

Abstract
Mendelian Primary Immunodeficiency Diseases (MPIDs) are rare disorders affecting distinct constituents of the innate and adaptive immune system. Although they are genetically heterogeneous a substantial group of MPIDs is due to mutations in genes affecting the NF-κB transcription pathway, essential for cell proliferation, cell survival, and involved in innate immunity and in inflammation. Many of these genes encode for crucial regulatory components of NF-κB pathway and their mutations are associated with immunological and developmental signs somehow overlapping in patients with MPIDs. At present nine different MPIDs listed in the OMIM are caused by mutations in at least nine different genes strictly involved in the NF-κB pathway that result in defects in immune responses.
We will report here on the distinct function of each causative gene, on the impaired NF-κB step and more in general on the molecular mechanisms underlining the pathogenesis of the disease. Overall, the MPIDs affecting NF-κB signalosome require a careful integrated diagnosis and appropriate genetic tests to be molecularly identified. Their discovery at an ever-increasing rate will help to establish common therapeutic strategy for a subclass of immunodeficient patients.
Keywords: MPIDs, mutations, NF-κB, immunodeficiency
1. Introduction
The Nuclear Factor-κB (NF-κB) family of transcription factors regulates diverse biological processes, including many aspects of immunological functions.1 Both innate and adaptive immune responses as well as the development and maintenance of the cells and tissues that comprise the immune system are, at multiple steps, under the control of the NF-κB family of transcription factors.2 The NF-κB family includes the structurally homologous transcription factors NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB and c-Rel (Figure 1).3
Figure 1.
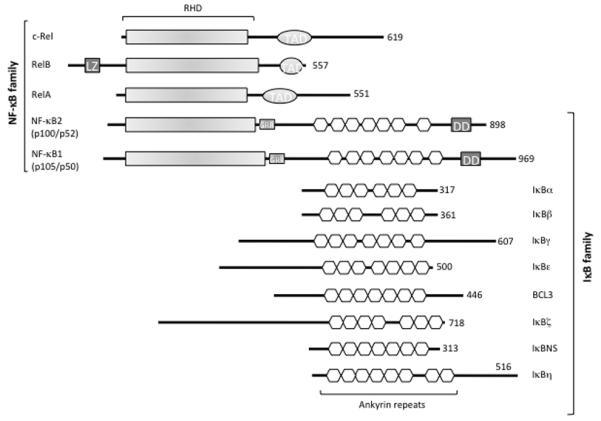
The NF-κB/IκB family. The protein domains of the NF-κB and IκB families are shown. TAD, Transcription Activation Domain; LZ, Leucine Zipper domain of RelB; GRR, Glycine-Rich Region; DD, Death Domain. The number of amino acids in each protein is shown on the right.
These transcription factors share a Rel Homology Domain (RHD) necessary for DNA binding, dimerization, and interaction with the inhibitor. They can form homo- and heterodimers and can bind to a variety of related target DNA sequences called κB sites to modulate gene expression. The p65, RelB, and c-Rel proteins contain C-terminal Transcription Activation Domains (TADs) that enable co-activator recruitment and target gene expression (Figure 1). As p50 and p52 lack TADs, they can activate transcription by forming heterodimers with p65, RelB, or c-Rel, or by recruiting other TAD-containing proteins. However, as homodimers lacking the ability to drive transcription, they can repress transcription trough the binding to DNA.1
In the resting cells, NF-κB dimers are retained in the cytoplasm by the Inhibitor of NF-κB proteins (IκBs), which consist of IκBα, IκBβ, IκBγ, IκBε, Bcl3, IκBζ, p100, p105, IκBns, and, recently, IκBη (Figure 1). All known IκB proteins contain multiple ankyrin repeats which mediate the association between IκB and NF-κB dimers. The typical IκBα, -β and -ε molecules contain six ankyrin repeats, while the other IκBs contain seven or eight repeats (Figure 1). The function of the IκB proteins is to prevent the NF-κB DNA binding: the ankyrin repeats interact with the RHD of the NF-κB proteins thus masking their Nuclear Localization Sequence (NLS) and preventing nuclear translocation.5 The release of NF-κB dimers from the IκB proteins depends on the activation of the IκB Kinase (IKK) complex, which consists of two catalytically active kinases, IκB Kinase-β (IKKβ also called IKK2) and IκB Kinase-α (IKKα also called IKK1), and of the regulatory subunit NF-κB Essential MOdulator (NEMO, also called IKKγ; Figure 2).
Figure 2.
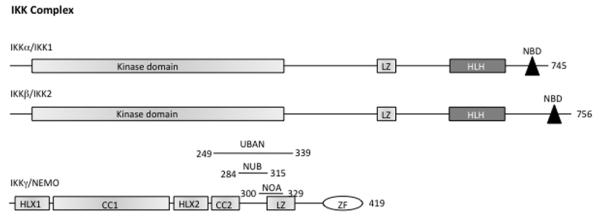
The IKK complex. The protein domains of the IKK complex are shown. CC, Coiled Coil; LZ, Leucine Zipper; HLH, Helix-Loop-Helix; NBD, NEMO Binding Domain; ZF, Zinc Finger. On NEMO protein is indicated: UBAN (Ubiquitin Binding in ABIN/NEMO) domain from aa. 249 to 339; NUB (NEMO Ubiquitin Binding) domain from aa. 284 to 315; NOA (NEMO-Optineurin-ABIN) domain from aa. 300 to 329. The number of amino acids in each protein is shown on the right.
A wide range of stimuli, including lipopolysaccharides (LPS), Interleukin-1 (IL-1), and Tumor Necrosis Factor α (TNF-α), cause the activation of the IKK complex, which leads to the phosphorylation of the IκB proteins (e.g., IκBα at Ser32 and Ser36 and IκBβ at Ser19 and Ser23). The phosphorylated IκB proteins are subsequently ubiquitinated and degraded, allowing the nuclear translocation of NF-κB and the activation of target genes transcription (Figure 3).
Figure 3.
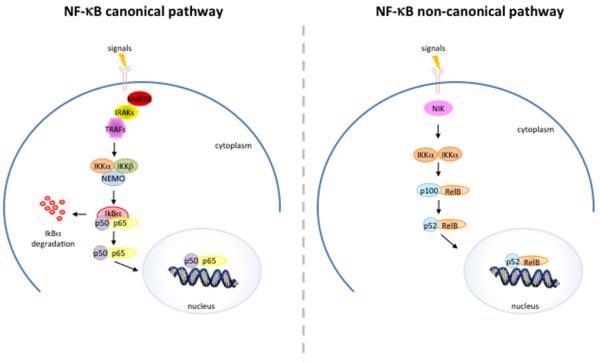
Schematic representation of canonical and non-canonical NF-κB activating pathway.
Although the activity of NF-κB is regulated by nuclear translocation, its transcriptional activity is further regulated by post-translational modifications.3 These regulatory modifications, including phosphorylation, ubiquitination, nitrosylation, and acetylation have distinct functional consequences and play a key role in determining the duration and strength of NF-κB nuclear activity as well as its transcriptional output.6 For example, the acetylation of p65 at K218 and K221 inhibits IκBα binding and enhances DNA-binding, whereas the acetylation of p65 at K122 and K123 inhibits its transcriptional activating activity.7
An alternative pathway leading to NF-κB activation called the non-canonical pathway also exists, which depends on the IKKα-mediated phosphorylation of p100 associated with RelB.3
The activation of p100/RelB complexes occurs during the development of lymphoid organs responsible for the generation of B and T lymphocytes. Only a small number of stimuli are known to activate NF-κB via this pathway and these factors include lymphotoxin B (LTβ) and B cell activating factor (BAFF). This pathway utilizes a complex consisting of two IKKα subunits, but not NEMO. Ligand induced activation results in the activation of NF-κB inducing kinase (NIK), which phosphorylates and activates the IKKα complex, which in turn phosphorylates p100 leading to the processing and liberation of the p52/RelB active heterodimer.4
Impaired NF-κB activation due to the identified genetic alterations in molecules involved in NF-κB pathway is responsible for some types of MPIDs.8
Here, we report an overview on MPIDs due to mutations in components proximally linked to the NF-κB activation pathway that result in defects in immune responses,7 providing information about the impact of each mutation on the impairment of NF-κB.
2. Autosomal Dominant Ectodermal Dysplasia with ImmunoDeficiency, AD-EDA-ID
Autosomal Dominant Ectodermal Dysplasia with ImmunoDeficiency, AD-EDA-ID (OMIM 612132) is a rare primary immunodeficiency associated with ectodermal dysplasia,8 due to heterozygous mutations of the NFKBIA gene, localized on chromosome 14 and encoding the inhibitory protein of the NF-κB pathway, IκBα (Figure 3; Table 1).9-13
Table 1.
List of primary immunodeficiency diseases associated with impaired NF-κB signaling including genetic defect and clinical aspects.
| DISEASE | AFFECTED CELL | ASSOCIATED FEATURES | GENETIC DEFECT | OMIM NUMBER |
|---|---|---|---|---|
| AD-EDA-ID, autosomal dominant. | Lymphocytes, monocytes. | Anhidrotic ectodermal dysplasia, T cell defect, various infections. | Mutation of NFKBIA gene encoding IκBα, an inhibitor of NF-κB. | 612132 |
| XL-EDA-ID, X-linked. | Lymphocytes, monocytes. | Anhidrotic ectodermal dysplasia, specific antibody deficiency (lack of Ab response to polysaccharides), mycobacteria and pyogenes infections. | Mutation of NEMO gene encoding IKKγ, the regulatory subunit of IKK complex. | 300291 |
| IKK2 deficiency, autosomal recessive. | Lymphocytes, monocytes. | Recurrent bacterial, viral, and fungal infections. | Mutation of IKBKB gene encoding IKK2, a catalytic subunit of IKK complex. | 615592 |
| IRAK4 deficiency, autosomal recessive. | Lymphocytes, granulocytes, monocytes. | Bacterial infections (pyogenes). | Mutation of IRAK4 gene encoding a component of TLR-and IL-1R-signaling pathway. | 607676 |
| MyD88 deficiency, autosomal recessive. | Lymphocytes, granulocytes, monocytes. | Bacterial infections (pyogenes). | Mutation of MYD88 encoding a component of the TLR and IL-1R. | 612260 |
| TRIF deficiency, autosomal recessive and autosomal dominant. | CNS resident cells and fibroblasts. | Herpes simplex virus 1 encephalitis. | Mutations of TRIF gene encoding an adaptor for TLR3 (Toll-like receptor 3)-and TLR4-mediated signalling. | 614850 |
| TRAF3 deficiency, autosomal dominant. | CNS resident cells and fibroblasts. | Herpes simplex virus 1 encephalitis. | Mutation of TRAF3 encoding a member of the TNF receptor associated factor (TRAF) protein family. | 614849 |
| HOIL1 deficiency, autosomal recessive. | Lymphocytes, granulocytes, monocytes, fibroblasts. | Bacterial infections (pyogenes). | Mutation of HOIL1 gene encoding a component of LUBAC. | 610924 |
| NFKB2 deficiency, autosomal dominant | Lymphocytes. | Recurrent infections, hypogammaglobulinemia and decreased numbers of B cells, switched memory B cells, and NK cells. | Mutations in NFKB2 gene, an essential component of the non-canonical NF-κB pathway. | 615577 |
Immunological aspects
The immunological phenotype of patients with IκBα deficiency is responsible for their broad susceptibility to infections with invasive pyogenic bacteria (meningitis, sepsis, arthritis, osteomyelitis, and abscesses), environmental mycobacteria, and, to a lesser extent, parasites, viruses, and fungi.9-16 Moreover, the patients suffer from a profound combined immunodeficiency with hypogammaglobulinemia with no specific antibodies; some also have low proportions of memory CD4 and CD8 T cells and no T-Cell Receptor (TCR) γ/δ T cells and display a severe impairment of T-cell proliferation in response to anti-CD3.14-17
Genetic aspects
Six heterozygous mutations in the NFKBIA gene have been identified in AD-EDA-ID patients. The pathogenic mutations have a dominant effect and they are called “hypermorphic” mutations, because enhance the inhibitory capacity of IκBα impairing the phosphorylation and degradation of IκBα and resulting in the partial retention of the NF-κB dimers in the cytoplasm.
Molecular aspects
The IκBα protein, a member of the serine/threonine protein kinase family, contains phosphorylation sites at its N-terminal, ankyrin repeat domains (Figure 1) in its central portion, and, at its C-terminal, a repeated peptidic sequence rich in proline, glutamic acids, serine, and threonine (rPEST) domains.9 IκBα inhibits the activation of NF-κB while its phosphorylation at the level of Ser32 and Ser36 triggers IκBα ubiquitination, leading to proteasomal degradation (Figure 3). This event causes the nuclear translocation of NF-κB and subsequent activation of its target genes.
The first case of AD-EDA-ID, in which a p.Ser32Ile mutation was identified was reported by Courtois et al.8 This mutation abrogates the phosphorylation of IκBα Ser32, required for the ubiquitination and proteasomal degradation of IκBα. Other IκBα mutations, p.Gln9X10, p.Glu14X11, and p.Trp11X12, cause a premature termination of protein translation and a re-start from Met37 of IκBα, resulting in a IκBα protein that is N-terminally truncated and lacks both of the critical serine residues, Ser32 and Ser36. The p.Ser36Tyr mutation results in a defective IκBα degradation and impaired NF-κB activation.18 As well as p.Met37Lys, it is capable of blocking NF-κB activation due to the gain-of-function of the IκBα protein.19
3. X-Linked Anhidrotic Ectodermal Dysplasia with ImmunoDeficiency, XL-EDA-ID
X-Linked Anhidrotic Ectodermal Dysplasia with ImmunoDeficiency (XL-EDA-ID, OMIM 300291) is a rare primary immunodeficiency associated with a developmental disorder due to mutations in the X linked gene named NF-κB Essential MOdulator (NEMO called also IKBKG) that encodes for the regulatory subunit of the IKK complex (Figure 2), essential for the canonical activation of NF-κB (Figure 3, Table 1).3,8
Immunological aspects
The broad immunological phenotypes of XL-EDA-ID patients are responsible for their susceptibility to infections with invasive pyogenic bacteria (S. pneumoniae, H. influenzae, and S. aureus), and mycobacterial (M. avium and M. kansasii), fungal, and/or viral diseases.16 The clinical and immunological phenotypes attributed to the NEMO mutations are characterized by: a dysregulated immunoglobulin synthesis or hyper-immunoglobulin M (hyper-IgM) syndrome; a defective antipolysaccharide antibody synthesis (antipneumococcal antibody and isohemagglutinin); reduced LPS and IL-1 family protein responses, and defective Natural Killer (NK) cell activity.16,20-22 Recently, a genotype/phenotype correlation has been identified.23
Genetic aspects
All patients with XL-EDA-ID are males. The first NEMO mutations impairing NF-κB activation in XL-EDA-ID patients were described in 200024 and 200125. Up to 100 male patients with about 43 different mutations of NEMO have been reported.26 The NEMO mutations in XL-EDA-ID patients are defined “hypomorphic” because they lead to an impairment of NF-κB signaling, but not to its abolition.27 Indeed, the NEMO loss-of-function mutations are lethal for males in utero.28,29
Molecular aspects
The NEMO protein consists essentially of a series of domains: Coiled-Coil (CC) 1 in the N-terminal segment, Helix-Loop-Helix 2 (HLX2) in the middle segment, and the CC2-Leucine Zipper (LZ) regulatory domain in the C-terminal segment. NEMO also has a Zinc Finger (ZF) domain at its C-terminal end (Figure 2).30 The function of NEMO depends on its dimerization and its ability to interact with linear or K63-linked polyubiquitin chains.31-34 This function requires the CC2-LZ domain, which is involved in NEMO dimerization and contains an ubiquitin-binding site called NOA/UBAN/NUB (NEMO-optineurin-ABIN/ubiquitin binding in ABIN and NEMO/NEMO ubiquitin binding), and the ZF domain, which bears a second ubiquitin-binding site.35,36
The degree of impairment of the NF-κB pathway depends on the NEMO mutated domain.26,27 The mechanisms by which some mutations associated with XL-EDA-ID affect NEMO’s structural and functional integrity have been investigated. The p.Ala288Gly mutation, which affects the CC2 domain, has no effect on the protein level but destabilizes the NEMO oligomers, altering the assembly of the IκB kinase complex and consequently impairing the canonical activation of NF-κB.37 The p.Asp311Asn and p.Asp311Gly mutations on the NOA ubiquitin-binding site of NEMO, impair NEMO-ubiquitin binding, with no detectable effect on NEMO expression and folding.38 The p.Glu315Ala and p.Arg319Gln, that affect the LZ domain, disrupt the formation of the salt bridge normally formed between residues Glu315 and Arg319 without affecting NEMO protein production.39,40 Moreover, the folding defect of the p.Glu315Ala mutant is responsible for the defect in binding to the ubiquitin chains.41 The p.Cys417Phe substitution modifies the structure of the C-terminal end of the ZF α-helix and decreases its stability, which leads to a defect in NF-κB activation. On the other hand, p.Cys417Arg does not affect the expression of the NEMO protein but impairs c-Rel activation in response to CD40 ligation.42 Moreover, mutations in the ZF domain are very common and are associated with some of the more severe phenotypes (e.g. ectodermal dysplasia with immune deficiency and osteopetrosis).
4. Autosomal recessive IKK2 deficiency
Autosomal recessive IKK2 deficiency (OMIM 615592) is a primary immunodeficiency disorder due to mutations in the IKBKB gene, a central component of the IKK complex in the canonical NF-κB signaling pathway (Figure 3, Table 1).8
Immunological aspects
The patients present within the first months of life with numerous bacterial, fungal, and viral infections, including candidiasis, pneumonia, bacteremia, sepsis, meningitis, and osteomyelitis. Multiple and variable organisms have been isolated from these patients, including Escherichia coli, Mycobacterium avium, Listeria monocytogenes, pneumococcus, Serratia marcescens, and Klebsiella. Other symptoms include chronic diarrhea and failure to thrive.43 These patients have normal B-cell and T-cell counts but very low levels of immunoglobulins, as well as a severe defect in immune-cell activation that affects both innate and adaptive immune-receptor pathways.43
Genetic aspects
Recently, mutations in the IKBKB gene have been discovered to be the cause of immunodeficiences.43 In four patients with Severe Combined ImmunoDeficiency (SCID) a homozygous duplication, c.1292dupG, in the IKBKB gene resulting in a complete loss of protein function has been identified.43
Molecular aspects
IKK2-deficient patient fibroblasts show an impaired phosphorylation of IκBα in response to TNF–α stimulation. Degradation of IκBα upon IL–1β stimulation is marginally affected, whereas degradation in response to Toll-Like Receptor (TLR)-5 stimulation by flagellin is absent, indicating distinct requirements for IKK2. The IL-6 response to TNF–α is normal, but it is reduced in response to LPS, and acts through TLR4. The finding of an impaired response to TNF-α, as well as to TLR4 or TLR5 stimulation, indicates an additional innate immunological defect in these patients. Moreover, the NF-κB binding to DNA after TNF-α stimulation is considerably decreased in patient cells.43
5. Autosomal recessive IRAK-4 deficiency
IL-1R-associated kinase (IRAK)-4 deficiency is an autosomal recessive primary immunodeficiency (OMIM 607676) that impairs NF-κB activation in the TLR signaling pathway.8,44,45
Immunological aspects
Patients affected by IRAK-4 deficiency present recurrent infections by the S. pneumoniae, S. aureus, and P. aeruginosa bacteria, and are also susceptible to infections with fungi (C. albicans) and other opportunistic infections. Blood cells from these patients fail to produce IL-1β, IL-6, IL-8, IL-12, TNF-α, or interferon (IFN)-γ in response to IL-1β, IL-18, or known TLR agonists, while their response to TNF-α is unaffected.46,47
The impact of IRAK-4 deficiency may vary from cell to cell (only blood cells and fibroblasts have been tested in IRAK-4 deficient patients). IRAK-4 deficient patients show apparently normal T- and B-cell responses, but a few patients seem to have a poor antibody response to carbohydrates, suggesting that T-independent B-cell responses might be affected.15
Genetic aspects
Autosomal recessive IRAK-4 deficiency was first discovered in 200348 and since then, up to 49 patients have been identified.46 It is caused by homozygous or compound heterozygous mutations in the IRAK4 gene: two missense (p.Arg12Cys and p.Arg391His49); five frameshift (p.Pro42fsX450, p.Ala211fsX251, p.Asn175fsX3152, p.Thr208fsX1253, p.Leu274fsX1454); and three nonsense (p.Tyr48X51, p.Gln293X54, p.Glu402X55). All the mutations other than the missense mutations were predicted to be loss-of-expression and loss-of-function, as they create a premature termination codon or delete a large segment of the gene.15,49-55
Molecular aspects
IRAK-4 is a member of the IRAK family of protein kinases that play an essential role in NF-κB activation in the TLR and TCR signaling pathways.44,45 IRAK-4 interacts with both MyD88 and IRAK-1, and its catalytic activity is required for IRAK-1 activation. Once hyperphosphorylated by IRAK-4, IRAK-1 associates with TNF receptor-associated factor (TRAF) 6, triggering the activation of both the NF-κB and Mitogen-Activated Protein Kinase (MAPK) pathways. Like other IRAKs, IRAK-4 contains two structural domains: a Death Domain (DD) that mediates the molecular recognition of other DD-containing proteins, and a catalytic Kinase Domain (KD).48,56 Moreover, in cells derived from patients, both the NF-κB and p38 activating signaling pathways were defective, suggesting that the immudeficiency caused by IRAK-4 mutations additionally involves a perturbed MAPK signaling.57
6. Autosomal recessive MyD88 deficiency
Autosomal recessive MyD88 deficiency (OMIM 612260) Is a rare primary immunodeficiency due to the Myeloid Differentiation primary response 88 (MyD88) gene, involved in the NF-κB canonical pathway in TLRs and in IL1Rs.8
Immunological aspects
Patients suffer from recurrent pyogenic bacterial infections, including invasive pneumococcal disease. The immunological phenotype of patients reported with this MyD88 deficiency is similar to that of Myd88-deficient mice, but the infectious phenotype is different. Indeed, MyD88-deficient patients are susceptible to S. aureus, P. aeruginosa, and S. pneumoniae, but are normally resistant to most other infectious agents. In contrast, Myd88-deficient mice have been shown to be susceptible to most common bacteria, viruses, fungi, and parasites.58
Genetic aspects
Autosomal recessive MyD88 deficiency was first discovered in 200858 and up to 24 cases have been reported.58-60 New mutations in the MyD88 gene have been reported: a homozygous in-frame MyD88 deletion (p.E52del), compound heterozygous missense mutations (p.L93P; p.R196C), and a homozygous missense mutation (p.R196C) have been identified.58 The deletion and missense mutations affected conserved residues.
Molecular aspects
MyD88 is a key downstream adapter for most TLRs and IL-1Rs that are essential for protective immunity to a small number of pyogenic bacteria. Functional analysis using patient fibroblasts and the expression of wild-type or mutant alleles in cell lines has demonstrated that p.E52del, p.R196C and p.L93P mutations result in a loss-of-function and lead to a complete MyD88 deficiency.58
The MyD88 protein has been detected in SV40-transformed fibroblasts in different amounts: in trace amounts for patients with a p.E52del/p.E52del mutations, small amounts for patients with L93P/R196C, and normal amounts for patients with p.R196C/p.R196C, suggesting that the patients have a functional MyD88 deficiency, with low or normal MyD88 protein levels.58
7. New immunodeficiency impairing the canonical NF-κB signaling pathway
Recently, mutations in new genes have been associated with primary immunodeficiency with functional defects in the canonical and non-canonical NF-κB signaling pathway.8
Autosomal Recessive and Autosomal Dominant TRIF deficiency (OMIM 614850)
Three unrelated cases of autosomal recessive and autosomal dominant TRIF deficiency affected by herpes simplex encephalitis (HSE) were reported.61 The autosomal recessive form of the disease has been found to be due to a homozygous nonsense mutation (p.R141X) that results in a complete absence of the Toll/IL-1R (TIR) domain-containing adaptor inducing IFN-β (TRIF) protein. Both the TLR3- and the TRIF-dependent TLR4 signaling pathways are abolished. The autosomal dominant form of disease has been found to be due to two heterozygous missense mutations (p.P625L and p.S186L) resulting in a dysfunctional protein. In this form of the disease, the TLR3 signaling pathway is impaired, whereas the TRIF-dependent TLR4 pathway is unaffected.
Autosomal Dominant TRAF3 deficiency (OMIM 614849)
Another immunodeficiency associated with a clinical phenotype of HSE is the autosomal dominant TRAF3 deficiency.61 Tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3) functions downstream of multiple receptors that induce IFN-α, IFN-β and IFN-λ production, including TLR3. The missense TRAF3 mutation (p.R118W) that proves to be responsible for the autosomal dominant predisposition to HSE has been reported.62 Previous studies have identified the p.R118W mutation of TRAF3 as a somatic mutation involved in multiple myeloma.63,64
Autosomal Recessive HOIL1 Deficiency (OMIM 610924)
A new fatal inherited disorder characterized by chronic autoinflammation, invasive bacterial infections and muscular amylopectinosis has been identified: autosomal recessive HOIL1 deficiency.65 Patients from two kindreds carry biallelic loss-of-expression and loss-of-function mutations in Heme-Oxidized Irp2 Ubiquitin Ligase 1 (HOIL1 also called RBCK1), a component of the Linear Ubiquitination Chain Assembly Complex (LUBAC): p.Q185X and p.L41fsX7.
These mutations produce an impairment of the stability of the LUBAC complex resulting in an impaired NF-κB-driven gene transcription and cytokine production in response to TNF and IL-1β. In particular, NF-κB activation in response to IL-1β is compromised in the patients’ fibroblasts. In contrast, the patients’ mononuclear leukocytes, particularly the monocytes, are hyper-responsive to IL-1β. The consequences of HOIL-1 and LUBAC deficiencies for IL-1β responses thus differ between cell types, consistent with the unique association of autoinflammation and immunodeficiency in these patients.65
Autosomal dominant NFKB2 deficiency (OMIM 615577)
Recently, a heterozygous 1-bp deletion (c.2564delA) in the NFKB2 gene, resulting in a frameshift and premature termination (p.K855SfsX7) was identified.66 The mutation caused a truncation in the C terminus of the protein, removing the conserved phosphorylation sites required for activation of p100 to p52. Another heterozygous mutation, c.2557C-T transition, in the NFKB2 gene, resulting in an p.R853X nonsense mutation, was described.66 This patient was identified from a cohort of 33 individuals with CVID who were tested for variants in the NFKB2 gene. The mutation caused a truncation in the C terminus of the protein, removing the conserved phosphorylation sites required for activation of p100 to p52. Liu et al. (2014) identified a heterozygous 8-bp deletion (c.2593_2600del) resulting in a frameshift and premature termination (p.D865VfsX17). The protein expressed from the mutant allele was unable to be phosphorylated at regulatory residue 866, which abolished the proper processing and activation of the NF-κB signaling pathway with a consequent defect in B-cell differentiation and T follicular helper cells development.67
8. Conclusions
Defects in the NF-κB activation pathway have been linked to several human diseases including the primary immunodeficiencies. NF-κB dimers are involved in the development and function of the immune system, with their activation affecting various immunity- and inflammation-associated genes such as acute-phase reactants, cytokines, chemokines, growth factors and receptors, adhesion molecules, and regulators of apoptosis and cellular proliferation. The rapid advances in gene identification technology, such as whole genome sequencing and the examinations through an integrated diagnostics of affected individuals help to clarify the infectious phenotypes associated with these genetic defects initiating the forward genetic dissection of NF-κB-mediated immunity. In fact, the effect of several mutations in different components of the NF-κB signaling pathway demonstrates the crucial role of this pathway in human immunity to infection (Table 1). The clinical features of the MPIDs vary in severity based on the residual function of the mutated protein.
Further in vitro characterization of the NF-κB signaling pathways in MPID patients should improve the definition of candidate genes in other patients with unexplained infectious diseases.
Acknowledgments
We are grateful to the Incontinentia Pigmenti International Foundation (IPIF, [http://www.ipif.org/]), the association France Incontinentia Pigmenti (FIP, [http://incontinentia-pigmenti.fr/]), the Italian Incontinentia Pigmenti ASSociation (IPASSI, [http://www.incontinentiapigmenti.it/]), DHITECH, Progetto Formazione PON n°01-02342 for the fellowship to M.I.C and the Basilicata Innovazione [http://www.basilicatainnovazione.it] for supporting M.P.
Footnotes
Conflict of interest
The authors declare no conflict of interests.
References
- 1.Hayden MS, Ghosh S. NF-κB in immunobiology. Cell Res. 2011;21:223–244. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh S, Hayden MS. Celebrating 25 years of NF-κB research. Immunol Rev. 2012;246:5–13. doi: 10.1111/j.1600-065X.2012.01111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun SC. The noncanonical NF-κB pathway. Immunol Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinz M, Arslan SÇ, Scheidereit C. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol Rev. 2012;246:59–76. doi: 10.1111/j.1600-065X.2012.01102.x. [DOI] [PubMed] [Google Scholar]
- 6.Huang B, Yang XD, Lamb A, Chen LF. Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway. Cell Signal. 2010;22:1282–1290. doi: 10.1016/j.cellsig.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ea CK, Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc Natl Acad Sci U S A. 2009;106:18972–18977. doi: 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162. doi: 10.3389/fimmu.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Courtois G, Smahi A, Reichenbach J, Döffinger R, Cancrini C, Bonnet M, et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest. 2003;112:1108–1115. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohnishi H, Miyata R, Suzuki T, Nose T, Kubota K, Kato Z, et al. A rapid screening method to detect autosomal-dominant ectodermal dysplasia with immune deficiency syndrome. J Allergy Clin Immunol. 2012;129:578–580. doi: 10.1016/j.jaci.2011.09.042. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Granados E, Keenan JE, Kinney MC, Leo H, Jain N, Ma CA, et al. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat. 2008;29:861–868. doi: 10.1002/humu.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald DR, Mooster JL, Reddy M, Bawle E, Secord E, Geha RS. Heterozygous N-terminal deletion of IkappaBalpha results in functional nuclear factor kappaB haploinsufficiency, ectodermal dysplasia, and immune deficiency. J Allergy Clin Immunol. 2007;120:900–907. doi: 10.1016/j.jaci.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 13.Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M, van Dongen J, et al. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes. J Exp Med. 2004;200:559–568. doi: 10.1084/jem.20040773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puel A, Yang K, Ku CL, von Bernuth H, Bustamante J, Santos OF, et al. Heritable defects of the human TLR signalling pathways. J Endotoxin Res. 2005;11:220–224. doi: 10.1179/096805105X37367. [DOI] [PubMed] [Google Scholar]
- 15.Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin Microbiol Rev. 2011;24:490–497. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawai T, Nishikomori R, Heike T. Diagnosis and treatment in anhidrotic ectodermal dysplasia with immunodeficiency. Allergol Int. 2012;61:207–217. doi: 10.2332/allergolint.12-RAI-0446. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham-Rundles C, Ponda PP. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol. 2005;5:880–892. doi: 10.1038/nri1713. [DOI] [PubMed] [Google Scholar]
- 18.Yoshioka T, Nishikomori R, Hara J, Okada K, Hashii Y, Okafuji I, et al. Autosomal Dominant Anhidrotic Ectodermal Dysplasia with Immunodeficiency Caused by a Novel NFKBIA Mutation, p.Ser36Tyr, Presents with Mild Ectodermal Dysplasia and Non-Infectious Systemic Inflammation. J Clin Immunol. 2013;33:1165–1174. doi: 10.1007/s10875-013-9924-z. [DOI] [PubMed] [Google Scholar]
- 19.Schimke LF, Rieber N, Rylaarsdam S, Cabral-Marques O, Hubbard N, Puel A, et al. A novel gain-of-function IKBA mutation underlies ectodermal dysplasia with immunodeficiency and polyendocrinopathy. J Clin Immunol. 2013;33:1088–1099. doi: 10.1007/s10875-013-9906-1. [DOI] [PubMed] [Google Scholar]
- 20.Nishikomori R, Akutagawa H, Maruyama K, Nakata-Hizume M, Ohmori K, Mizuno K, et al. X-linked ectodermal dysplasia and immunodeficiency caused by reversion mosaicism of NEMO reveals a critical role for NEMO in human T-cell development and/or survival. Blood. 2004;103:4565–4572. doi: 10.1182/blood-2003-10-3655. [DOI] [PubMed] [Google Scholar]
- 21.Dupuis-Girod S, Cancrini C, Le Deist F, Palma P, Bodemer C, Puel A, et al. Successful allogeneic hemopoietic stem cell transplantation in a child who had anhidrotic ectodermal dysplasia with immunodeficiency. Pediatrics. 2006;118:e205–11. doi: 10.1542/peds.2005-2661. [DOI] [PubMed] [Google Scholar]
- 22.Mancini AJ, Lawley LP, Uzel G. X-linked ectodermal dysplasia with immunodeficiency caused by NEMO mutation: early recognition and diagnosis. Arch Dermatol. 2008;144:342–346. doi: 10.1001/archderm.144.3.342. [DOI] [PubMed] [Google Scholar]
- 23.Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–1177. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) Am J Hum Genet. 2000;67:1555–1562. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doffinger R, Smahi A, Bessia C, Feinberg J, Durandy A, Bodemer C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 26.Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin Microbiol Rev. 2011;26:490–497. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puel A, Picard C, Ku CL, et al. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol. 2004;16:34–41. doi: 10.1016/j.coi.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 28.Fusco F, Pescatore A, Steffann J, Royer G, Bonnefont JP, Ursini MV. Clinical Utility Gene Card for: incontinentia pigmenti. Eur J Hum Genet. 2013;21 doi: 10.1038/ejhg.2012.227. doi: 10.1038/ejhg.2012.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fusco F, Fimiani G, Tadini G, Michele D, Ursini MV. Clinical diagnosis of incontinentia pigmenti in a cohort of male patients. J Am Acad Dermatol. 2007;56:264–267. doi: 10.1016/j.jaad.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Cordier F, Vinolo E, Veron M, Delepierre M, Agou F. Solution structure of NEMO zinc finger and impact of an anhidrotic ectodermal dysplasia with immunodeficiency-related point mutation. J Mol Biol. 2008;377:1419–1432. doi: 10.1016/j.jmb.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 31.Zeng W, Xu M, Liu S, Sun L, Chen ZJ. Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol Cell. 2009;36:315–325. doi: 10.1016/j.molcel.2009.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Véron M, et al. NEMO specifically recognizes K63-linked polyubiquitin chains through a new bipartite ubiquitin binding domain. EMBO J. 2009;28:2885–2895. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 34.Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Cordier F, Grubisha O, Traincard F, Véron M, Delepierre M, Agou F. The zinc finger of NEMO is a functional ubiquitin-binding domain. J Biol Chem. 2009;284:2902–2907. doi: 10.1074/jbc.M806655200. [DOI] [PubMed] [Google Scholar]
- 36.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Vinolo E, Sebban H, Chaffotte A, Israël A, Courtois G, Véron M, et al. A point mutation in NEMO associated with anhidrotic ectodermal dysplasia with immunodeficiency pathology results in destabilization of the oligomer and reduces lipopolysaccharide- and tumor necrosis factor-mediated NF-kB activation. J Biol Chem. 2006;281:6334–6348. doi: 10.1074/jbc.M510118200. [DOI] [PubMed] [Google Scholar]
- 38.Hubeau M, Ngadjeua F, Puel A, Israel L, Feinberg J, Chrabieh M, et al. New mechanism of X-linked anhidrotic ectodermal dysplasia with immunodeficiency: impairment of ubiquitin binding despite normal folding of NEMO protein. Blood. 2011;118:926–935. doi: 10.1182/blood-2010-10-315234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–1759. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, et al. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Curr Opin Immunol. 2008;20:39–48. doi: 10.1016/j.coi.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 41.Lo YC, Lin SC, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, et al. Structural basis for recognition of diubiquitins by NEMO. Mol Cell. 2009;33:602–615. doi: 10.1016/j.molcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Temmerman ST, Ma CA, Borges L, Kubin M, Liu S, Derry JM, et al. Impaired dendritic-cell function in ectodermal dysplasia with immune deficiency is linked to defective NEMO ubiquitination. Blood. 2006;108:2324–2331. doi: 10.1182/blood-2006-04-017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pannicke U, Baumann B, Fuchs S, Henneke P, Rensing-Ehl A, Rizzi M, et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N Engl J Med. 2013;369:2504–2514. doi: 10.1056/NEJMoa1309199. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki N, Saito T. IRAK-4 a shared NF-kappaB activator in innate and acquired immunity. Trends Immunol. 2006;27:566–572. doi: 10.1016/j.it.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H, et al. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J Exp Med. 2007;204:1013–1024. doi: 10.1084/jem.20061523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picard C, von Bernuth H, Ku CL, Yamamoto M, Matsui K, Kato H, et al. Inherited human IRAK-4 deficiency: an update. Immunol Res. 2007;38:347–352. doi: 10.1007/s12026-007-0006-2. [DOI] [PubMed] [Google Scholar]
- 47.Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 48.Picard C, Puel A, Bustamante J, Ku CL, Casanova JL. Primary immunodeficiencies associated with pneumococcal disease. Curr Opin Allergy Clin Immunol. 2003;3:451–459. doi: 10.1097/00130832-200312000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Hoarau C, Gérard B, Lescanne E, Henry D, François S, Lacapère JJ, et al. TLR9 activation induces normal neutrophil responses in a child with IRAK-4 deficiency: involvement of the direct PI3K pathway. J Immunol. 2007;179:4754–4765. doi: 10.4049/jimmunol.179.7.4754. [DOI] [PubMed] [Google Scholar]
- 50.Takada H, Yoshikawa H, Imaizumi M, Kitamura T, Takeyama J, Kumaki S, et al. Delayed separation of the umbilical cord in two siblings with interleukin-1 receptor-associated kinase 4 deficiency: rapid screening by flow cytometer. J Pediat. 2006;148:546–548. doi: 10.1016/j.jpeds.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 51.Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204:2407–2422. doi: 10.1084/jem.20070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Enders A, Pannicke U, Berner R, Henneke P, Radlinger K, Schwarz K, et al. Two siblings with lethal pneumococcal meningitis in a family with a mutation in interleukin-1 receptor-associated kinase 4. J Pediatr. 2004;145:698–700. doi: 10.1016/j.jpeds.2004.06.065. [DOI] [PubMed] [Google Scholar]
- 53.Medvedev AE, Lentschat A, Kuhns DB, Blanco JC, Salkowski C, Zhang S, et al. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J Exp Med. 2003;198:521–531. doi: 10.1084/jem.20030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuhns DB, Long Priel DA, Gallin JI. Endotoxin and IL-1 hyporesponsiveness in a patient with recurrent bacterial infections. J Immunol. 1997;158:3959–3964. [PubMed] [Google Scholar]
- 55.Cardenes M, von Bernuth H, Garcia-Saavedra A, Santiago E, Puel A, Ku CL, et al. Autosomal recessive interleukin-1 receptor-associated kinase 4 deficiency in fourth-degree relatives. J Pediatr. 2006;148:549–551. doi: 10.1016/j.jpeds.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 56.Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci USA. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 58.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–696. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.von Bernuth H, Picard C, Puel A, Casanova JL. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunol. 2012;42:3126–3135. doi: 10.1002/eji.201242683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaschignard J, Levy C, Chrabieh M, Boisson B, Bost-Bru C, Dauger S, et al. Invasive pneumococcal disease in children can reveal a primary immunodeficiency. Clin Infect Dis. 2014;59:244–251. doi: 10.1093/cid/ciu274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sancho-Shimizu V, Pérez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest. 2011;121:4889–4902. doi: 10.1172/JCI59259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33:400–411. doi: 10.1016/j.immuni.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13:1178–1186. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen K, Coonrod EM, Kumanovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-kappa-B pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 2013;93:812–824. doi: 10.1016/j.ajhg.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MA. Novel NFKB2 mutation in early-onset CVID. J. Clin. Immun. 2014;34:686–690. doi: 10.1007/s10875-014-0064-x. [DOI] [PubMed] [Google Scholar]