

Abstract
Halorhodopsin from Natronomonas pharaonis (pHR) functions as a light-driven halide ion pump. In the presence of halide ions, the photochemical reaction of pHR is described by the scheme: K→ L1 → L2 → N → O → pHR′ → pHR. Here, we report light-induced structural changes of the pHR-bromide complex observed in the C2 crystal. In the L1-to-L2 transition, the bromide ion that initially exists in the extracellular vicinity of retinal moves across the retinal Schiff base. Upon the formation of the N state with a bromide ion bound to the cytoplasmic vicinity of the retinal Schiff base, the cytoplasmic half of helix F moves outward to create a water channel in the cytoplasmic interhelical space, whereas the extracellular half of helix C moves inward. During the transition from N to an N-like reaction state with retinal assuming the 13-cis/15-syn configuration, the translocated bromide ion is released into the cytoplasmic medium. Subsequently, helix F relaxes into its original conformation, generating the O state. Anion uptake from the extracellular side occurs when helix C relaxes into its original conformation. These structural data provide insight into the structural basis of unidirectional anion transport.
Introduction
Halorhodopsin (HR) is a retinylidene protein that uses light energy to transport chloride ions from the extracellular side to the cytoplasmic side of the cell membrane (1–4). The HR homolog found in Natronomonas pharaonis (pHR) participates in light-driven ATP synthesis (5–7). Because this electrogenetic ion pump was reported to be useful as a neuron silencer (8,9), many researchers have made efforts to design a novel ion pump with more useful optical properties (10–13). It is certain that innovation in the field of optogenetics will be accelerated by the accumulation of structural data on ion-transporting rhodopsins.
To date, pHR and the HR homolog from Halobacterium salinarum (sHR) have been crystallized (14,15). Their crystallographic data show that the active site structure is well conserved, i.e., retinal is covalently linked via a protonated Schiff base to the ε-amino group of a lysine residue in the seventh transmembrane helix (helix G), and the primary anion-binding site (site I) is located in the extracellular vicinity of the retinal Schiff base. A peculiar structural feature of pHR is seen in that the N-terminal polypeptide folds into a short amphipathic helix (A′), which, together with a long BC loop, forms a hydrophobic cap that covers the extracellular surface. This cap structure is absent in sHR, which instead possesses a secondary anion-binding site near the extracellular surface (16). This structural difference may be correlated with a significant difference in the reaction kinetics of anion uptake from the extracellular medium (17,18). A remarkable difference is also seen in the spectral properties, i.e., the removal of chloride ions from pHR is accompanied by a purple-to-blue transition (a 20 nm red shift of the visible band of retinal), whereas a 10 nm blue shift is induced upon anion removal from sHR (19,20). The latter spectral shift may be related to the coexistence of two retinal isomers in the dark-adapted state (21). By contrast, pHR contains the trans isomer as the major component (22). This property has simplified the analysis of the anion-transporting photocycle of pHR, which is usually described by the following scheme: pHR + hν → K→ L1 → L2 → N → O → pHR′ → pHR (18,23,24). From time-resolved absorption and electric measurements, it was suggested that anion release into the cytoplasmic medium takes place during the N-to-O transition, whereas anion uptake from the extracellular medium occurs in the decay of the O state (24–26). (Although pHR′ was introduced to explain the biphasic decay of O observed in bacterioruberin-free samples of pHR, this reaction state was not always detected in the anion-pumping cycle of the trimeric pHR-bacterioruberin complex, the structure of which is shown in Fig. S1 in the Supporting Material; our unpublished data.)
The anion-depleted blue form of pHR is considered to mimic the O state (27). A previous structural analysis has shown that the removal of a halide ion from site I is accompanied by the inward movement of the extracellular half of helix C (28). This result suggests that the extracellular half of helix C functions as a valve by which the anion uptake process is regulated. On the other hand, the details of the anion-release process have remained unclear. According to the generally accepted concept of ion transport, it has been postulated that the opening of the cytoplasmic interhelical space takes place during the anion release process (29). To quantitatively analyze this postulated structural change, we need to solve a technical problem as to how efficiently a key reaction state with a largely deformed conformation is trapped in such a three-dimensional crystal that the target proteins are densely packed. This same problem has been encountered in crystallographic analyses of reaction intermediates of other photoactive proteins. For example, electron-micrographic studies of bacteriorhodopsin (bR) in the purple membrane suggested that the extracellular half of helix F moves outward upon the formation of the N state (30–32), whereas this movement is inhibited in high-quality three-dimensional crystals of bR (33,34). One possible answer to the previous problem was provided by a recent crystallographic study of light-induced structural changes in the pHR-azide complex, i.e., the C2 crystal, the asymmetric unit of which contains three subunits with different motional freedoms, was shown to be useful for structural investigations into the reaction state in which the cytoplasmic half of helix F is largely deformed (35). However, as the anion-pumping capability is abolished in the pHR-azide complex, which instead works as an outward-directed proton pump (36), the structure of this reaction state provides no direct information about the anion transport mechanism.
In this study, we investigated light-induced structural changes in the trimeric pHR assembly using a C2 crystal that was soaked in a postcrystallization solution containing bromide ions. It was found that slow cooling of the C2 crystal after illumination at 240 K was effective at trapping the N state in a subunit with the EF loop facing to the open space in the unit cell. Meanwhile, rapid cooling of the C2 crystal after illumination at 240 K was effective at trapping the O state in a different subunit. Furthermore, diffraction data from the crystals that were illuminated at 200 K provided information about the reaction states occurring in the early stage of the photoreaction cycle. The integration of the structural data of these reaction states enabled a discussion about the molecular mechanism of anion translocation by pHR.
Materials and Methods
Preparation and crystallization of pHR
The claret membrane of N. pharaonis strain KM-1 was prepared as previously described (37). For the crystallization of pHR, the claret membrane was partially delipidated with 0.5% Tween 20 in 0.1 M NaCl (pH 7). The crystallization of pHR was performed as previously described (15).
For the structural investigation of the resting state of the pHR-bromide complex, a C2 crystal of pHR that was prepared in the presence of chloride ions was soaked in a postcrystallization solution consisting of 0.01–0.1 M NaBr, 3.0 M (NH4)2SO4, 0.1 M HEPES (pH 7), and 30% trehalose for ∼10 min and then flash-cooled with liquid propane at its melting temperature (Tmelt = 83 K). In the investigated range of the bromide concentration (0.01–0.1 M), the primary anion-binding site was fully occupied by a bromide ion. However, the occupancy of a bromide ion at the secondary anion-binding site, which is located in the intersubunit crevice, varied notably with the bromide concentration. For the structural investigation of the reaction states occurring at an early stage of the photocycle, the frozen crystal was transiently warmed to 200 K, illuminated with red light (λ = 635 nm, ∼1 mW/mm2) for 1 min, and then cooled to 100 K in the dark. For the structural investigation of the reaction states occurring in the late half of the photocycle, the frozen crystal in 0.01–0.1 M NaBr, 3.0 M (NH4)2SO4 or 1.5 M Na-citrate, 0.1 M HEPES at pH 7, and 30% trehalose was warmed to 240 K, illuminated with red light for ∼1 min, and then cooled to 100 K at a cooling rate of 2.3 K/s. Alternatively, a crystal was precooled to 240 K under a flow of cold nitrogen gas, illuminated with red light, and then flash-cooled with liquid propane.
Measurement of absorption spectra and kinetics
Light-induced absorption changes in the C2 crystal of pHR were measured using a microspectrophotometer, in which monochromatic light from a double monochromator was passed through a pinhole with a diameter of 0.05 mm and focused on a small area of the crystal (38).
Data collection and scaling
The x-ray diffraction measurements were performed at the beamline SPring8-BL38B1, where a frozen crystal kept at 100 K was exposed to a monochromatic x-ray beam at a wavelength of 1.0 Å with an x-ray flux rate of ∼2 × 1012 photons/mm2/s. Diffraction data were collected using a charge-coupled device (CCD) detector, with an oscillation range of 1° and an x-ray flux of ∼6 × 1012 photons/mm2 per image. For collection of a full data set, a single crystal was exposed to an x-ray flux of ∼1 × 1015 photons/mm2; using RADDOSE (39), the radiation dose absorbed by the crystal during the data collection was estimated to be ∼0.5 MGy. This flux of x-rays caused no significant x-ray-induced change in the protein conformation (Fig. S2). Indexing and integration of diffraction spots were carried out with Mosflm 6.1 (40). The scaling of data was done using SCALA in the CCP4 program suite (41). Crystal parameters and data collection statistics are summarized in Table S1.
Structural refinement
The model building of the resting state of the bromide-bound purple form of pHR was performed using the previously reported model of pHR (pdb id: 3FEB) as an initial search model. Structural refinement was done using CNS-1.2 (42) and XtalView-4.0 (43). For the structural investigation of the reaction states of the pHR-bromide complex, the diffraction data set (Flight) from an illuminated crystal was compared with that (Fdark) from a crystal that was flash-cooled in dim light, and the difference electron density map between the reaction state and the resting state (|Flight| − |Fdark| map) was evaluated using the phase derived from the structural model of the resting state of pHR-bromide complex.
When the N (or O) state was preferentially trapped in subunit B (or C) within the asymmetric unit, the structural analysis was performed based on the approximation that two conformers (a reaction state and the resting state) were contained in each subunit. The calculated amplitude Fobs was evaluated as follows: |Fcalc| = Σi {αi⋅|Fi_React| + (1−αi)⋅|Fi_Rest|}, where Fi_React and Fi_Rest are the structure factors of the reaction state and the resting state, respectively, in the i-th subunit. The initial model of each reaction state was built on the basis of the |Flight| − |Fdark| map, whereas the structure of the second conformer in each subunit was assumed to be identical to that of the resting state in a crystal that was flash-cooled in dim light. At each value of αi, the structure of the reaction state in the i-th subunit was refined by the simulated annealing method, whereas the position/orientation of the second conformer was adjusted by the rigid body refinement. In a new cycle of search for the optimal αi values, the structural refinement of the reaction state in the i-th subunit was performed using the previously determined optimal αj (j ≠ i) values of the reaction states in the other subunits. Such a refinement cycle was repeated until no large decrease in the Rcryst value (at the optimal αj values) was observed. For a crystal that was soaked in a solution containing 0.1 M bromide ions and slowly cooled after illumination at 240 K, the crystallographic R value decreased to 0.213 when the optimal values of αi (0.50, 0.65, and 0.52 for subunits A, B, and C, respectively) were used (Fig. S3).
Results
Fig. 1 shows light-induced absorption changes that were observed in the C2 crystal that had been soaked in a solution containing 0.01 M NaBr and cooled to 240 K. As the b axis of the C2 crystal was orientated in parallel to the optical path of the measuring light beam, the visible absorption band of retinal (λmax ∼578 nm) was more intense as compared with the vibronic bands (at 542, 506, and 470 nm) of bacterioruberin, which is bound to the intersubunit crevice within the pHR trimer (15). When the C2 crystal with this orientation was illuminated with orange light (>540 nm, ∼100 mW/cm2), the absorbance at 580 nm decreased and, conversely, significant absorption increases were observed at 650 and 520 nm (the blue line in Fig. 1 b). This absorption change suggested the accumulation of two reaction states; the absorption increase at 640 nm is attributable to accumulation of the O state with a red-shifted absorption spectrum (λmax ∼600 nm), whereas the absorption increase at 500 nm can be explained by the accumulation of a reaction state(s) with a blue-shifted absorption spectrum (λmax ∼520 nm). When the absorption spectrum was recorded 3 min after illumination, the absorption increase at 640 nm was no longer observed (the red line in Fig. 1 b). It appeared that, in the C2 crystal, the O state decayed more rapidly than the blue-shifted reaction state. This observation can be explained by supposing that the photoreaction kinetics of pHR are greatly influenced by the crystal lattice force. It will be shown later that the C2 crystal shrinks under illumination, and that this shrinkage is accompanied by a significant retardation of the N-to-O transition in one subunit within the asymmetric subunit, whereas this retardation is not significant in other subunits.
Figure 1.
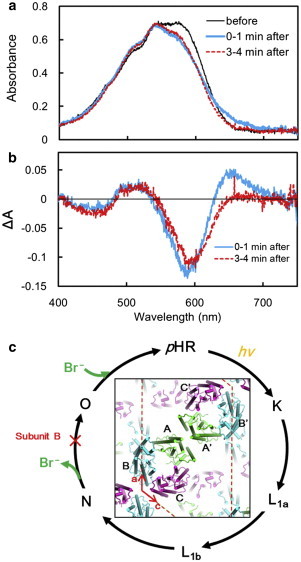
Light-induced absorption changes in the C2 crystal at 240 K. (a) The C2 crystal was cooled to 240 K under a flow of cold nitrogen gas and its absorption spectrum was recorded before illumination (black line), 0–1 min (blue line), and 3–4 min (red line) after illumination with orange light (>540 nm, ∼100 mW/cm2). For this measurement, a crystal adhered to a lower glass in the sitting-drop crystallization kit was soaked in a postcrystallization solution consisting of 0.01 M NaBr, 3.0 M ammonium sulfate, 0.1 M HEPES at pH7, and 30% trehalose, and mounted on an x-y stage attached to an optical microscope; the crystal orientation was then adjusted so that the b axis was parallel to the optical path of the measuring light beam. (b) Difference absorption spectra derived by subtracting the absorption spectrum of the unilluminated crystal from the spectra recorded after illumination. (c) The anion-transporting photocycle of pHR. (Inset) Protein packing in the C2 crystal, viewed along the b axis. To see this figure in color, go online.
Light-induced structural changes at 240 K
For the structural investigation of the reaction states of the pHR-bromide complex, the C2 crystal was soaked in a solution containing 0.01–0.1 M bromide ions, illuminated at 240 K with red light at 635 nm, and then cooled to 83–100 K at various cooling rates. Fig. 2 shows the |Flight| – |Fdark| difference maps that were evaluated by comparing the diffraction amplitudes (|Flight|) observed for illuminated crystals with those (|Fdark|) observed for unilluminated crystals. It is clear that the three subunits within the asymmetric unit underwent different structural changes under illumination, and that the contents of the reaction states trapped in the individual subunits were dependent on the bromide concentration as well as the cooling rate after illumination.
Figure 2.
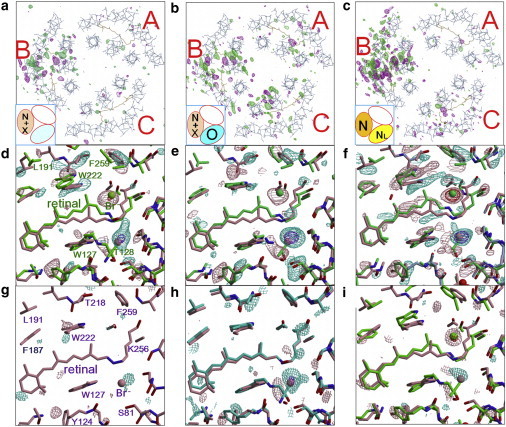
Light-induced structural changes in the trimeric pHR-bromide complex. |Flight| – |Fdark| difference maps observed for the C2 crystal that had been soaked in a solution containing 0.01 M (a and b) or 0.1 M NaBr (c). The crystal was cooled in the dark to 100 K at a cooling rate of 2.3 K/s after illumination with red light at 240 K (a), cooled rapidly to 83 K after illumination at 240 K (b), or slowly cooled in red light from 240 to 100 K (c). Each |Flight| – |Fdark| difference map was evaluated by comparing the diffraction amplitudes (|Flight|) observed for an illuminated crystal with those (|Fdark|) observed for an unilluminated crystal, contoured at 3.2 σ (positive densities in purple, negative in green), and overlaid on the structural model of the resting state (a–c). In the middle and lower panels, the difference map around the retinal chromophore in subunit B (d–f) or subunit C (g–i) were contoured at 3 σ (positive densities in purple, negative in sky blue) and 8 σ (positive densities in red, negative in dark blue) and overlaid on the structural models of the resting state (pink) and the major reaction state (green or cyan) trapped in subunit B or subunit C. To see this figure in color, go online.
When the C2 crystal soaked in a solution containing 0.01 M bromide ions was cooled to 100 K at a cooling rate of 2.3 K/s after illumination at 240 K (this cooling was done in the dark), for example, a significant structural change was observed only in the subunit (subunit B) with the EF loop facing to the open space in the unit cell (Fig. 2 a). The |Flight| – |Fdark| difference map within this subunit exhibited a strong negative peak at site I and a strong positive peak in the cytoplasmic vicinity of the retinal Schiff base (see Fig. 2 d). This suggests that the bromide ion initially existing at site I moved to a cavity that was transiently created between the Schiff base and the side chain of Ile-134. Hereafter, the latter ion binding site is called site i134. When the crystal was rapidly cooled to 83 K with liquid propane after illumination at 240 K, on the other hand, significant structural changes were observed in all the subunits (Fig. 2 b). In this case, the |Flight| – |Fdark| difference map within subunit C (see Fig. 2 h) exhibited a strong negative peak at site I, whereas no strong positive peak was seen at any other position within subunit C; i.e., a reaction state with no bromide inside this subunit was trapped. Together with the result of the light-induced absorption changes (Fig. 1), the |Flight| – |Fdark| difference maps shown in Fig. 2, a and b, suggests that rapid cooling after illumination is effective at trapping the O state in subunit C, whereas slow cooling after illumination is effective at trapping the blue-shifted reaction state(s) in subunit B.
Structure of the O state
We first constructed the structural model of the O state that was preferentially trapped in subunit C within the C2 crystal that was rapidly cooled after illumination at 240 K. When the electron density (2|Fo|-|Fc|) map was analyzed on the approximation that only two states (the O state and the resting state) were contained in subunit C, the content of the O state was estimated to be ∼50%. The major structural difference between the O state and the resting state was seen in the anion uptake pathway (Fig. 3). In the O state, 1) the extracellular half of helix C deforms in such a manner that the side chain of Thr-126 comes to the position where the bromide ion occupied in the resting state and 2) the side chain of Glu-234 is reorientated toward Tyr-124. These structural differences are similar to those seen between the chloride-bound purple form and the anion-depleted blue form (28). In other words, the structure of the O state is nearly identical to that of the anion-depleted blue form, except that the O state has a bromide ion at the second anion-binding site (site II) that is located near the cytoplasmic end of bacterioruberin in the intersubunit crevice (Fig. S1).
Figure 3.
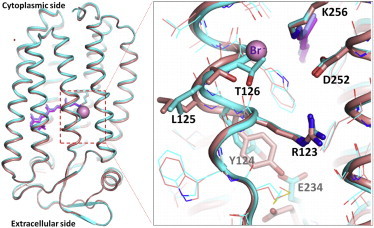
The structure of the O state. The structure of the O state trapped in subunit C (cyan) is compared with that of the resting state (salmon). The pink sphere represents the bromide ion bound to site I in the resting state. To see this figure in color, go online.
Structure of the N state
We next tried to determine the structure of the blue-shifted reaction state by analyzing the electron density (2|Fo|-|Fc|) map in subunit B within the C2 crystal that was slowly cooled after illumination at 240 K. However, it turned out that besides the reaction state with a bromide ion bound to site i134 (we refer to this as the N state), the O state and/or another reaction state with no bromide ion inside the protein was also trapped in this subunit. In such a case, it was difficult to accurately determine the structure of the N state.
After many trials, we found that the N state was preferentially trapped in subunit B when the crystal that had been soaked in a solution containing 0.1 M bromide ions was illuminated at 240 K and cooled to 100 K at a cooling rate of 2.3 K/s under illumination. In this case, the |Flight| – |Fdark| difference map in subunit B had such a strong positive peak at site i134 that its absolute amplitude was comparable to that of a strong negative peak seen at site I (Fig. 2 h). This difference map suggests that the N state is the major reaction state trapped in subunit B. When the structural analysis was performed on the approximation that only two states (the resting state and the N state) were contained in subunit B, the content of the N state trapped in this subunit was estimated to be 65% (Fig. S3). This high content of the N state made it possible to construct a structural model of the N state at a resolution of 2.2 Å.
In Fig. 4, the structural model of the N state trapped in subunit B is compared with that of the resting state of the pHR-bromide complex. It was found that the retinal chromophore in the N state takes on a planar 13-cis/15-anti configuration so that the C13 methyl of retinal pushes the indole ring of Trp-222 upward (Fig. 4 b). The upward movement of this tryptophan is accompanied by large movements of most residues in the cytoplasmic half of helix F (Thr-218, Lys-215, Phe-211, and so on). Retinal isomerization also induces a large rotation of the side chain of Ile-134, which is coupled with outward movements of the side chain of Phe-259 in helix G and the main chains of Leu-217 and Thr-218 in helix F (see Fig. 4 d). Because of these movements, a long water channel is created in the cytoplasmic interhelical space. The innermost region of this channel corresponds to site i134, where a bromide ion interacts directly with the positively charged Schiff base and Ser-130 OH. This bromide ion also interacts with a water molecule (Wat601), which is in turn connected to a linear water cluster (Wat501, Wat602–606) extending to the cytoplasmic membrane surface. It is noteworthy that, in the resting state, only one water molecule (Wat501) was detected in the cytoplasmic interhelical region.
Figure 4.
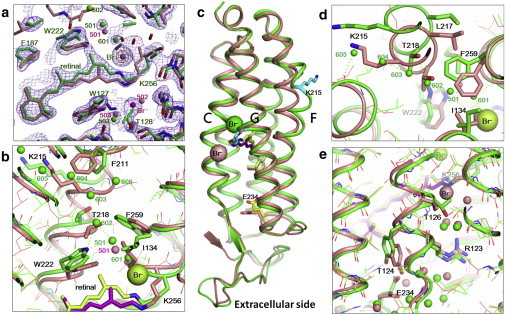
Structure of the N state trapped in subunit B. (a) 2 Fo – Fc map of a mixture of the N state (65%) and the resting state (45%) that was trapped in subunit B when the C2 crystal that had been soaked in a solution containing 0.1 M bromide ions was slowly cooled after illumination at 240 K; this map was contoured at 1.4 σ (blue) and 7.0 σ (red), and overlaid on the structural model of a mixture of the N state (green) and the resting state (salmon). (b) Structural change in the cytoplasmic interhelical space. The small green spheres represent water molecules in the N state. (c) The overall structure of the N state trapped in subunit B (green) is compared with that of the resting state (salmon). (d) Deformation of the cytoplasmic half of helix F viewed from the cytoplasmic side. (e) Deformation of the extracellular half of helix C. To see this figure in color, go online.
A considerable structural difference between the N state and the resting state was also seen on the extracellular side (Fig. 4 e). In the N state, the middle moiety of helix C deforms in such a manner that the side chain of Thr-126 moves toward Asp-252, and the side chain of Glu-234 is oriented toward Tyr-124. These differences were also seen between the O state and the resting state. In other words, there is no great structural difference in the extracellular half between the N state and the O state. This implies that the structure of the anion-release pathway is closed during the N-to-O transition, whereas the anion uptake pathway remains unaltered.
Another important structural change occurring during the N-to-O transition is a sliding movement of helix G toward the cytoplasmic side. When the structure of the N state generated in subunit B is compared with those of O and the resting state, it becomes clear that the relative position of helix G is shifted toward the extracellular side in the N state (Fig. S4). This shift seems to be coupled with reisomerization of retinal from the all-trans to the 13-cis/15-anti configuration. It will be discussed later if this sliding movement is responsible for the light-induced crystal shrinkage along the b axis.
A reaction state with the 13-cis/15-syn retinal-Lys-256 chain
A previous study on the photocycle of pHR provided evidence for two O-kinetic states in equilibrium with a temperature-dependent L-like state and a chloride-dependent N-like state, respectively (23). In this study, we observed that besides the N state, a reaction state with no bromide ion at site i134 also accumulated in subunit B under illumination at 240 K when the bromide ion concentration in the soaking solution was not high. This reaction state (we refer to it as X), in which the cytoplasmic interhelical space is open, is distinct from the O state mentioned in Fig. 3.
To get the structural information of X, we analyzed the diffraction data (FOn_0.01M) from the crystal that was soaked in a solution containing 0.01 M bromide ions, illuminated at 240 K, and rapidly cooled to 83 K (Fig. 2 a). It was suggested that this illuminated crystal contained three states (N, X, and the resting state) in subunit B. In this case; the contribution from subunit B to the diffraction amplitude FOn_0.01M (= Σi=1,2,3 Fi) is expressed by using the equation:
| (1) |
where α2 and β2 are the occupancies of N and X in subunit B, respectively; F2_N, F2_X, and F2_Rest represent the Fi=2 amplitudes from N, X, and the resting state, respectively, that would be expected if subunit B is fully occupied by one of these states. The occupancies α2 and β2 can be estimated by comparing FOn_0.01M with the diffraction data (FOn_0.1M) from the crystal that was illuminated at a higher bromide ion concentration (0.1 M NaBr), in which the N state was preferentially trapped in subunit B (i.e., Fi=2 was approximated by 0.65 F2_N + 0.35 F2_Rest).
First, the |FOn_0.01M| − |FOn_0.1M | difference map has no peak at site I, suggesting that β2 is ∼0.35 (Fig. 5 a). This difference map, which is identical substantially to the difference map between N and X, suggests that the protein structure of X is not much different from that of N. The major differences between X and N are seen in the following features: 1) in X, the side chain of Ile-134 is directed toward the retinal Schiff base and no bromide ion exists at site i134; 2) the water distribution in the cytoplasmic interhelical space is altered; 3) the main chain of Lys-256 is less distorted in X (Fig. 5 a). The last feature can be explained by supposing that the retinal configuration in X is different from that seen in N. On the other hand, the 2Fo-Fc map that is derived from the quantity 2.2 × |FOn_0.01M| – 1.2 × |FOn_0.1M| has no significant peak at site i134, suggesting that the contribution of N to this map was nearly zero (i.e., α2 = 0.295) (Fig. 5 b). This 2Fo-Fc map was expected to represent a mixture of X (65%) and the resting state (35%). Because the b axis of the crystal shrank noticeably under illumination (Table S1), the structure of X was determined by the same procedure as used for the structural determination of N. The result of analysis suggests that in X, the retinal-Lys-256 chain takes on the 13-cis/15-syn configuration and the Schiff base interacts directly with Asp-252 (Fig. 5 c).
Figure 5.
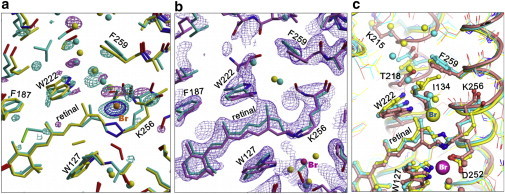
Structure of the X state accumulating in subunit B under illumination at a low bromide ion concentration. (a) The |FOn_0.01M| – |FOn_0.1M| difference map, contoured at 3.4 σ (positive, purple; negative, sky blue) and 6 σ (negative, dark blue), is overlaid on the models of X (cyan) and N (yellow) trapped in subunit B. Here, FOn_0.01M and FOn_0.1M are the diffraction amplitudes from the illuminated crystals that had been soaked in 1.5 M Na-citrate solutions containing 0.01 and 0.1 M, respectively, NaBr. The former crystal (crystal id: 1107 in Table S1) was rapidly cooled to 83 K after illumination at 240 K; the other crystal (crystal id: 974 in Table S1) was slowly cooled from 240 to 100 K in red light. The occupancy of the resting state in subunit B was suggested to be similar (∼35%) in these illuminated crystals; namely, the contribution of the resting state to the |FOn_0.01M| − |FOn_0.1M| map was nearly zero. (b) The 2Fo-Fc map that was derived from the quantity 2.2|FOn_0.01M| – 1.2|FOn_0.1M| is contoured at 1.3 σ (blue) and overlaid on the models of X (cyan) and the resting state (purple) in subunit B. This electron density map represents the structural feature that would be expected for a mixture of X (65%) and the resting state (25%) in subunit B. Although the two crystals illuminated at different bromide ion concentrations (0.01 and 0.1 M NaBr) contained different amounts (∼30% and 65%, respectively) of N in subunit B, the contribution of this N state to the 2Fo-Fc map was cancelled out when the quantity 2.2|FOn_0.01M| – 1.2|FOn_0.1M| was used. (c) The structures of X (cyan), N (yellow), and the resting state (salmon) are compared. To see this figure in color, go online.
Possible interpretations of this observation are 1) X is a reaction state occurring between N and O; 2) X is in dynamic equilibrium with N, whereas N decays into O; and 3) X is an artificial reaction state generated in the C2 crystal. It is currently difficult to decide which interpretation is correct. But, because it is natural to consider that in the N-to-O transition the anion release pathway should be open, we adopt the first interpretation here. In this case, it would be expected that the reisomerization process of retinal occurring in the late half of photocycle is divided into two steps; i.e., the first step is the isomerization around the Schiff base linkage and the second step is the double-isomerization around the C13=C14 double bond and the Schiff base linkage. It is important to point out that the transition from N to X, during which the Schiff base NH bond is reorientated toward the nearby aspartate (Asp-252), prevents the formation of an M-like state with the deprotonated Schiff base, which has never been observed to occur during the anion pumping cycle. The possible reaction pathways from X to pHR, which may be dependent on the anion concentration, are shown in Fig. 6, where X is renamed as N′. (From absorption kinetic data of the pHR-bacterioruberin complex in membrane suspensions, it is suggested that at high bromide ion concentrations (>1 M), the X(N′) state decays rapidly into a long-living state (pHR′) whose absorption spectrum is similar to that of N (H. Kawaguchi and T. Kouyama, unpublished data)).
Figure 6.
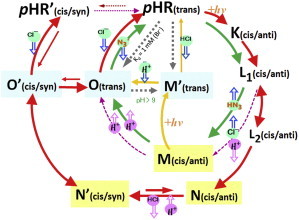
A reaction scheme of the anion-pumping cycle of pHR in complex with halide ions (red arrows). The retinal-Lys-256 chain takes on the 13-cis/15-anti configuration in the early stages (K, L1, L2, and N) of the anion-pumping cycle, whereas it takes on the 13-cis/15-syn configuration in N′(=X), O′, and pHR′ and the 13-trans/15-anti configuration in pHR, O, and M′. The cytoplasmic interhelical space is open in N, N′, and M, whereas it is closed in the other states. The open blue and pink arrows indicate ion movements occurring at the extracellular side and at the cytoplasmic side, respectively, of the protein. At a very high anion concentration, the transition from O′ to pHR′ may be faster than the O′-to-O transition so that pHR′ is transiently generated. In the absence of anions, O, O′, and N′ occur at neutral pH and M′ occurs at high pH in the dark. The nonpumping cycle of pHR-halide ion complex (purple arrows), the inward-directed proton-pumping cycle driven by two photons (orange arrows), and the outward-directed proton-pumping cycle of the pHR-azide complex (green arrows) are also shown. To see this figure in color, go online.
Effects of the crystal lattice force on the photoreaction kinetics
The absorption kinetics data of aqueous suspensions of claret membrane at pH 7 show that the lifetime of the N state of the pHR-bromide complex is much shorter (τ = 2.5 ms at 24°C) than that of the O state, unless the bromide ion concentration is very high (>1 M). Hence, a question arises as to why a large amount of N can accumulate in the C2 crystal under illumination. To answer this question, we need to take into account a possible effect of the crystal lattice force on the reaction kinetics. It was previously shown that the decay of the M state of bacteriorhodopsin in the P622 crystal slowed down significantly (>100 times) when the intermembrane space was reduced by increasing the concentration of the precipitant (34).
In this study, we observed that the unit cell of the C2 crystal shrank noticeably (∼1.5%) along the b axis when a large amount of N was trapped in subunit B. It is suggested that the major cause of this crystal shrinkage is a light-induced sliding movement of helix G in subunit B, the cytoplasmic end of which makes contact with helix A′ in a neighboring subunit that is related by a crystallographic twofold screw axis (Fig. S4). Conversely, it would be expected that light-induced crystal shrinkage exerts a nonnegligible effect on the reaction kinetics in subunit B. That is, as the crystal lattice force becomes stronger in the direction normal to the membrane (nearly parallel to the b axis), helix G in subunit B is pushed more strongly toward the extracellular side and, as a consequence, the formation of the O state, during which a sliding movement of helix G toward the cytoplasmic side takes place, is inhibited.
The crystal shrinkage mentioned previously affected the protein structure and/or the reaction kinetics in other subunits. With respect to subunit A, in which no reaction state was trapped, a better explanation of the electron density map was achieved by supposing that the resting state in subunit A underwent a small conformational change in a passive way. In subunit C, on the other hand, a different reaction state than the O state was trapped (Fig. 2 i). This reaction state was suggested to have a bromide ion at site i134. However, unlike the N state trapped in subunit B, the cytoplasmic interhelical space was only partially opened (Fig. S5). It is noteworthy that the EF loop of subunit C is involved in protein-protein interactions between two trimers related by a twofold screw axis. Hence, it seems possible that the reaction state trapped in subunit C represents an early substate of N (NL), the relaxation of which would be inhibited when the obstacle against a structural change in the EF loop becomes larger.
It has been shown that the transition from N to O is reversible (18,23). From the dependence of a dynamic equilibrium between N and O on the chloride ion concentration, the apparent dissociation constant of the chloride ion to site i134 (i.e., the anion-binding site generated in the N state) was estimated to be ∼1 M (44). In this study, we observed that a mixture of N and X (a reaction state with no bromide ion at site i134) was trapped in subunit B when the crystal in a solution containing 0.01 M bromide ions was illuminated at 240 K and cooled to 100 K (Fig. 2 e). From this observation, the apparent dissociation constant of the bromide ion to site i134 was estimated to be ∼10 mM. A possible explanation for this low dissociation constant is that the dynamic equilibrium between N and O and/or between X and O is influenced by the crystal lattice force. This is consistent with the observation that a much smaller amount of X or N than O accumulated in subunit C when the C2 crystal was illuminated at a low bromide ion concentration. It should be noted that a mixture of N and X generated in subunit B decayed much more slowly as compared with the O state generated in subunit C. Together with these structural data, the spectral data shown in Fig. 1 suggest that the spectral property of X is similar to that of N or the resting state.
Light-induced structural changes at 200 K
To elucidate bromide movements in the early stage of the photoreaction cycle, we investigated the structure of a reaction state(s) that was trapped when the C2 crystal was cooled to 100 K after illumination at 200 K with red light at 635 nm (∼1 mW/mm2). At this low temperature, only reaction states with blue-shifted absorption spectra accumulated under illumination. The |Flight| – |Fdark| difference map between an illuminated crystal and an unilluminated crystal showed that the light-induced structural change was confined to the central part of the protein. Inspection of this difference map showed that two reaction states were trapped (Fig. 7). One of them, designated as the L1 state, takes on such a conformation that the bromide ion still exists on the extracellular side of the Schiff base, but the formation of L1 is accompanied by a small movement of a bromide ion within site I, which explains the pair of strong positive and negative peaks seen around site I. Similar ion movement was reported to occur upon the formation of the L1 state of sHR (16). The other reaction state, designated as the L2 state, possesses a bromide ion at a transiently generated binding site (termed as site s130) between Ser-130 and the retinal Schiff base; its formation explains the strong positive peak seen in the vicinity of the main chain of Ser-130. In this reaction state, the middle moiety of helix C is suggested to deform to create the binding site s130. But, as this binding site is not large, it seems necessary to postulate a significant backward reaction from L2 state to the L1 state. This result is in line with the previous report that the OH group of Ser-130 plays an important role for the ion transport (45).
Figure 7.
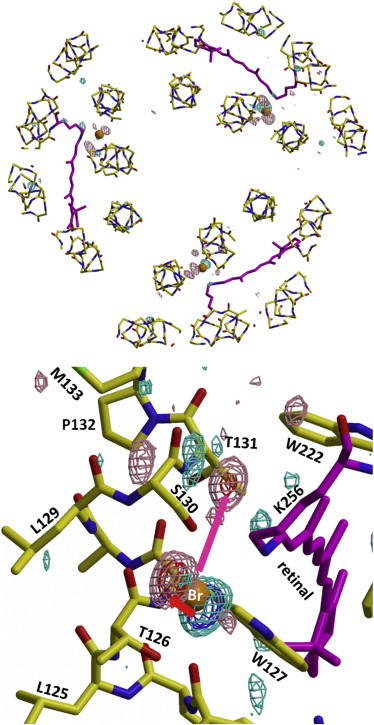
Light-induced structural change in the trimeric pHR-bromide complex at 200 K. (a) Structural change in the trimeric pHR-bromide complex that was observed when the C2 crystal that had been soaked in a solution containing 0.01 M NaBr was illuminated with red light (635 nm, 1 mW/mm2) at 200 K and then cooled to 100 K. The |Flight| – |Fdark| difference map was evaluated by comparing the diffraction data from the illuminated crystal with those from an unilluminated crystal, contoured at 4.5 σ (positive densities in purple, negative in sky blue) and overlaid on the structural model of the trimeric pHR-bromide complex. (b) An enlarged view of the |Flight| – |Fdark| difference map around the retinal Schiff base of subunit A; this map was contoured at 4 σ (positive densities in purple, negative in sky blue) and 6.5 σ (positive densities in red, negative in dark blue), and overlaid on the structural model of the resting state of pHR. To see this figure in color, go online.
Discussion
The reaction scheme of pHR that is derived from the integration of the currently available structural data is summarized as follows. 1) In the resting state, a halide ion is bound to the primary anion-binding site (site I) that is located between Thr-126 OH and the retinal Schiff base. 2) The photoisomerization of retinal into the 13-cis/15-anti configuration is followed by a small movement of the halide ion within site I. 3) In the L1-to-L2 transition, the halide ion existing in site I moves to site s130 that is created between Ser-130 and the Schiff base. 4) In the L2-to-N transition, the halide ion at site s130 moves to site i134 that is created by the rotation of the side chain of Ile-134. Simultaneously, the extracellular half of helix C moves inward, leading to significant shrinkage at site I. Coupled with these movements or within a short delay, the cytoplasmic half of helix F largely deforms, creating a long water channel in the cytoplasmic interhelical space. 5) Upon formation of X, in which the cytoplasmic interhelical space is still open, the halide ion in site i134 is released into the cytoplasmic medium. In this state, the retinal-Lys-256 chain takes on the 13-cis/15-syn configuration and the protonated Schiff base interacts with the nearby aspartate (Asp-252). 6) In the X-to-O transition, reisomerization of retinal to the all-trans configuration takes place and the cytoplasmic interhelical space is closed. 7) In the recovery process of the resting state, helix C relaxes into its original conformation and a bromide ion is translocated from the extracellular surface to site I.
The basic concept involved in this reaction scheme is that the extracellular half of helix C and the cytoplasmic half of helix F function as two independent valves for unidirectional ion transport (Fig. 8). Basically, similar structural changes have been argued to take place during the photoreaction of the light-driven proton pump bacteriorhodopsin (BR) (30–32,46).
Figure 8.
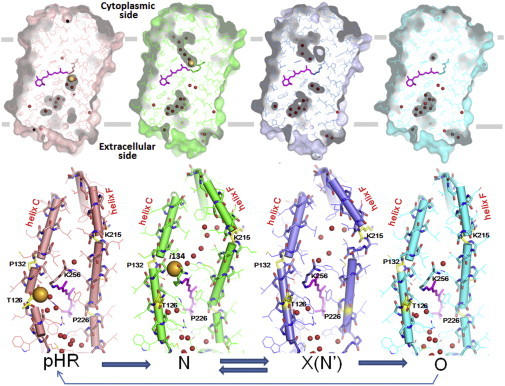
Conformational changes during the anion-transporting cycle of pHR. In the resting state (left panels), the cytoplasmic half of helix C has a kink at Thr-216 to accommodate a halide ion in the extracellular vicinity of the retinal Schiff base. In the N state (middle panels), the extracellular half of helix C moves inward, a halide ion is translocated to the extracellular vicinity of the Schiff base, and the cytoplasmic half of helix F has a kink at Lys-215 to create a water channel in the cytoplasmic interhelical space. In the X(N′) state, the translocated halide ion is released into the cytoplasmic medium and the retinal Schiff base interacts with the nearby aspartate (Asp-252). In the O state (right panel), the cytoplasmic interhelical space is closed, whereas the extracellular half of helix C remains to take on an unbent conformation. The small red spheres represent water molecules. To see this figure in color, go online.
An interesting structural feature of the N state is the formation of a narrow water channel in the cytoplasmic interhelical space (Fig. 4). This water channel is a potential candidate for the anion release pathway from site i134 to the cytoplasmic medium. But, as this transiently generated water channel is surrounded mostly by hydrophobic residues, it would be difficult for a nonhydrated halide ion to escape from the protonated Schiff base on the millisecond timescale at room temperature. The rapid anion release observed under physiological conditions can be explained by postulating the following scenario: 1) the halide ion at site i134 receives a proton from a positively charged residue (i.e., Lys-215) existing near the cytoplasmic medium and 2) the resulting HCl or HBr escapes from the protonated Schiff base and diffuses along the water channel to the cytoplasmic medium (47). This scenario is in line with a previous mutagenesis study showing that the reaction kinetics of pHR are greatly perturbed by the replacement of Lys-215 (48). It is also supported by our observation that, in the presence of 4 M NaBr, the formation rate of the O state becomes slower as the medium pH is increased above pH 10 (Fig. S6).
In fact, the above scenario was already suggested by a previous crystallographic study of the pHR-azide complex, which showed that the cytoplasmic half of helix F greatly deforms when an M-like state (Malk) is generated in subunit B under illumination at pH 9 (35). It is interesting to note that the structure of Malk is similar to that of the N state of the pHR-bromide complex, except that the bromide ion at site i134 is replaced by a water molecule in Malk. Because the pHR-azide complex works as a light-driven proton pump (36), this similarity implies that the same molecular valves (i.e., the extracellular half of helix C and the cytoplasmic half of helix F) are used for the inward-directed transport of halide ions in the pHR-halide ion complex and the outward-directed transport of protons in the pHR-azide complex.
N. pharaonis contains another retinylidene protein, phoborhodopsin (sensory rhodopsin-2), which functions as a receptor of negative phototaxis by forming a complex with a membrane-embedded transducin, pHtrII (49). Kamo and colleagues (50) have previously shown that the pHR double mutant, P240T/F250Y, can form a complex with pHtrII and that the L-to-N transition of this pHR mutant is inhibited after the complex formation with pHtrII (51). When the structure of the N state of pHR is taken into account, their observations suggest that the outward movement of the extracellular half of helix F is a key event for transmission of the light signal to the transducer. It would be interesting to ask whether similar light-induced structural changes as shown in Fig. 8 occur in other rhodopsins. For bovine rhodopsin, the outward movement of the cytoplasmic moiety of helix VI was suggested to occur upon the formation of Meta II (52). It has recently been shown that channelrhodopsin, which functions as a light-gated cation channel and is frequently used for light-induced neuron activation, can be converted to a light-gated chloride channel by replacement of a glutamate in the central gate with positively charged residues (53). It seems likely that the molecular mechanism of this light-gated chloride channel is not much different from that of the light-driven chloride pump. If this is the case, it would not be difficult to modify pHR into such a light-gated chloride channel that exhibits an absorption maximum in the red region.
Recent crystallographic studies of bacteriorhodopsin have shown that the conformation of the active site is modified by a very low dose of x-rays (54–56). Although no significant x-ray-induced structural change was detected in this study (Fig. S2), we cannot exclude the possibility that the retinal chromophore in pHR was partially modified during the data collection. It is currently difficult to determine the undamaged structures of the reaction states of pHR at high resolutions. We need to overcome this difficulty for a quantitative discussion about the retinal bond twists in key reaction states (especially in the X state), which will become an important issue.
Author Contributions
T.K., H.K., and T.N. designed the experiments. T.K. performed all structural modeling and collected the spectral data. H.K. and T.N. prepared crystals and collected the diffraction data. H.K. and M.M. contributed other essential material. T.K. prepared the manuscript. All authors discussed, interpreted, and approved the article.
Acknowledgments
We thank the staffs of BL38B1 of SPring-8 (Harima, Japan) for their technical assistance during data collection. This work was supported by a grant-in-aid from the Ministry of Education, Science, and Culture of Japan.
Editor: Andreas Engel.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Accession Numbers
Crystallographic coordinates of a mixture of the N state and the resting state of the pHR-bromide complex are deposited in the Protein Data Bank with accession code 4QRY.
Supporting Material
References
- 1.Mukohata Y., Kaji Y. Light-induced ATP synthesis dependent on halorhodopsin-pH regulation. Arch. Biochem. Biophys. 1981;208:615–617. doi: 10.1016/0003-9861(81)90551-8. [DOI] [PubMed] [Google Scholar]
- 2.Schobert B., Lanyi J.K. Halorhodopsin is a light-driven chloride pump. J. Biol. Chem. 1982;257:10306–10313. [PubMed] [Google Scholar]
- 3.Essen L.O. Halorhodopsin: light-driven ion pumping made simple? Curr. Opin. Struct. Biol. 2002;12:516–522. doi: 10.1016/s0959-440x(02)00356-1. [DOI] [PubMed] [Google Scholar]
- 4.Ernst O.P., Lodowski D.T., Kandori H. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 2014;114:126–163. doi: 10.1021/cr4003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bivin D.B., Stoeckenius W. Photoactive retinal pigments in haloalkaliphilic bacteria. J. Gen. Microbiol. 1986;132:2167–2177. doi: 10.1099/00221287-132-8-2167. [DOI] [PubMed] [Google Scholar]
- 6.Lanyi J.K., Duschl A., Oesterhelt D. The primary structure of a halorhodopsin from Natronobacterium pharaonis. Structural, functional and evolutionary implications for bacterial rhodopsins and halorhodopsins. J. Biol. Chem. 1990;265:1253–1260. [PubMed] [Google Scholar]
- 7.Avetisyan A.V., Kaulen A.D., Feniouk B.A. Photophosphorylation in alkalophilic halobacterial cells containing halorhodopsin: chloride-ion cycle? Biochemistry (Mosc.) 1998;63:625–628. [PubMed] [Google Scholar]
- 8.Han X., Boyden E.S. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS ONE. 2007;2:e299. doi: 10.1371/journal.pone.0000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang F., Wang L.P., Deisseroth K. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 10.Seki A., Miyauchi S., Kamo N. Heterologous expression of Pharaonis halorhodopsin in Xenopus laevis oocytes and electrophysiological characterization of its light-driven Cl− pump activity. Biophys. J. 2007;92:2559–2569. doi: 10.1529/biophysj.106.093153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gradinaru V., Zhang F., Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chuong A.S., Miri M.L., Boyden E.S. Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat. Neurosci. 2014;17:1123–1129. doi: 10.1038/nn.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochbaum D.R., Zhao Y., Cohen A.E. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods. 2014;11:825–833. doi: 10.1038/nmeth.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolbe M., Besir H., Oesterhelt D. Structure of the light-driven chloride pump halorhodopsin at 1.8 A resolution. Science. 2000;288:1390–1396. doi: 10.1126/science.288.5470.1390. [DOI] [PubMed] [Google Scholar]
- 15.Kouyama T., Kanada S., Ihara K. Crystal structure of the light-driven chloride pump halorhodopsin from Natronomonas pharaonis. J. Mol. Biol. 2010;396:564–579. doi: 10.1016/j.jmb.2009.11.061. [DOI] [PubMed] [Google Scholar]
- 16.Gmelin W., Zeth K., Oesterhelt D. The crystal structure of the L1 intermediate of halorhodopsin at 1.9 angstroms resolution. Photochem. Photobiol. 2007;83:369–377. doi: 10.1562/2006-06-23-RA-947. [DOI] [PubMed] [Google Scholar]
- 17.Váró G., Zimányi L., Lanyi J.K. Photocycle of halorhodopsin from Halobacterium salinarum. Biophys. J. 1995;68:2062–2072. doi: 10.1016/S0006-3495(95)80385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Váró G., Brown L.S., Lanyi J.K. Light-driven chloride ion transport by halorhodopsin from Natronobacterium pharaonis. 1. The photochemical cycle. Biochemistry. 1995;34:14490–14499. doi: 10.1021/bi00044a027. [DOI] [PubMed] [Google Scholar]
- 19.Ogurusu T., Maeda A., Yoshizawa T. Light-induced reaction of halorhodopsin prepared under low salt conditions. J. Biochem. 1981;90:1267–1273. doi: 10.1093/oxfordjournals.jbchem.a133591. [DOI] [PubMed] [Google Scholar]
- 20.Scharf B., Engelhard M. Blue halorhodopsin from Natronobacterium pharaonis: wavelength regulation by anions. Biochemistry. 1994;33:6387–6393. doi: 10.1021/bi00187a002. [DOI] [PubMed] [Google Scholar]
- 21.Kamo N., Hazemoto N., Mukohata Y. Light and dark adaptation of halorhodopsin. Arch. Biochem. Biophys. 1985;238:90–96. doi: 10.1016/0003-9861(85)90144-4. [DOI] [PubMed] [Google Scholar]
- 22.Zimányi L., Lanyi J.K. Fourier transform Raman study of retinal isomeric composition and equilibration in halorhodopsin. J. Phys. Chem. B. 1997;101:1930–1933. [Google Scholar]
- 23.Chizhov I., Engelhard M. Temperature and halide dependence of the photocycle of halorhodopsin from Natronobacterium pharaonis. Biophys. J. 2001;81:1600–1612. doi: 10.1016/S0006-3495(01)75814-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ludmann K., Ibron G., Váró G. Charge motions during the photocycle of pharaonis halorhodopsin. Biophys. J. 2000;78:959–966. doi: 10.1016/S0006-3495(00)76653-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hackmann C., Guijarro J., Siebert F. Static and time-resolved step-scan Fourier transform infrared investigations of the photoreaction of halorhodopsin from Natronobacterium pharaonis: consequences for models of the anion translocation mechanism. Biophys. J. 2001;81:394–406. doi: 10.1016/S0006-3495(01)75708-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoue K., Kubo M., Terazima M. Reaction dynamics of halorhodopsin studied by time-resolved diffusion. Biophys. J. 2009;96:3724–3734. doi: 10.1016/j.bpj.2008.12.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato M., Kanamori T., Nitta K. Stopped-flow analysis on anion binding to blue-form halorhodopsin from Natronobacterium pharaonis: comparison with the anion-uptake process during the photocycle. Biochemistry. 2002;41:2452–2458. doi: 10.1021/bi011788g. [DOI] [PubMed] [Google Scholar]
- 28.Kanada S., Takeguchi Y., Kouyama T. Crystal structures of an O-like blue form and an anion-free yellow form of pharaonis halorhodopsin. J. Mol. Biol. 2011;413:162–176. doi: 10.1016/j.jmb.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 29.Haupts U., Tittor J., Oesterhelt D. General concept for ion translocation by halobacterial retinal proteins: the isomerization/switch/transfer (IST) model. Biochemistry. 1997;36:2–7. doi: 10.1021/bi962014g. [DOI] [PubMed] [Google Scholar]
- 30.Subramaniam S., Lindahl M., Henderson R. Protein conformational changes in the bacteriorhodopsin photocycle. J. Mol. Biol. 1999;287:145–161. doi: 10.1006/jmbi.1999.2589. [DOI] [PubMed] [Google Scholar]
- 31.Vonck J. Structure of the bacteriorhodopsin mutant F219L N intermediate revealed by electron crystallography. EMBO J. 2000;19:2152–2160. doi: 10.1093/emboj/19.10.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersson M., Malmerberg E., Neutze R. Structural dynamics of light-driven proton pumps. Structure. 2009;17:1265–1275. doi: 10.1016/j.str.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Chen D., Lanyi J.K. Structural changes in the N and N′ states of the bacteriorhodopsin photocycle. Biophys. J. 2009;96:2779–2788. doi: 10.1016/j.bpj.2008.12.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto M., Hayakawa N., Kouyama T. Crystal structures of different substrates of bacteriorhodopsin’s M intermediate at various pH levels. J. Mol. Biol. 2009;393:559–573. doi: 10.1016/j.jmb.2009.08.047. [DOI] [PubMed] [Google Scholar]
- 35.Nakanishi T., Kanada S., Kouyama T. Large deformation of helix F during the photoreaction cycle of Pharaonis halorhodopsin in complex with azide. Biophys. J. 2013;104:377–385. doi: 10.1016/j.bpj.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulcsár A., Groma G.I., Váró G. Characterization of the proton-transporting photocycle of pharaonis halorhodopsin. Biophys. J. 2000;79:2705–2713. doi: 10.1016/S0006-3495(00)76508-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ihara K., Narusawa A., Kouyama T. A halorhodopsin-overproducing mutant isolated from an extremely haloalkaliphilic archaeon Natronomonas pharaonis. FEBS Lett. 2008;582:2931–2936. doi: 10.1016/j.febslet.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 38.Hayakawa N., Kasahara T., Kouyama T. Effect of xenon binding to a hydrophobic cavity on the proton pumping cycle in bacteriorhodopsin. J. Mol. Biol. 2008;384:812–823. doi: 10.1016/j.jmb.2008.09.075. [DOI] [PubMed] [Google Scholar]
- 39.Zeldin O.B., Gerstel M., Garman E.F. RADDOSE-3D: time- and space-resolved modelling of dose in macromolecular crystallography. J. Appl. Cryst. 2013;46:1225–1230. [Google Scholar]
- 40.Steller I., Bolotovsky B., Rossmann E.G. An algorithm for automatic indexing of oscillation images using Fourier analysis. J. Appl. Cryst. 1997;D30:1036–1040. [Google Scholar]
- 41.Dodson E.J., Winn M., Ralph A. Collaborative Computational Project, number 4: providing programs for protein crystallography. Methods Enzymol. 1997;277:620–633. doi: 10.1016/s0076-6879(97)77034-4. [DOI] [PubMed] [Google Scholar]
- 42.Brünger A.T., Adams P.D., Warren G.L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.McRee D.E. Academic Press; San Diego, CA: 1993. Practical Protein Crystallography. [Google Scholar]
- 44.Shibasaki K., Shigemura H., Demura M. Role of Thr-218 in the light-driven anion pump halorhodopsin from Natronomonas pharaonis. Biochemistry. 2013;52:9257–9268. doi: 10.1021/bi401295e. [DOI] [PubMed] [Google Scholar]
- 45.Sato M., Kikukawa T., Nitta K. Roles of Ser-130 and Thr-126 in chloride binding and photocycle of pharaonis halorhodopsin. J. Biochem. 2003;134:151–158. doi: 10.1093/jb/mvg123. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J., Yamazaki Y., Kouyama T. Crystal structure of the O intermediate of the Leu-93→Ala mutant of bacteriorhodopsin. Proteins. 2012;80:2384–2396. doi: 10.1002/prot.24124. [DOI] [PubMed] [Google Scholar]
- 47.Kouyama T., Murakami M. Structural divergence and functional versatility of the rhodopsin superfamily. Photochem. Photobiol. Sci. 2010;9:1458–1465. doi: 10.1039/c0pp00236d. [DOI] [PubMed] [Google Scholar]
- 48.Sato M., Kubo M., Demura M. Role of putative anion-binding sites in cytoplasmic and extracellular channels of Natronomonas pharaonis halorhodopsin. Biochemistry. 2005;44:4775–4784. doi: 10.1021/bi047500f. [DOI] [PubMed] [Google Scholar]
- 49.Gordeliy V.I., Labahn J., Engelhard M. Molecular basis of transmembrane signalling by sensory rhodopsin II-transducer complex. Nature. 2002;419:484–487. doi: 10.1038/nature01109. [DOI] [PubMed] [Google Scholar]
- 50.Sudo Y., Yamabi M., Kamo N. Importance of specific hydrogen bonds of archaeal rhodopsins for the binding to the transducer protein. J. Mol. Biol. 2006;357:1274–1282. doi: 10.1016/j.jmb.2006.01.061. [DOI] [PubMed] [Google Scholar]
- 51.Hasegawa C., Kikukawa T., Kamo N. Interaction of the halobacterial transducer to a halorhodopsin mutant engineered so as to bind the transducer: Cl− circulation within the extracellular channel. Photochem. Photobiol. 2007;83:293–302. doi: 10.1562/2006-06-09-RA-916. [DOI] [PubMed] [Google Scholar]
- 52.Choe H.W., Kim Y.J., Ernst O.P. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 53.Wietek J., Wiegert J.S., Hegemann P. Conversion of channelrhodopsin into a light-gated chloride channel. Science. 2014;344:409–412. doi: 10.1126/science.1249375. [DOI] [PubMed] [Google Scholar]
- 54.Matsui Y., Sakai K., Kouyama T. Specific damage induced by X-ray radiation and structural changes in the primary photoreaction of bacteriorhodopsin. J. Mol. Biol. 2002;324:469–481. doi: 10.1016/s0022-2836(02)01110-5. [DOI] [PubMed] [Google Scholar]
- 55.Borshchevskiy V., Round E., Gordeliy V. Low-dose X-ray radiation induces structural alterations in proteins. Acta Crystallogr. D Biol. Crystallogr. 2014;70:2675–2685. doi: 10.1107/S1399004714017295. [DOI] [PubMed] [Google Scholar]
- 56.Wickstrand C., Dods R., Neutze R. Bacteriorhodopsin: would the real structural intermediates please stand up? Biochim. Biophys. Acta. 2015;1850:536–553. doi: 10.1016/j.bbagen.2014.05.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.