli D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-alpha) J Biol Chem. 2004;279:27849–27854. doi: 10.1074/jbc.M404087200. [DOI] [PubMed] [Google Scholar]
- 57.Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy SP, Kendall DA, Bennett AJ. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br J Pharmacol. 2007;152:734–743. doi: 10.1038/sj.bjp.0707478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.