

Abstract
Exuberant fibroproliferation is a common complication after injury for reasons that are not well understood1. One key component of wound repair that is often overlooked is mechanical force, which regulates cell-matrix interactions through intracellular focal adhesion components, including focal adhesion kinase (FAK)1,2. Here we report that FAK is activated after cutaneous injury and that this process is potentiated by mechanical loading. Fibroblast-specific FAK knockout mice have substantially less inflammation and fibrosis than control mice in a model of hypertrophic scar formation. We show that FAK acts through extracellular-related kinase (ERK) to mechanically trigger the secretion of monocyte chemoattractant protein-1 (MCP-1, also known as CCL2), a potent chemokine that is linked to human fibrotic disorders3–5. Similarly, MCP-1 knockout mice form minimal scars, indicating that inflammatory chemokine pathways are a major mechanism by which FAK mechanotransduction induces fibrosis. Small-molecule inhibition of FAK blocks these effects in human cells and reduces scar formation in vivo through attenuated MCP-1 signaling and inflammatory cell recruitment. These findings collectively indicate that physical force regulates fibrosis through inflammatory FAK–ERK–MCP-1 pathways and that molecular strategies targeting FAK can effectively uncouple mechanical force from pathologic scar formation.
Traditional cytokine-based paradigms for fibrosis largely overlook the role of cell-matrix interactions and physical cues in disease pathogenesis4–7. However, clinicians and anatomists have suspected for over a century that mechanical forces influence the fibrotic response to cutaneous injury8, with numerous studies having suggested the key role of reducing wound tension to decrease scar formation9–11. Most recently, our group showed that mechanical offloading of incisions can decrease hypertrophic scar (HTS) formation in humans, further underscoring the crucial role of the mechanical environment in cutaneous pathology12. Despite these longstanding clinical observations, the molecular pathways linking physical force with fibrosis remain unknown.
We previously showed that mechanical force induced HTS-like fibrosis in a mouse model of cutaneous scarring13. To elucidate candidate pathways driving early scar formation, we performed a genome-wide microarray analysis on wild-type mouse scars that had been subjected to human-like amounts of skin tension (0.15–0.27 N (mm2)−1) between days 4 and 14 after injury13. Construction of transcriptome networks around mechanically regulated genes implicated a central role for FAK as a transducer of both inflammatory and physical signals (Supplementary Fig. 1a–c and Supplementary Table 1). To test this concept (Supplementary Fig. 1d), we examined whether FAK is activated after either incisional injury or loading of unwounded skin. Notably, we found that cutaneous injury activated FAK and that mechanical force substantially potentiated this effect (Supplementary Fig. 1e), leading us to hypothesize that FAK is a key mediator of both inflammation and fibrosis.
Although current mechanotransduction research implicates a role for both integrin-matrix interactions and FAK in the cellular response to force1,2,6,7, the importance of these mechanisms on the scale of complex tissues and organs is unclear. Previous studies using keratinocyte-specific FAK knockout mice reported no wound healing phenotype14,15, suggesting that dermal FAK signaling may be more vital for cutaneous repair than epidermal FAK. Accordingly, we focused on the role of FAK specifically in dermal fibroblasts, the end effector for fibrosis and scar formation. To generate adult dermal fibroblast-restricted FAK knockout mice, floxed FAK mice (Ptk2loxP/loxP)16 were crossed with tamoxifen-inducible Col1α2-Cre mice (Col1a2-Cre+/−)17. FAK knockout progeny (Ptk2loxP/loxP; Col1a2-Cre+/−) were viable, fertile, produced at expected Mendelian ratios and showed no overt pathologic phenotype (Supplementary Fig. 2).
We then applied the HTS model to FAK knockout mice and, although their normal incisional healing was not impaired (Supplementary Fig. 3a–c), scar formation and matrix density were less than that observed in wild-type mice (Fig. 1a–c). FAK knockout scars had less dermal area (69%; P = 0.002) and cellularity (48%; P < 0.001) compared to scars from wild-type mice at day 10 after injury. These effects were associated with impaired proliferation (Ki67 index: 5.5 ± 1.4 for FAK knockout compared to 13.0 ± 1.7 for wild-type mice (means ± s.e.m.), P < 0.001), but apoptosis was similar in FAK knockout and wild-type mice (percentage of apoptotic cells: 6.2 ± 1.1 for FAK knockout compared to 3.6 ± 0.6 for wild-type mice (means ± s.e.m.), P = 0.07), which is consistent with our recent report suggesting that activation of survival pathways is not a primary factor in scar formation18. In vitro motility and contraction assays showed aberrant matrix mechanosensing in FAK knockout fibroblasts (Supplementary Fig. 3d–f), suggesting that FAK is necessary to transmit mechanotransduction cues that induce fibrosis.
Figure 1.
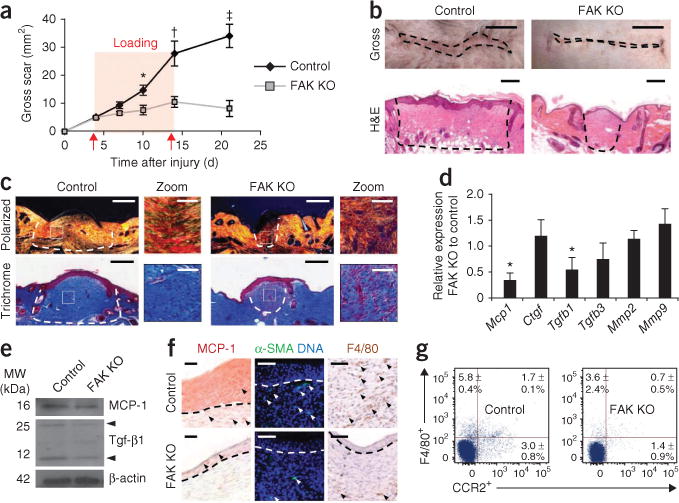
HTS model. (a) Surface scar formation. n = 6. (b) Images of scars at 10 d after injury. Scale bars, top, 0.5 cm; bottom, 200 μm. (c) Polarized light and trichrome-stained images. n = 6. Scale bars, 200 μm; zoom scale bars, 50 μm. (d) Quantitative RT-PCR (qRT-PCR) analysis of wound cytokines. n = 9. (e) Scar cytokine densitometry. Arrowheads point to the monomer and dimer forms of Tgf-β1. n = 3. (f) Immunolocalization of MCP-1, α-SMA+ cells and F4/80+ macrophages. Scale bars, left column, 20 μm; middle and right columns, 50 μm. n = 6. (g) F4/80+ and CCR2+ flow cytometry. Quadrant values represent the percentage of total scar cells. n = 4. Values are means ± s.e.m. *P < 0.05, †P < 0.01, ‡P < 0.001. The dashed lines outline the scar.
Because inflammatory mechanisms are strongly implicated in fibrosis1,4,19, we examined whether FAK modulates cytokine/chemokine signaling, as suggested by our microarray analysis (Supplementary Table 2). Consistent with traditional paradigms for fibrosis1,4,5, transcriptional and protein concentrations of transforming growth factor-β1 (Tgf-β1) were significantly lower in FAK knockout scars compared to wild-type scars (densitometry: 0.7 ± 0.1 for FAK knockout compared to 1.4 ± 0.2 for wild type (means ± s.e.m.), P < 0.05) (Fig. 1d,e). Notably, concentrations of MCP-1, a chemokine highly associated with inflammatory cell recruitment and fibrotic skin diseases3–5,20–22, were also less in FAK knockout wounds than in wild-type wounds (densitometry: 0.4 ± 0.05 for FAK knockout compared to 0.6 ± 0.07 for wild type (means ± s.e.m.), P < 0.05) (Fig. 1d,e), implicating a key role for MCP-1 in scar mechanotransduction. FAK knockout scars had lower numbers of myofibroblasts positive for α smooth muscle actin (α-SMA+) than wild-type scars (cells per high power field (hpf): 1.6 ± 0.4 for FAK knockout compared to 5.8 ± 0.5 for wild type (means ± s.e.m.), P = 0.003) and lower numbers of F4/80+ macrophages compared to wild-type scars (cells per hpf: 3.0 ± 0.9 for FAK knockout compared to 7.6 ± 0.8 for wild-type (means ± s.e.m.), P = 0.03) (Fig. 1f), cell populations that have been extensively linked to MCP-1 (refs. 4,5). In addition, overall wound expression of Ccr2 (the surface receptor for MCP-1) was less in FAK-deficient wounds compared to wild-type scars (Fig. 1g), indicating a defect in MCP-1–mediated cell trafficking.
To clarify the role of fibroblast-specific secretion of MCP-1, FAK knockout fibroblasts were subjected to mechanical strain and showed less expression of Mcp-1 compared to wild-type fibroblasts (Fig. 2a). In situ hybridization studies confirmed lower amounts of fibroblast Mcp-1 transcripts in vivo in FAK knockout scars compared to wildtype scars (Fig. 2b). Finally, intradermal administration of recombinant mouse MCP-1 restored the fibrotic phenotype of wild-type mice in FAK-deficient wounds (Fig. 2c–f), confirming the key role of fibroblast secretion of MCP-1 during scar formation. We then applied our HTS model to global MCP-1 knockout mice, which showed 70% less scar formation relative to wild-type control scars (at day 10 after injury, P = 0.001) (Supplementary Fig. 4a,b). These MCP-1 knockout scars also had significantly less recruitment of macrophages, which are key regulators of matrix remodeling4,5,23, than wild-type scars (F4/80+ cells per hpf: 0.3 ± 0.2 for FAK knockout compared to 5.3 ± 0.6 for wild type (means ± s.e.m.), P < 0.001) (Supplementary Fig. 4c). Together, these findings indicate that MCP-1–dependent inflammatory pathways have a major role in scar mechanotransduction.
Figure 2.
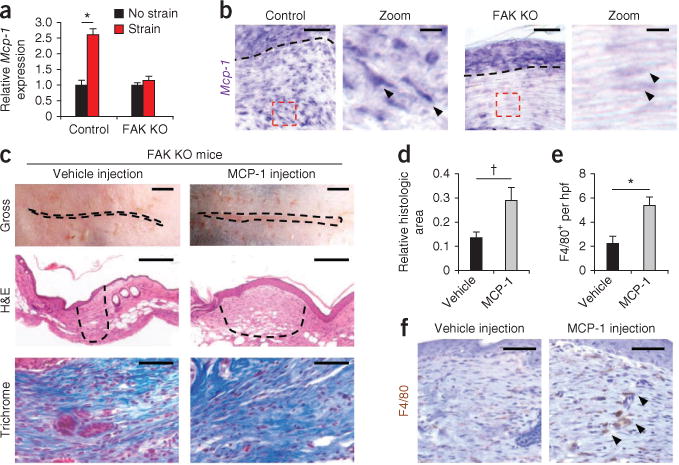
Fibroblast-specific MCP-1 pathways. (a) Mcp-1 transcription in strained fibroblasts. n = 6. (b) Mcp-1 in situ hybridization at day 10 after injury (Mcp-1 transcripts are stained purple). Arrowheads point to spindle-shaped fibroblasts in the hpf (dashed red boxes). The dashed black lines indicate the basement membrane. Scale bars for the zoom images, 10 μm. (c) Analysis of fibrosis after intradermal injection of recombinant mouse MCP-1. The dashed lines outline the scar. Scale bars, top row, 0.25 cm; middle row, 200 μm. (d) Scar area at day 10 after injury. n = 6. (e,f) Quantification (e) and images (f) of F4/80+ macrophages in FAK knockout scars treated with vehicle or MCP-1. The arrowheads point to the macrophages. Values are means ± s.e.m. Scale bars, 50 μm unless otherwise noted. *P < 0.001, †P < 0.05.
To fully elucidate the intracellular signaling events connecting FAK with MCP-1 secretion, we analyzed known downstream mediators of FAK, including thymoma viral proto-oncogene 1 (Akt) and the mitogen-activated protein kinases (MAPKs) ERK, p38 (also known as MAPK14) and JNK (also known as MAPK8)1,13,24 (Supplementary Fig. 5a). Only ERK was both activated by mechanical stimuli and differentially regulated by FAK (Supplementary Fig. 5b), corroborating previous in vitro findings that implicated ERK as a key target of FAK mechanotransduction24. Together, these data underscore the crucial role of FAK-ERK pathways in wound mechanosensing and indicate that MCP-1 may be a key effector of FAK-ERK–mediated fibrosis.
To substantiate the importance of these findings in human tissues, we examined FAK pathways in vitro in human fibroblasts. After the application of strain, FAK was activated within 5 mins and was sustained during the static strain period (Fig. 3a,b). ERK was phosphorylated 10 mins after the application of strain, and its activation was crucially dependent on FAK, thus recapitulating the FAK-ERK mechanotransduction axis that we observed in vivo. In conjunction with established surrogate assays for wound repair, small-molecule inhibition of FAK (using PF573228) was used to investigate mechanically regulated fibroblast function in vitro13,25. We found that treatment of human fibroblasts with PF573228 effectively inhibited the formation of focal adhesions and impaired their spread morphology (Supplementary Fig. 6a–d). Additionally, cell motility (Fig. 3c), contraction and α-SMA expression (Fig. 3d,e) were all less in FAK-inhibitor treated fibroblasts compared to control fibroblasts. Together, these findings suggest that small-molecule therapies targeting FAK can potentially affect human fibrotic disease.
Figure 3.
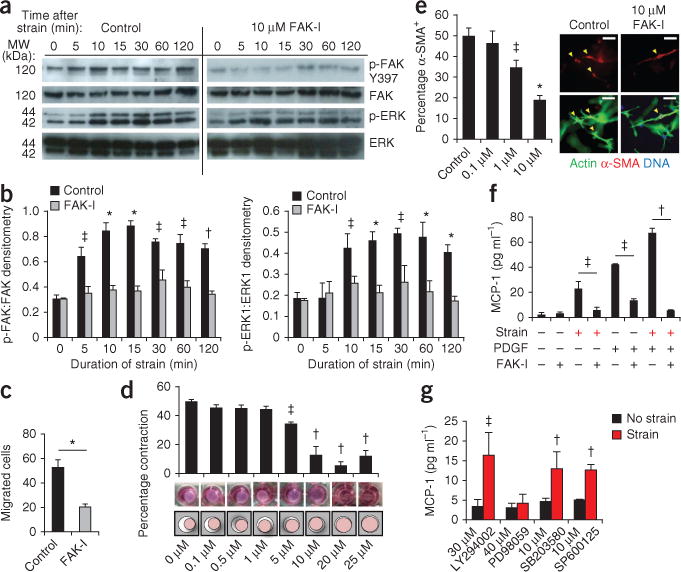
FAK-mediated mechanoresponsive pathways in human fibroblasts. (a,b) Representative immunoblots and quantification of static strain-induced FAK and ERK activation in untreated human fibroblasts (Control) and those treated with the FAK inhibitor (FAK-I) PF573228. n = 3. (c) Fibroblast motility in a scratch migration assay. n = 6. (d,e) Fibroblast contraction (d) and α-SMA+ expression (arrowheads) (e) in three dimensional collagen lattices. Concentrations of PF573228 are indicated along the x-axes of the bar graphs. n = 3. Scale bars, 50 μm. (f) Synergistic (strain plus 10 ng ml−1 platelet-derived growth factor) induction of MCP-1 secretion. n = 4. (g) Strain-induced MCP-1 secretion with small-molecule inhibition of FAK (PF573228), Akt (LY294002), ERK (PD98059), p38 (SB203580) or JNK (SP600125). n = 4. Values are means ± s.e.m. *P < 0.001, †P < 0.01, ‡P < 0.05.
We then examined how these early intracellular events were connected with profibrotic pathways in human tissues. In vitro, both mechanical and inflammatory stimuli induced robust MCP-1 secretion (Fig. 3f). When we applied both stimuli simultaneously, there was a potentiating effect on MCP-1 secretion. Small-molecule blockade of FAK was sufficient to prevent either stimulus from activating MCP-1 (Fig. 3f), suggesting that FAK is a crucial node in chemokine signaling. In addition, inhibition of ERK (but not Akt, p38 or JNK) blocked strain-induced MCP-1 release (Fig. 3g), highlighting the key role of the FAK-ERK-MCP1 axis in mechanotransduction and inflammation in human fibroblasts.
To evaluate the therapeutic potential of small-molecule anti-FAK therapies, we administered PF573228 daily to cutaneous wounds in the mouse HTS model. Consistent with the in vitro and FAK knockout data, pharmacologic blockade of FAK resulted in substantially less scar formation in vivo compared to control animals (Fig. 4a). In scars 10 days after injury, gross scar area was 170% less with FAK inhibition compared to controls (P = 0.003) (Fig. 4b) and scar matrix density was also less in scars with FAK inhibition compared to controls (Supplementary Fig. 6e,f). Additionally, epithelial thickness, epithelial proliferation and dermal proliferation were 35% (P = 0.004), 57% (P < 0.001) and 28% (P = 0.01) lower, respectively, in FAK-inhibitor–treated scars compared to controls (Supplementary Fig. 6g). PF573228 treatment was also associated with less mechanical activation of ERK and MCP-1 compared to controls (Fig. 4c–e and Supplementary Fig. 7a–c), replicating the phenotype observed in the transgenic mice.
Figure 4.
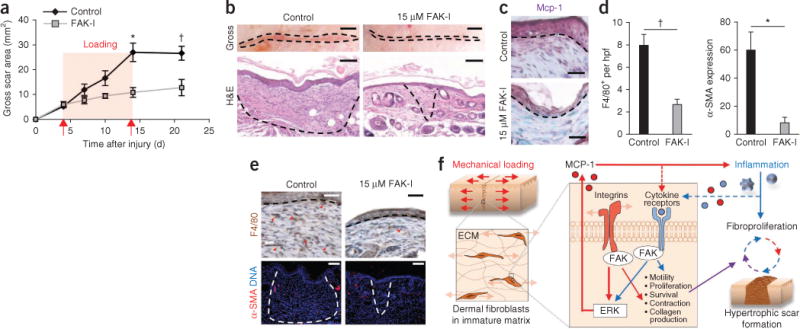
Intradermal treatment with PF573228. (a) Surface scar formation with treatment with 15 μM PF573228. n = 6. (b) Images of scars at day 10 after injury. The dashed lines indicate scar area. Scale bars, top row, 0.25; micrographs, 100 μm. n = 6. (c) Mcp-1 immunolocalization (purple color). The dashed lines indicate the basement membrane. Scale bars, 50 μm. (d,e) Quantification (d) and micrographs (e) of F4/80+ macrophages and α-SMA expression. n = 6. Scale bars, top row, 50 μm; bottom row, 100 μm. Values are means ± s.e.m. *P < 0.01, †P < 0.05. (f) Schematic of the proposed vicious cycle of hypertrophic scarring driven by mechanical activation of local and systemic fibroproliferative pathways through fibroblast FAK.
It is possible that in addition to these chemokine-mediated mechanisms, FAK may also control fibrosis by directly activating fibroblast collagen production26–28, which would explain the marginally higher amount of fibrosis observed in loaded compared to unloaded MCP-1 knockout wounds (Supplementary Fig. 4b). In vitro inhibition of either FAK or ERK significantly blocked collagen production in human fibroblasts (Supplementary Fig. 7d), and the ratio of collagen I (thicker) to collagen III (thinner)1 gene expression was 18% lower in these fibroblasts after inhibition compared to before inhibition (P = 0.076) (Supplementary Fig. 7e). Similarly, the ratio of collagen I to collagen III expression was 34% (P = 0.040) and 51% (P = 0.036) less in FAK knockout scars compared to wild-type scars and FAK-inhibitor–treated scars compared to control scars, respectively, corroborating the histologic analyses that showed a paucity of scar matrix after FAK blockade. This suggests that a minor secondary effect of FAK mechanotransduction is the direct modulation of collagen fibrillogenesis, as has been previously described in the kidney29.
Based on these studies, we propose a model for load-induced fibrosis whereby mechanical force activates both MCP-1 secretion and collagen production through FAK to perpetuate a ‘vicious cycle’ of fibroproliferation after injury (Fig. 4f). Although other mechanoresponsive cell types and cytokines are undoubtedly involved in scar mechanotransduction, these findings collectively provide a framework to understand how mechanical stimuli trigger local and systemic responses to induce scar hypertrophy. More broadly, these results suggest that targeted strategies to uncouple mechanical force from inflammation and fibrosis may prove clinically successful across diverse organ systems.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturemedicine/.
ONLINE METHODS
Mice
The Administrative Panel on Laboratory Animal Care at Stanford University approved all mouse procedures. We generated transgenic mice with fibroblast-restricted FAK deletion by crossing fibroblast-specific procollagen-α2(I)–Cre recombinase mice17 to homozygous floxed FAK mice (B6.129-Ptk2tm1Lfr/Mmcd, purchased from the UC Davis Mutant Mouse Regional Resource Center). We backcrossed mice at least eight generations on a C57BL/6 background and identified transgenic progeny using published methods14. Tamoxifen induction was performed through daily intraperitoneal injections for 5 d 1 week before wounding17. We used female mice aged 8–12 weeks for all experiments; FVB/NJ and MCP-1 knockout (B6.129S4-Ccl2tm1Rol) mice were purchased from The Jackson Laboratory.
Microarray
We harvested wild-type mouse tissue at day 6 or day 14 after injury and after 48 h or 10 d of mechanical loading, respectively (n = 4 mice per group per time point). RNA was hybridized to the GeneChip microarray (Affymetrix). We analyzed the data using GeneSpring GX 11.0 (Agilent Technologies) and the Significance Analysis of Microarray toolkit in Microsoft Excel. We performed hierarchical clustering using MATLAB (MathWorks). We constructed pathway networks using Ingenuity Pathways Analysis (Ingenuity Systems).
Fibroblast harvest
We isolated primary dermal mouse fibroblasts after overnight incubation in trypsin (Gibco/Invitrogen) followed by 1 mg ml−1 Liberase TL (Roche). We obtained human fibroblasts from fresh tissue specimens following protocols approved by the institutional review board at Oregon Health and Sciences University. We incubated specimens overnight in 5 mg ml−1 dispase (Roche) followed by 0.5 mg ml−1 collagenase type I (Roche) and 0.2 mg ml−1 trypsin. We used human cells from passages four to eight.
In vitro strain studies
We applied equibiaxial static strain to human fibroblasts as previously published24. After 24 h of strain, we collected media for MCP-1 (CCL2) ELISA (R&D Systems) or Sircol total collagen assay (Biocolor). Commercially available small molecules were dissolved in DMSO (Sigma-Aldrich) and used to target FAK (PF573228, Tocris Biosciences), Akt (LY294002), ERK (PD98059), p38 (SB203580) and JNK (SP600125). Platelet-derived growth factor (10 ng ml−1) was used for ligand activation of FAK. We purchased all molecules from Sigma-Aldrich unless otherwise noted.
Live/dead assay
We used a live/dead assay (Calbiochem/EMD Chemicals) after 24 h of exposure to PF573228 in vitro.
In vitro wound healing
We assessed fibroblast motility using a scratch migration assay13.
Fibroblast-populated collagen lattices
We performed fibroblast contraction studies as previously published25.
Mouse HTS model
We used a mouse HTS model as previously published by our group13.
Microscopy and immunohistochemistry
We incubated paraformaldehydefixed cells with primary antibody (pFAK-Y397 or FAK 1:200, BD Biosciences) followed by Alexa Fluor 488–conjugated secondary antibody (Molecular Probes/Invitrogen). We performed cytoskeletal staining with phalloidin-AF488 or Cy3-conjugated antibody against α-SMA (Sigma-Aldrich). We performed H&E, picrosirius red and trichrome staining (Sigma-Aldrich) on paraffin-embedded sections. We used primary antibodies against FAK, MCP-1, F4/80, Ki67 and α-SMA. Immunostaining was developed with VIP, NovaRed or diaminobenzidine (Vector Laboratories) or the secondary fluorescent antibodies AF488 or AF594. We performed apoptotic staining using the In situ Cell Death Detection Kit (Roche). We purchased all antibodies from Abcam unless otherwise noted. We processed images for publication using Adobe Photoshop CS3 (Adobe Systems), and two blinded investigators performed image quantification using ImageJ software (US National Institutes of Health).
Gene expression
We performed qRT-PCR as previously published30. We used TaqMan Gene Expression Assays (Applied Biosystems) for in vitro qRT-PCR. The primers used are listed in Supplementary Table 3.
In situ hybridization
We amplified the MCP-1 template from mouse complementary DNA by PCR using sequence-specific primers including the T7 or T3 RNA promoter region to make anti-sense and sense probes, respectively. We transcribed Riboprobe in the presence of Digoxigenin-11-dUTP (Roche), which we detected using the DIG Nucleic Acid Detection Kit (Roche).
Immunoblotting
We performed immunoblotting as previously described13. We used antibodies to the following: FAK (BD Biosciences), p-FAK Tyr397, p-FAK Ser732, p-ERK Thr202/Tyr204 and ERK, p-Akt Ser473 and Akt (Cell Signaling Technology), p-p38 Tyr182 and p38, p-JNK Thr183/Tyr185 and JNK, MCP-1 and β-actin. We obtained all antibodies from Abcam unless otherwise noted.
Intradermal injections
We administered intradermal injections (300 μl) of DMSO, 15 μM of PF573228 or 150 μM of PF573228 to unwounded mouse skin or unloaded incisions. For the HTS model experiments, we injected 15 μM of PF573228 or recombinant mouse Mcp-1 (1 μg ml−1; R&D Systems) daily between days 4 and 14 after injury.
Flow cytometry
We digested scars at 10 d after injury and incubated them with rat monoclonal antibodies against F4/80 (eBioscience) and CCR2 (R&D Systems) as previously described30.
Statistical analyses
We performed statistical analyses using a Student’s unpaired t test or one-way analysis of variance for multiple comparisons (MATLAB). P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank B. de Crombrugghe (The University of Texas MD Anderson Cancer Center, Houston, Texas) for providing the procollagen-α2(I)-Cre transgenic mice, J. Rajadas for experimental insight, F. You for harvesting human fibroblasts and Y. Park for histologic processing. This work was supported by the Oak Foundation, the Hagey Family Endowed Fund in Stem Cell Research and Regenerative Medicine and a United States Armed Forces Institute of Regenerative Medicine grant (DOD #W81XWH-08-2-0033).
Footnotes
AUTHOR CONTRIBUTIONS
V.W.W. and G.C.G. designed the research. V.W.W., M.J. and J.P. analyzed the microarray data. J.P. and I.N.V. generated the FAK knockout mice. V.W.W. and K.C.R. performed the in vitro human fibroblast experiments. V.W.W. and S.A. performed the small molecule injection experiments. V.W.W., K.C.R., M.S., J.P.G. and E.R.N. performed and analyzed the in vivo data. V.W.W. and K.L. performed and analyzed the biomechanics data. A.A.K. provided reagents. V.W.W., K.C.R., M.J. and G.C.G. analyzed data. K.C.R., J.P.G., M.J. and M.T.L. helped prepare the manuscript. V.W.W. and G.C.G. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Accession codes. Microarray data have been deposited in the Gene Expression Omnibus database with series accession number GSE26390.
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 2.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira AM, et al. Diminished induction of skin fibrosis in mice with MCP-1 deficiency. J Invest Dermatol. 2006;126:1900–1908. doi: 10.1038/sj.jid.5700302. [DOI] [PubMed] [Google Scholar]
- 4.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingber DE. Mechanobiology and diseases of mechanotransduction. Ann Med. 2003;35:564–577. doi: 10.1080/07853890310016333. [DOI] [PubMed] [Google Scholar]
- 7.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langer K. On the anatomy and physiology of the skin. I. The cleavability of the cutis (Translated from Langer, K (1861) Zur Anatomie und Physiologie der Haut I Uber die Spaltbarkeit der Cutis Sitzungsbericht der Mathematischnaturwissenschaftlichen Classe der Kaiserlichen Academie der Wissenschaften, 44, 19.) Br J Plast Surg. 1978;31:3–8. [PubMed] [Google Scholar]
- 9.Elliot D, Mahaffey PJ. The stretched scar: the benefit of prolonged dermal support. Br J Plast Surg. 1989;42:74–78. doi: 10.1016/s0007-1226(89)90117-3. [DOI] [PubMed] [Google Scholar]
- 10.Chantarasak ND, Milner RH. A comparison of scar quality in wounds closed under tension with PGA (Dexon) and Polydioxanone (PDS) Br J Plast Surg. 1989;42:687–691. doi: 10.1016/0007-1226(89)90082-9. [DOI] [PubMed] [Google Scholar]
- 11.Durkaya S, et al. Do absorbable sutures exacerbate presternal scarring? Tex Heart Inst J. 2005;32:544–548. [PMC free article] [PubMed] [Google Scholar]
- 12.Gurtner GC, et al. Improving cutaneous scar by controlling the mechanical environment: large animal and phase I studies. Ann Surg. 2011;254:217–225. doi: 10.1097/SLA.0b013e318220b159. [DOI] [PubMed] [Google Scholar]
- 13.Aarabi S, et al. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 2007;21:3250–3261. doi: 10.1096/fj.07-8218com. [DOI] [PubMed] [Google Scholar]
- 14.Essayem S, et al. Hair cycle and wound healing in mice with a keratinocyte-restricted deletion of FAK. Oncogene. 2006;25:1081–1089. doi: 10.1038/sj.onc.1209130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLean GW, et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beggs HE, et al. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–514. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng B, Zhang Z, Black CM, de Crombrugghe B, Denton CP. Ligand-dependent genetic recombination in fibroblasts: a potentially powerful technique for investigating gene function in fibrosis. Am J Pathol. 2002;160:1609–1617. doi: 10.1016/S0002-9440(10)61108-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paterno J, et al. Akt-mediated mechanotransduction in murine fibroblasts during hypertrophic scar formation. Wound Repair Regen. 2011;19:49–58. doi: 10.1111/j.1524-475X.2010.00643.x. [DOI] [PubMed] [Google Scholar]
- 19.Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009–1017. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- 20.Shibusawa Y, Negishi I, Tabata Y, Ishikawa O. Mouse model of dermal fibrosis induced by one-time injection of bleomycin-poly(L-lactic acid) microspheres. Rheumatology (Oxford) 2008;47:454–457. doi: 10.1093/rheumatology/ken058. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, et al. Toll-like receptors expressed by dermal fibroblasts contribute to hypertrophic scarring. J Cell Physiol. 2011;226:1265–1273. doi: 10.1002/jcp.22454. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto T. Pathogenic role of CCL2/MCP-1 in scleroderma. Front Biosci. 2008;13:2686–2695. doi: 10.2741/2875. [DOI] [PubMed] [Google Scholar]
- 23.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, et al. Increased transcriptional response to mechanical strain in keloid fibroblasts due to increased focal adhesion complex formation. J Cell Physiol. 2006;206:510–517. doi: 10.1002/jcp.20486. [DOI] [PubMed] [Google Scholar]
- 25.Ehrlich HP. The fibroblast-populated collagen lattice. A model of fibroblast collagen interactions in repair. Methods Mol Med. 2003;78:277–291. doi: 10.1385/1-59259-332-1:277. [DOI] [PubMed] [Google Scholar]
- 26.Wang JH, Thampatty BP, Lin JS, Im HJ. Mechanoregulation of gene expression in fibroblasts. Gene. 2007;391:1–15. doi: 10.1016/j.gene.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dun ZN, et al. Specific shRNA targeting of FAK-influenced collagen metabolism in rat hepatic stellate cells. World J Gastroenterol. 2010;16:4100–4106. doi: 10.3748/wjg.v16.i32.4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leucht P, Kim JB, Currey JA, Brunski J, Helms JA. FAK-mediated mechanotransduction in skeletal regeneration. PLoS ONE. 2007;2:e390. doi: 10.1371/journal.pone.0000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashida T, et al. MAP-kinase activity necessary for TGFβ1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J Cell Sci. 2007;120:4230–4240. doi: 10.1242/jcs.03492. [DOI] [PubMed] [Google Scholar]
- 30.Wong VW, et al. Engineered pullulan-collagen composite dermal hydrogels improve early cutaneous wound healing. Tissue Eng Part A. 2011;17:631–644. doi: 10.1089/ten.tea.2010.0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.